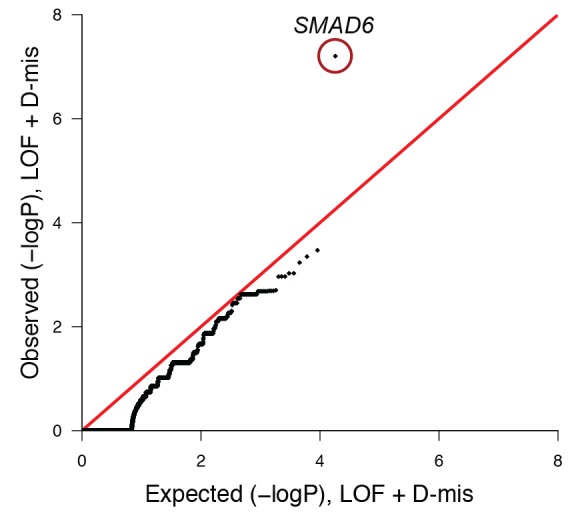
Figure 3. Quantile-quantile plots of observed versus expected p-values comparing the burden of rare LOF and damaging (LOF + D-mis) variants in protein-coding genes in craniosynostosis cases.
Rare (allele frequency <2 × 10–5 in the ExAC03 database) loss of function (LOF) and damaging missense (D-mis) variants were identified in 191 probands. The probability of the observed number of variants in each gene occurring by chance was calculated from the total number of observed variants and the length of the coding region of each gene using the binomial test. The distribution of observed P-values compared to the expected distribution is shown. (a) Q-Q plot for rare LOF variants in each gene from a total of 1135 LOF variants identified in probands. The distribution of observed p-values closely conforms to expectation with the exception of SMAD6, which shows p=1.1 × 10–15 and 156-fold enrichment in cases. (b) Q-Q plot for rare damaging (LOF + D-mis) variants in each gene from a total of 3156 damaging variants in probands. Again, SMAD6 deviates greatly from the expected distribution, with p<10–20 and 91-fold enrichment.
DOI: http://dx.doi.org/10.7554/eLife.20125.012

Figure 3—figure supplement 1. Quantile-quantile plots comparing all transmitted, damaging variants in protein-coding genes in 191 probands with midline craniosynostosis to the expected binomial distribution.

Figure 3—figure supplement 2. Principal-component analysis of 191 probands and 3337 European autism controls.

Figure 3—figure supplement 3. Quantile-quantile plot of observed versus expected p-values comparing the burden of damaging (LOF + D-mis) variants in protein-coding genes in craniosynostosis cases and controls.