

Abstract
MOF (males absent on the first) was initially identified as a dosage compensation factor in Drosophila that acetylates lysine 16 of histone H4 (H4K16ac) and increased gene transcription from the single copy male X-chromosome. In humans, however, the ortholog of Drosophila MOF has been shown to interact with a range of proteins that extend its potential significance well beyond transcription. For example, recent results indicate MOF is an upstream regulator of the ATM (ataxia-telangiectasia mutated) protein, the loss of which is responsible for ataxia telangiectasia (AT). ATM is a key regulatory kinase that interacts with and phosphorylates multiple substrates that influence critical, cell-cycle control and DNA damage repair pathways in addition to other pathways. Thus, directly or indirectly, MOF may be involved in a wide range of cellular functions. This review will focus on the contribution of MOF to cellular DNA repair and new results that are beginning to examine the in vivo physiological role of MOF.
Keywords: MOF, DNA DSB, H4K16ac, NHEJ, HR, Oncogenesis
Introduction
MOF was originally identified in Drosophila as a component of the protein complexes known as male-specific lethal (MSL) (1, 2), which is only found in male flies, and the non-specific lethal (NSL) complex (3–5), which is found in both male and female flies. In Drosophila, MOF was initially found to function in dosage compensation whereby transcription of genes on the single male X-chromosome must be increased two-fold relative to females who have two X-chromosomes. This is achieved by MOF mediated acetylation of histone H4 Lys16 (H4K16ac), which alters chromatin-protein interactions as well as the local chromatin structure (6, 7). MOF is evolutionally conserved in mammals (8, 9) and is associated with transcription regulation and DNA damage repair as well as many additional cellular functions (10–14). Most immortalized human cell lines, whether derived from tumor or normal tissue, contain MOF levels either similar to or elevated above those found in normal cells (10).
There is growing scientific evidence that the accumulative DNA damage resulting from defective DNA repair can lead to neurological disorders such as Ataxia telangiectasia, Alzheimer’s disease, Nijmegen breakage syndrome etc. Recently it has been realized that histone modifications play an important and primary role in DNA damage response (DDR) by facilitating repair protein access to DNA breaks (15–18). Most interestingly, several studies indicate that pre-existing histone modifications play an important role in the recruitment of repair proteins. Schizosaccharomyces pombe Crb2 [SpCrb2] and Saccharomyces cerevisiae Rad9 [ScRad9]) require methylated histone H3 Lys79 (H3K79me) (19) or methylated histone H4 Lys20 (H4K20me) and/or CBP/p300-mediated acetylation of histone H3 lysine 56 (H3K56ac) (19–23) at DNA-damage sites to form repair protein foci. The aforesaid histone modifications are present on histones and are not altered in response to ionizing radiation (IR)-induced DNA damage. Genotoxic stress in human cells can result in mild increases in histone acetylation; however some studies have reported rapid deacetylation of histone H3 K9 (H3K9ac) and H3K56ac (24). Preexisting modifications can influence DNA damage responses at the level of signaling, chromatin relaxation to facilitate repair protein recruitment and the restoration of chromatin to the original native state (25–27). In human cells, histone modifications such as phosphorylation of H2AX at S139, acetylation of H4K16, methylation of H3K79 and H4K20 have been linked with damage signaling; H3K9, H4K16 and H2A(X) are linked with chromatin opening and modifications like phosphorylation of H4S1 and H2B S14, dephosphorylation of H2AX S139, acetylation of H3 K14, K23, K56 and H4 K5, K8, K12, K16, K91, deacetylation of H3/H4 Ks and H2A K119 (17, 18, 27). Here we will discuss mainly two of the many aspects of MOF: MOF function in DNA DSB repair and the physiological consequences of defective MOF expression.
MOF dependent genome-wide distribution of histone H4 Lysine16 acetylation sites
Histone post-translational modifications are critical determinants of chromatin structure, impacting multiple cellular processes including DNA transcription, replication, and repair (28). Acetylation of histone H4 at lysine 16 (H4K16ac) was initially identified in association with dosage compensation of the Drosophila male X-chromosome, which does not occur in mammals, Genome-wide mapping of the H4K16ac distribution in human cells identified 25,893 DNA regions, average length of 692 nucleotides, with elevated levels. Interestingly, although a majority of the sites localized within genes, only a relatively small fraction (~10%) was found near promoters (29). These studies revealed that H4K16 acetylation has a limited effect on transcription regulation in HEK293 cells, whereas H4K16ac has been demonstrated to have critical roles in regulating transcription in mouse embryonic stem cells (30). Thus, H4K16ac-dependent transcriptional regulation is likely subject to additional cell type specific controls or the distribution also reflects other functions like maintenance of genomic stability.
Role of MOF in DNA Damage Response (DDR)
MOF was identified as an ATM interacting partner by yeast two-hybrid screening; subsequently the interaction was verified by immunoprecipitation and its role in DDR reported by several investigators (10–13, 31–33). The role of MOF in genomic stability is highly conserved as its depletion leads to ionizing radiation sensitivity and defective DNA damage repair in Drosophila (14), mice (10, 12) and human cell lines (11, 13). Cell death results from the failure of IR-induced ATM activation, which is consistent with the observation that in human cells depleted of MOF, ATM dependent phosphorylation of H2AX is abrogated (33). A similar defective response was reported in mMof knock out mouse embryonic fibroblasts (10). Further linkage between H4K16 acetylation status and IR-induced H2AX phosphorylation was obtained by blocking deacetylation of H4K16ac, with either a drug inhibitor or by a gene knockout approach. Both approaches revealed that blocking H4K16ac deacetylation rescued the IR-induced phosphorylation of H2AX (33), supporting the argument that H4K16 acetylation is critical for the initial DNA damage response. Consistent with the importance of acetylation in DDR, Dobbin and coworkers have reported that the NAD(+)-dependent deacetylase SIRT1 is recruited to DSBs in post mitotic neurons (34) and that SIRT1 recruitment to breaks was ATM dependent. SIRT1, thus, seems to function as an apical transducer of the DSB response and may offer an important therapeutic avenue for treating neurodegeneration diseases (34).
Gupta and coworkers reported altered growth characteristics of haploinsufficient mMof in mouse embryonic fibroblast (MEF) cells that correlated with increased genomic instability (10). This is consistent with results in erythrocytes from mMof heterozygous null mice where a modestly increased ratio of normochromatic to polychromatic erythrocytes and an increased frequency of micronucleus were detected in comparison to wild type mice. The higher spontaneous chromosome aberration rate observed in cultured MEFs and in phytohemagglutinin-stimulated lymphocytes isolated from mMof heterozygous null mice are similarly indicative of MOF related genomic instability. A reduced level of IR-induced γ-H2AX focus appearance was also observed in heterozygous mMof cells compared to wild-type mMof cells. As expected, reduced levels of mMof increased genomic instability and decreased survival after IR or mitomycin C exposure, while total loss resulted in cell lethality in MEF cells (10).
Role of MOF in DNA repair by non-homologous end joining (NHEJ)
Inactivation of MOF has been reported to increase basal DNA damage levels as well as lead to higher residual post-irradiation DNA damage (11–13, 31, 32). Spontaneous DNA damage in MOF depleted cells could be attributed to lack of acetylation at H4K16 as well as some other functions where MOF plays a role in transcription (34). Since NHEJ mediated DNA damage repair is most prevalent in G0, G1 cells, depletion of MOF results higher residual chromosome damage in cells irradiated in G1 phase and analyzed at metaphase (11). ATM at T392 post-translationally phosphorylates MOF upon induction of DNA damage but this modification has no effect on DNA DSB repair by the NHEJ pathway, because the phosphorylation-deficient MOF mutant (MOF-T392A) had no impact on the repair of DNA DSB in G1 phase cells (11). It is very interesting that loss of MOF affects DNA repair in G1 phase cells (13), and that subsequent expression of mutant MOF-T392A rescues IR-induced DNA DSB repair in G1 phase cells, as this supports the argument that MOF, but not ATM dependent MOF phosphorylation at T-392, contributes to G1 specific DNA DSB repair (11). Sharma and coworkers have reported that H4K16ac is critical for DNA damage response (33). Interestingly, Gupta and coworkers found that mutant MOF retains the acetylation activity, thus it is obviously clear that acetylation activity of mutant MOF facilitates the initial DNA damage response as there was no major difference in the initial appearance of phosphorylated H2AX in wild type or mutant MOF post irradiation (11). The reason for mutant MOF having least effect on NHEJ could be because of its interaction with 53BP1, which is involved in NHEJ and suppressing HR. Mutant MOF (MOF-T392A) shows an increased interaction with 53BP1 post irradiation, thus resulting in the shift of the HR to NHEJ. Thus it is obvious that MOF phosphorylation does not impact on DNA DSB repair by HR to NHEJ in G0/G1 cells; however, it is essential for the repair of DNA DSB by HR in S/G2 cells. Although Li and coworkers reported that MOF depletion results in loss of 53BP1 as well as BRCA1 foci formation (32), expression of mutant MOF-T392A had no effect on IR-induced 53BP1 foci formation, but reduced frequency of BRCA1 foci formation (11). These results suggest a model in which phosphorylated MOF may be regulating repair pathway choice for DNA DSBs.
Role of MOF in DNA repair by homologous recombination
Gupta and coworkers reported that mutant MOF-T392A expression did not affect IR-induced γ-H2AX foci appearance, but delayed the disappearance of pS1981-ATM foci post-irradiation (11). However, mutant MOF (MOF-T392A) expression has been reported to affect DNA DSB repair in S and G2 phase cells, where homologous recombination is up-regulated. It is possible that post-translational modification of MOF by ATM may enhance the recruitment of HR related proteins to DSB sites in S- and G2-phase cells. ATM has been reported to be activated by DNA damage in all phases of the cell cycle (35) and has an essential role in DNA DSB repair in all cell cycle phases (36, 37). Since cells deficient in ATM are defective for DNA DSB repair in all phases, the role of MOF phosphorylation at T392 in the recruitment of DSB repair proteins appears to be specific to the homologous recombination pathway. Repair of DNA damage by homologous recombination repair is suppressed by 53BP1 and consistent with this observation there is an increased association of wild-type MOF with 53BP1 immediately post irradiation that subsequently decreases thereby allowing recruitment of HR related proteins (Figure 1).
Figure 1. Role of MOF phosphorylation in regulating DSB repair pathway choice.
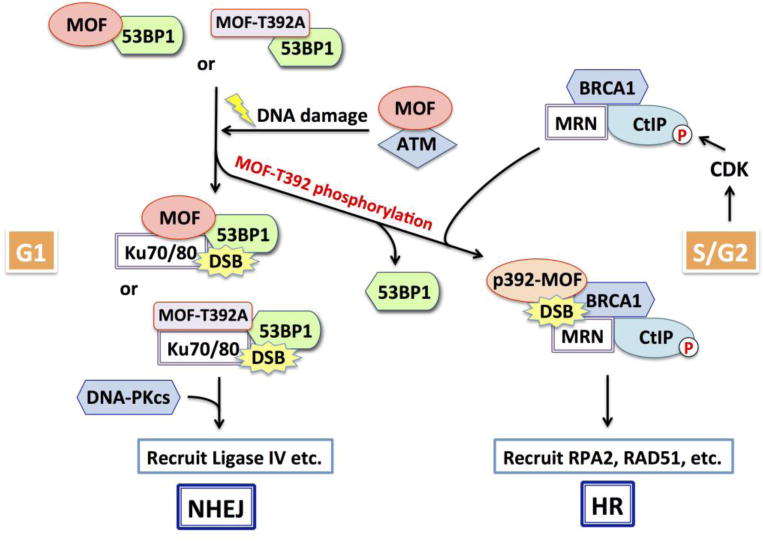
Cell-cycle phase is a critical determinant of the choice between DNA double strand break (DSB) repair by nonhomologous end-joining (NHEJ) or homologous recombination (HR). DSBs induce ATM-dependent MOF phosphorylation at T392. MOF-pT392 allows release of 53BP1 from DSB sites recruiting BRCA1 complex to initiate repair by HR. Unphosphorylated MOF or phosphorylation-deficient mutant MOF (MOF-T392A) impedes DNA repair by HR in S and G2 phases by blocking the release of DSB-associated 53BP1, resulting in enhanced 53BP1 retention and reduced BRCA1 association. In sum, ATM-mediated MOF-T392 phosphorylation modulates 53BP1 function to facilitate the subsequent recruitment of HR repair proteins thereby regulating DSB repair pathway choice during S and G2 phases.
In vivo role of MOF in post-mitotic and non-dividing cell survival
The majority of cells in mammals, except stem cells, are not actively proliferating, meaning that cultured cell lines are not exact models for tissue function. Kumar and coworkers used a tissue-specific gene deletion approach to test the role of MOF in non-proliferating, post-mitotic Purkinje cells and found that in the absence of MOF, Purkinje cells died by 50–60 days post birth, resulting in the mice developing motor defects similar to those seen in Ataxia Telangiectasia patients (38). However, T-cell-specific Mof deletion (Lck-driven Cre expression) resulted in less severe physiological outcomes, manifesting as defective T-cell differentiation and a moderate decline in overall survival (12). Furthermore mice with T-cell specific deletion had defective T-cell differentiation as was evident by an increased DN3 (CD44− CD25+) population (12), the cell stage during which T-cell receptor rearrangement takes place. In contrast, Sheikh and coworkers found that MOF loss in differentiated non-proliferating podocytes had no effect on normal kidney function or morphology in mice followed up to 5–6 months of age (39). However, following kidney injury by Adriamycin (doxorubicin) treatment, pyknotic nuclei and activated caspase-3 were detected in MOF depleted podocytes and an associated decrease in kidney function (39). Thus, in non-proliferating tissue cells the outcome of MOF loss can range from no immediate effect to cell death, depending on the cell type (28).
MOF is critical for embryogenesis
Embryonic stem cells are capable of indefinite self-renewal and differentiation into all cell lineages. Gupta and coworkers reported that ablation of the mouse Mof gene (mMof) by gene targeting resulted in early embryonic lethality and cell death, consistent with a role for MOF in stem cell maintenance (10). Similar studies by Thomas and coworkers (40) demonstrated that MOF is essential for embryonic development past the blastocyst stage. Li and coworkers (30) further supported the argument that MOF is critical for stem cell self renewal and pluripotency by demonstrating that Mof depleted mouse ESCs lose characteristic morphology, alkaline staining, and differentiation potential and such cells have aberrant expression of the Nanog, Oct4, and Sox2. Using genome-wide ChIP-sequencing and transcriptome analyses, Li and coworkers demonstrated that Mof is an integral component of the embryonic stem cell core transcriptional network (30) and also found an interconnection between Mof and H3K4me3 in embryonic stem cells (30). Furthermore, the close interactions between Mof and Wdr5/H3K4me3 contributes to the role of Mof in regulating NOS expression and their regulatory circuitry (30).
Not surprising considering the role of MOF in maintaining ESC, iPSCs contain high levels of MOF mRNA and MOF protein which are dramatically up-regulated following reprogramming (41). MOF overexpression has been reported to improve reprogramming efficiency, thus facilitating formation of iPSCs, and depletion of MOF impairs iPSCs generation (41). This is possibly due to MOF interaction with the H3K4 methyltransferase Wdr5 to promote endogenous Oct4 expression (stem cell factor) during the reprogramming process (41). The loss of MOF also results in autophagy as it is coupled to the reduction of H4K16ac, which is associated with the down regulation of autophagy-related genes (42). Thus MOF, through its production of H4K16ac, has multiple functions in dosage compensation dependent or independent transcription and the DNA damage response, two important factors for cell survival.
Highlights.
MOF acetylates lysine 16 of histone H4 (H4K16ac) and plays a role in transcription and the DNA damage response.
MOF interacts with a range of proteins that extend its potential significance well beyond transcription and DNA damage repair.
MOF affects ATM (ataxia-telangiectasia mutated) function and ATM-dependent MOF post-translational modification regulates DNA DSB pathway choice.
MOF is essential for embryonic development as well as post-mitotic and non-dividing cell survival.
MOF is critical for stem cell-renewal and pluripotency.
Acknowledgments
Research in the authors’ laboratory is supported by NIH grants (CA129537, CA154320 and GM109768).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
COI: Authors declare that they have no conflict of interest.
References
- 1.Laverty C, Lucci J, Akhtar A. The MSL complex: X chromosome and beyond. Curr Opin Genet Dev. 2010;20(2):171–8. doi: 10.1016/j.gde.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 2.Lucchesi JC, Kuroda MI. Dosage compensation in Drosophila. Cold Spring Harbor perspectives in biology. 2015;7(5) doi: 10.1101/cshperspect.a019398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cai Y, Jin J, Swanson SK, Cole MD, Choi SH, Florens L, et al. Subunit composition and substrate specificity of a MOF-containing histone acetyltransferase distinct from the male-specific lethal (MSL) complex. J Biol Chem. 2010;285(7):4268–72. doi: 10.1074/jbc.C109.087981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mendjan S, Taipale M, Kind J, Holz H, Gebhardt P, Schelder M, et al. Nuclear pore components are involved in the transcriptional regulation of dosage compensation in Drosophila. Mol Cell. 2006;21(6):811–23. doi: 10.1016/j.molcel.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 5.Raja SJ, Charapitsa I, Conrad T, Vaquerizas JM, Gebhardt P, Holz H, et al. The nonspecific lethal complex is a transcriptional regulator in Drosophila. Mol Cell. 2010;38(6):827–41. doi: 10.1016/j.molcel.2010.05.021. [DOI] [PubMed] [Google Scholar]
- 6.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311(5762):844–7. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 7.Pandita TK. Histone H4 lysine 16 acetylated isoform synthesis opens new route to biophysical studies. Proteomics. 2013;13(10–11):1546–7. doi: 10.1002/pmic.201300145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rea S, Xouri G, Akhtar A. Males absent on the first (MOF): from flies to humans. Oncogene. 2007;26(37):5385–94. doi: 10.1038/sj.onc.1210607. [DOI] [PubMed] [Google Scholar]
- 9.Neal KC, Pannuti A, Smith ER, Lucchesi JC. A new human member of the MYST family of histone acetyl transferases with high sequence similarity to Drosophila MOF. Biochim Biophys Acta. 2000;1490(1–2):170–4. doi: 10.1016/s0167-4781(99)00211-0. [DOI] [PubMed] [Google Scholar]
- 10.Gupta A, Guerin-Peyrou TG, Sharma GG, Park C, Agarwal M, Ganju RK, et al. The mammalian ortholog of Drosophila MOF that acetylates histone H4 lysine 16 is essential for embryogenesis and oncogenesis. Mol Cell Biol. 2008;28(1):397–409. doi: 10.1128/MCB.01045-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gupta A, Hunt CR, Hegde ML, Chakraborty S, Udayakumar D, Horikoshi N, et al. MOF Phosphorylation by ATM Regulates 53BP1-Mediated Double-Strand Break Repair Pathway Choice. Cell reports. 2014;8(1):177–89. doi: 10.1016/j.celrep.2014.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gupta A, Hunt CR, Pandita RK, Pae J, Komal K, Singh M, et al. T-cell-specific deletion of Mof blocks their differentiation and results in genomic instability in mice. Mutagenesis. 2013;28(3):263–70. doi: 10.1093/mutage/ges080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gupta A, Sharma GG, Young CS, Agarwal M, Smith ER, Paull TT, et al. Involvement of human MOF in ATM function. Mol Cell Biol. 2005;25(12):5292–305. doi: 10.1128/MCB.25.12.5292-5305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhadra MP, Horikoshi N, Pushpavallipvalli SN, Sarkar A, Bag I, Krishnan A, et al. The role of MOF in the ionizing radiation response is conserved in Drosophila melanogaster. Chromosoma. 2012;121(1):79–90. doi: 10.1007/s00412-011-0344-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pandita TK, Richardson C. Chromatin remodeling finds its place in the DNA double-strand break response. Nucleic Acids Res. 2009;37(5):1363–77. doi: 10.1093/nar/gkn1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Misri S, Pandita S, Kumar R, Pandita TK. Telomeres, histone code, and DNA damage response. Cytogenet Genome Res. 2008;122(3–4):297–307. doi: 10.1159/000167816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Attikum H, Gasser SM. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 2009;19(5):207–17. doi: 10.1016/j.tcb.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 18.van Attikum H, Gasser SM. The histone code at DNA breaks: a guide to repair? Nat Rev Mol Cell Biol. 2005;6(10):757–65. doi: 10.1038/nrm1737. [DOI] [PubMed] [Google Scholar]
- 19.Huyen Y, Zgheib O, Ditullio RA, Jr, Gorgoulis VG, Zacharatos P, Petty TJ, et al. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 2004;432(7015):406–11. doi: 10.1038/nature03114. [DOI] [PubMed] [Google Scholar]
- 20.Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, et al. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127(7):1361–73. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Das C, Lucia MS, Hansen KC, Tyler JK. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature. 2009;459(7243):113–7. doi: 10.1038/nature07861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanders SL, Portoso M, Mata J, Bahler J, Allshire RC, Kouzarides T. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell. 2004;119(5):603–14. doi: 10.1016/j.cell.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 23.Wysocka J, Swigut T, Milne TA, Dou Y, Zhang X, Burlingame AL, et al. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell. 2005;121(6):859–72. doi: 10.1016/j.cell.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 24.Tjeertes JV, Miller KM, Jackson SP. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J. 2009;28(13):1878–89. doi: 10.1038/emboj.2009.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar R, Horikoshi N, Singh M, Gupta A, Misra HS, Albuquerque K, et al. Chromatin modifications and the DNA damage response to ionizing radiation. Front Oncol. 2012;2:214. doi: 10.3389/fonc.2012.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gupta A, Hunt CR, Chakraborty S, Pandita RK, Yordy J, Ramnarain DB, et al. Role of 53BP1 in the Regulation of DNA Double-Strand Break Repair Pathway Choice. Radiation research. 2014;181(1):1–8. doi: 10.1667/RR13572.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hunt CR, Ramnarain D, Horikoshi N, Iyengar P, Pandita RK, Shay JW, et al. Histone modifications and DNA double-strand break repair after exposure to ionizing radiations. Radiation research. 2013;179(4):383–92. doi: 10.1667/RR3308.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horikoshi N, Hunt CR, Pandita TK. More complex transcriptional regulation and stress response by MOF. Oncogene. 2015 doi: 10.1038/onc.2015.373. [DOI] [PubMed] [Google Scholar]
- 29.Horikoshi N, Kumar P, Sharma GG, Chen M, Hunt CR, Westover K, et al. Genome-wide distribution of histone H4 Lysine 16 acetylation sites and their relationship to gene expression. Genome Integr. 2013;4(1):3. doi: 10.1186/2041-9414-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X, Li L, Pandey R, Byun JS, Gardner K, Qin Z, et al. The histone acetyltransferase MOF is a key regulator of the embryonic stem cell core transcriptional network. Cell Stem Cell. 2012;11(2):163–78. doi: 10.1016/j.stem.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taipale M, Rea S, Richter K, Vilar A, Lichter P, Imhof A, et al. hMOF histone acetyltransferase is required for histone H4 lysine 16 acetylation in mammalian cells. Mol Cell Biol. 2005;25(15):6798–810. doi: 10.1128/MCB.25.15.6798-6810.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li X, Corsa CA, Pan PW, Wu L, Ferguson D, Yu X, et al. MOF and H4 K16 acetylation play important roles in DNA damage repair by modulating recruitment of DNA damage repair protein Mdc1. Mol Cell Biol. 2010;30(22):5335–47. doi: 10.1128/MCB.00350-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharma GG, So S, Gupta A, Kumar R, Cayrou C, Avvakumov N, et al. MOF and histone H4 acetylation at lysine 16 are critical for DNA damage response and double-strand break repair. Mol Cell Biol. 2010;30(14):3582–95. doi: 10.1128/MCB.01476-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dobbin MM, Madabhushi R, Pan L, Chen Y, Kim D, Gao J, et al. SIRT1 collaborates with ATM and HDAC1 to maintain genomic stability in neurons. Nat Neurosci. 2013;16(8):1008–15. doi: 10.1038/nn.3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pandita TK, Lieberman HB, Lim DS, Dhar S, Zheng W, Taya Y, et al. Ionizing radiation activates the ATM kinase throughout the cell cycle. Oncogene. 2000;19(11):1386–91. doi: 10.1038/sj.onc.1203444. [DOI] [PubMed] [Google Scholar]
- 36.Pandita TK, Hittelman WN. The contribution of DNA and chromosome repair deficiencies to the radiosensitivity of ataxia-telangiectasia. Radiat Res. 1992;131(2):214–23. [PubMed] [Google Scholar]
- 37.Pandita TK, Hittelman WN. Initial chromosome damage but not DNA damage is greater in ataxia telangiectasia cells. Radiat Res. 1992;130(1):94–103. [PubMed] [Google Scholar]
- 38.Kumar R, Hunt CR, Gupta A, Nannepaga S, Pandita RK, Shay JW, et al. Purkinje cell-specific males absent on the first (mMof) gene deletion results in an ataxia-telangiectasia-like neurological phenotype and backward walking in mice. Proc Natl Acad Sci U S A. 2011;108(9):3636–41. doi: 10.1073/pnas.1016524108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheikh BNB-W W, Lucci J, Karpiuk O, Hild I, Hartleben B, Vornweg J, Helmstädter M, Sahyoun AH, Bhardwaj V, Stehle T, Diehl S, Kretz O, Voss AK, Thomas T, Manke T, Huber TB, Akhtar A. MOF maintains Transcriptional Programs regulating Cellular Stress Response. Oncogene. 2015 doi: 10.1038/onc.2015.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomas T, Dixon MP, Kueh AJ, Voss AK. Mof (MYST1 or KAT8) is essential for progression of embryonic development past the blastocyst stage and required for normal chromatin architecture. Mol Cell Biol. 2008;28(16):5093–105. doi: 10.1128/MCB.02202-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mu X, Yan S, Fu C, Wei A. The Histone Acetyltransferase MOF Promotes Induces Generation of Pluripotent Stem Cells. Cell Reprogram. 2015;17(4):259–67. doi: 10.1089/cell.2014.0102. [DOI] [PubMed] [Google Scholar]
- 42.Fullgrabe J, Lynch-Day MA, Heldring N, Li W, Struijk RB, Ma Q, et al. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature. 2013;500(7463):468–71. doi: 10.1038/nature12313. [DOI] [PMC free article] [PubMed] [Google Scholar]