

Abstract
Many studies have demonstrated that depressed myofilament Ca2+ sensitivity is common to dilated cardiomyopathy (DCM) in humans. However, it remains unclear whether a single determinant—such as myofilament Ca2+ sensitivity—is sufficient to characterize all cases of DCM because the severity of disease varies widely with a given mutation. Because dynamic features dominate in the heart muscle, alterations in dynamic contractile parameters may offer better insight on the molecular mechanisms that underlie disparate effects of DCM mutations on cardiac phenotypes. Dynamic features are dominated by myofilament cooperativity that stem from different sources. One such source is the strong tropomyosin binding region in troponin T (TnT), which is known to modulate crossbridge (XB) recruitment dynamics in a myosin heavy chain (MHC)-dependent manner. Therefore, we hypothesized that the effects of DCM-linked mutations in TnT on contractile dynamics would be differently modulated by α- and β-MHC. After reconstitution with the mouse TnT equivalent (TnTR134W) of the human DCM mutation (R131W), we measured dynamic contractile parameters in detergent-skinned cardiac muscle fiber bundles from normal (α-MHC) and transgenic mice (β-MHC). TnTR134W significantly attenuated the rate constants of tension redevelopment, XB recruitment dynamics, XB distortion dynamics, and the magnitude of length-mediated XB recruitment only in α-MHC fiber bundles. TnTR134W decreased myofilament Ca2+ sensitivity to a greater extent in α-MHC (0.14 pCa units) than in β-MHC fiber bundles (0.08 pCa units). Thus, our data demonstrate that TnTR134W induces a more severe DCM-like contractile phenotype against α-MHC than against β-MHC background.
Keywords: dilated cardiomyopathy, Troponin T, myosin heavy chain, contractile dynamics, thin filament function, myofilament cooperativity
Introduction
Dilated Cardiomyopathy (DCM), a disease caused by mutations in many sarcomeric proteins, is characterized by systolic dysfunction and ventricular dilatation (Kushner et al., 2006; Hershberger et al., 2009; Willott et al., 2010; Marston, 2011; Lu et al., 2013). In vitro studies of DCM-causing mutations in cardiac Troponin T (TnT) generally correlate depressed myofilament Ca2+ sensitivity to systolic dysfunction (Kushner et al., 2006; Hershberger et al., 2009), with some exceptions (Mirza et al., 2005). Therefore, it remains unclear whether a single determinant, such as myofilament Ca2+ sensitivity, is sufficient to characterize all cases of DCM because the severity of disease varies widely with a given mutation. Proper pumping actions of the heart dictates that—in addition to normal Ca2+ dynamics—both the magnitude and speed of contraction are not only sustained but adjusted properly on a beat-to-beat basis. Because contractile dynamics are strongly dependent on thin filament cooperativity, and such cooperativity is modulated by the central region (CR) of TnT (Schaertl et al., 1995; Tobacman et al., 2002; Gollapudi et al., 2013), mutations in the CR of TnT are expected to affect myofilament activation by modifying dynamic features of cardiac contractile activation. A better assessment of disparate cardiac phenotypes is made possible when studies account for the mutation-mediated effect on dynamic contractile function because dynamic aspects dominate heart function under physiological conditions.
The focus of this study is the DCM-related mutation, R131W (Mogensen et al., 2004), which lies within the CR (residues 80–180) of human TnT. We previously demonstrated that the CR of TnT plays an important role in tuning the dynamics of crossbridge (XB) recruitment in cardiac muscle by modulating cooperative mechanisms within thin filaments (Gollapudi et al., 2013). Such actions likely involve strong CR-Tropomyosin (Tm) interactions that take place near the overlap junction of adjacent Tm dimers (Jackson et al., 1975; Pearlstone and Smillie, 1977; Palm et al., 2001; Hinkle and Tobacman, 2003; Gollapudi et al., 2013). Therefore, the R131W mutation in TnT may perturb CR-Tm interactions to modulate XB recruitment dynamics. Given that the TnT-mediated function is dependent on the myosin heavy chain (MHC) isoform (Ford et al., 2012; Chandra et al., 2015; Gollapudi et al., 2015; Gollapudi and Chandra, 2016), we hypothesized that the effects of DCM-linked mutations in TnT on contractile dynamics would be differently modulated by α- and β-MHC. To better understand the molecular mechanisms that lead to contractile dysfunction, especially in relevance to humans, it is important to consider the differential impact of α- and β-MHC isoforms on contractile dynamics. This is because previous studies have demonstrated that the effects of cardiomyopathy mutations in TnT on steady-state and/or dynamic contractile features are differently modulated by α- and β-MHC isoforms (Ford et al., 2012; Chandra et al., 2015; Gollapudi et al., 2015; Gollapudi and Chandra, 2016).
To test our hypothesis, we generated a recombinant mouse TnT equivalent (TnTR134W) of the human DCM mutation, R131W. Various indices of steady-state and dynamic contractile function were measured in normal (α-MHC) and transgenic mouse (β-MHC) cardiac muscle fiber bundles reconstituted with wild-type (WT) TnT (TnTWT) or TnTR134W. Dynamic contractile features mediated by TnTR134W were altered only in α-MHC fiber bundles, despite desensitization of myofilaments to Ca2+ to a different degree in α- and β-MHC fiber bundles. For instance, TnTR134W attenuated rate constants of tension redevelopment, XB recruitment dynamics, XB distortion dynamics and the magnitude of length-mediated XB recruitment only in α-MHC fiber bundles. We will discuss the correlation between altered contractile dynamics and a more severe DCM-like contractile phenotype against α-MHC than against β-MHC background.
Materials and methods
Animal treatment protocols
3–4 month-old male mice were used in this study. WT C57BL/6N-strain (non-TG, NTG) mice were acquired from Simonsen's laboratories (Gilroy, CA). β-MHC TG mice were a generous gift from Dr. Jil Tardiff, University of Arizona, Tucson, AZ. The generation and characterization of β-MHC TG mice was as previously described (Krenz et al., 2003, 2007; Krenz and Robbins, 2004; Gollapudi et al., 2015). Mice were carefully handled to minimize pain and suffering, as per the established guidelines of the National Academy of Sciences Guide for the Care and Use of Laboratory Animals. All procedures used for the treatment of mice were approved by the board of Washington State University Institutional Animal Care and Use Committee.
Purification of recombinant mouse cardiac Tn subunits
c-myc tagged mouse TnTWT and TnTR134W genes were synthesized (GenScript USA Inc., Piscataway, NJ) after codon optimization for enhanced protein expression. c-myc tagged TnTWT served as the control in our study. Recombinant mouse TnT (TnTWT and TnTR134W), mouse TnI, and mouse TnC were generated and purified, as described previously (Guo et al., 1994; Pan and Johnson, 1996; Chandra et al., 1999; Gollapudi and Chandra, 2012; Mamidi et al., 2013). Recombinant proteins were cloned into the T7 promoter-based pSBETa vector and expressed in BL21*DE3 cells (Novagen, Madison, WI) for protein synthesis. All proteins were purified using ion-exchange chromatography techniques. TnTWT and TnTR134W were purified by anion-exchange chromatography on a DEAE fast Sepharose column (Chandra et al., 1999; Gollapudi and Chandra, 2012; Mamidi et al., 2013; Gollapudi et al., 2015), TnI was purified by cation-exchange chromatography on a CM Sepharose column (Guo et al., 1994; Gollapudi and Chandra, 2012; Mamidi et al., 2013; Gollapudi et al., 2015), and TnC was purified by anion-exchange chromatography on a DE-52 column (Pan and Johnson, 1996; Gollapudi and Chandra, 2012; Mamidi et al., 2013; Gollapudi et al., 2015). All eluted fractions containing pure proteins were pooled and dialyzed extensively against deionized water containing 15 mM β-mercaptoethanol, lyophilized, and stored at −80°C for long-term use.
Preparation of detergent-skinned cardiac muscle fiber bundles and Tn reconstitution
Left ventricular papillary muscle bundles were isolated from deeply-anesthetized (Isoflurane) mice, and further dissected into smaller muscle fiber bundles (~0.15 mm in cross-section and 2.0–2.5 mm in length) in high-relaxing (HR) solution (Chandra et al., 2006, 2007; Gollapudi et al., 2015). The HR solution contained 20 mM 2,3-butanedione monoxime (BDM), 50 mM N,N-bis (2-hydroxyethyl)-2-amino-ethane-sulfonic acid (BES), 20 mM ethylene glycol tetra-acetic acid (EGTA), 6.29 mM magnesium chloride (MgCl2), 6.09 mM disodium hydrate salt of adenosine triphosphate (Na2ATP), 30.83 mM potassium propionate (K-Prop), 10 mM sodium azide (NaN3), 1.0 mM dithiothreitol (DTT), and 4 mM benzamidine hydrochloric acid (Benz HCl). Fresh protease inhibitors [in μM: 5 bestatin, 2 E-64, 10 leupeptin, 1 pepstatin, and 200 phenylmethylsulfonyl fluoride (PMSF)] were also included in the HR solution. The pH of the HR solution was adjusted to 7.0 and the ionic strength to 180 mM. The smaller muscle fiber bundles were detergent-skinned overnight at 4°C in HR solution containing 1% Triton X-100.
Recombinant Tn subunits were reconstituted into detergent-skinned muscle fiber bundles, as described elsewhere (Chandra et al., 2006, 2007; Mamidi et al., 2013; Gollapudi et al., 2015). Briefly, TnTWT or TnTR134W (0.9 mg/ml, W/V) and TnI (1.0 mg/ml, W/V) were solubilized in an extraction buffer (buffer 1) containing the following (in M): 0.05 Tris-HCl (pH 8.0), 6.0 Urea, 1.0 KCl. High salt and urea in the extraction buffer were removed by successive dialysis against buffers 2-4, whose compositions are listed below.
Buffer 2 (in M): 0.050 Tris-HCl, 4 urea, 0.7 KCl (pH 8.0 at 4°C)
Buffer 3 (in M): 0.050 Tris-HCl, 2 urea, 0.5 KCl (pH 8.0 at 4°C)
Buffer 4 (in mM): 50 BES, 200 KCl, 10 BDM, 6.27 MgCl2, 5 EGTA (pH 7.0 at 20°C)
All buffers (1-4) included several protease inhibitors (0.2 mM PMSF, 2 mM Benz HCl, 1 mM DTT, and 0.01% NaN3). Any undissolved protein in the extraction buffer was removed by spinning it at 3000 rpm for 15 min. Detergent-skinned fiber bundles were treated with the extraction buffer containing TnTWT+TnI or TnTR134W+TnI for ~3 h at room temperature (22°C) with gentle stirring. Muscle fiber bundles were then washed twice (10 min each) using buffer 4 and incubated overnight at 4°C in HR solution containing TnC (3.0 mg/ml, W/V).
Western blot analysis
Reconstituted muscle fiber bundles were solubilized in a muscle protein extraction buffer containing the following: 2.5% SDS, 10% glycerol, 50 mM tris base (pH 6.8 at 4°C), 1 mM DTT, 1 mM PMSF, 4 mM Benz HCl, and a fresh cocktail of phosphatase (PhosSTOP) and protease inhibitors (E 64, Leupeptin, and Bestatin). The final concentration of all solubilized protein samples was adjusted to 2 mg/ml using the protein loading dye (125 mM Tris-HCl (pH 6.8), 20% glycerol, 2% SDS, 0.01% bromophenol blue, and 50 mM β-mercaptoethanol). 5 μg of each protein sample was loaded and run on an 8% SDS-gel for optimal separation of c-myc tagged recombinant and endogenous TnT (Gollapudi et al., 2012, 2013; Mamidi et al., 2013). Proteins were then transferred to a polyvinylidene difluoride membrane and TnT was probed using a monoclonal anti-TnT primary antibody (M401134, Fitzgerald Industries Int, Concord, MA), followed by HRP-labeled anti-mouse secondary antibody (RPN 2132, Amersham Biosciences, Piscataway, NJ). The percentage incorporation of the exogenous Tn was determined by the densitometric analysis of the TnT band profiles on the Western blot using ImageJ software (acquired from NIH at http://rsbweb.nih.gov/ij/).
PCA solutions and their compositions
For tension measurements, muscle fiber bundles were exposed to various solutions with pCa (= −log of [Ca2+]free) ranging from 9.0 to 4.3. The compositions of pCa 9.0 and 4.3 solutions were based on the program by Fabiato and Fabiato (1979), and are listed below.
pCa 9.0 (in mM): 50 BES, 5 NaN3, 10 phosphoenol pyruvate (PEP), 10 EGTA, 0.024 calcium chloride (CaCl2), 6.87 MgCl2, 5.83 Na2ATP, and 51.14 K-Prop
pCa 4.3 (in mM): 50 BES, 5 NaN3, 10 PEP, 10 EGTA, 10.11 CaCl2, 6.61 MgCl2, 5.95 Na2ATP, and 31 K-Prop
In addition, the pCa 9.0 and 4.3 solutions contained 0.5 mg/ml pyruvate kinase, 0.05 mg/ml lactate dehydrogenase, along with fresh protease inhibitors [(in μM): 10 leupeptin, 1000 pepstatin, 100 PMSF, 20 diadenosine penta-phosphate, 10 oligomycin]. The pH and ionic strength of pCa 9.0 and 4.3 solutions were adjusted to 7.0 and 180 mM, respectively.
Measurements of steady-state isometric tension and ATPase activity
Simultaneous measurements of steady-state isometric tension and ATPase activity were made, as described previously (de Tombe and Stienen, 1995; Stienen et al., 1995; Chandra et al., 2006, 2007; Gollapudi et al., 2015). T-shaped aluminum clips were used to attach muscle fiber bundles between a motor arm (322C, Aurora Scientific Inc., Ontario, Canada) and a force transducer (AE 801, Sensor One Technologies Corp., Sausalito, CA). The sarcomere length (SL) of the muscle fiber bundles was set to 2.3 μm in HR solution by laser diffraction. Each fiber bundle was subjected to two cycles of maximal activation (pCa 4.3) and relaxation (pCa 9.0), and the SL was re-adjusted to 2.3 μm if necessary. The cross-sectional area (CSA) and the initial muscle length (ML) corresponding to the SL of 2.3 μm were measured for each preparation. Muscle fiber bundles were then bathed in various solutions with pCa ranging from 9.0 to 4.3, one at a time, in a constantly-stirred chamber. The fiber-elicited responses in steady-state force and ATPase activity were recorded on a computer at a sampling frequency of 1 kHz. All measurements were made at 20°C.
Measurements of steady-state ATPase activity under isometric conditions were based on an enzymatically coupled assay, as described previously (de Tombe and Stienen, 1995; Stienen et al., 1995; Chandra et al., 2006, 2007; Gollapudi et al., 2015). Tension cost was determined as the slope of the linear relationship between steady-state tension and ATPase activity at various pCa (de Tombe and Stienen, 1995; Stienen et al., 1995).
Mechano-dynamic studies
Fully activated muscle fiber bundles were subjected to various amplitude stretch/release perturbations. First, we wanted to test if force scaled linearly with muscle length changes; therefore, we subjected the muscle fiber bundles to varying amplitudes of length changes (±0.1% to ±2.0%). Experimental data showing the averaged relationship between force changes (ΔT) and ML changes (ΔL) in Figure 1 clearly indicates that force scales linearly with ML. We used the previously established protocol (Ford et al., 2010) to record force responses to varying amplitude length changes (±0.5, ±1.0, ±1.5, and ±2.0% of ML). Force and length data were sampled at 2 kHz. A nonlinear recruitment-distortion (NLRD) model was fitted to this family of force responses to estimate the following four model parameters: the magnitude of instantaneous increase in stiffness caused by a sudden increase in ML (ED); the rate by which the sudden ML-induced increase in stiffness decays to a minimum (c); the rate by which a new steady-state force is attained due to the recruitment of new force-bearing XBs, following an increase in ML (b); and the magnitude of increase in the steady-state stiffness caused by the ML-mediated increase in the number of newly-recruited force-bearing XBs (ER). More details on step perturbation protocol and the physiological significance of NLRD model parameters are provided in our previously published works (Ford et al., 2010; Gollapudi et al., 2012; Chandra et al., 2015).
Figure 1.
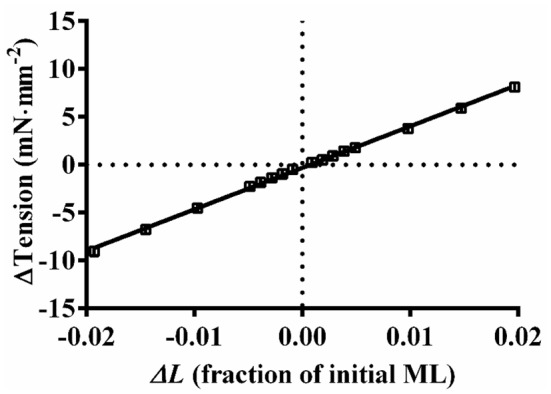
Relationship between changes in the steady-state force responses and the imposed muscle length changes. Five individual mouse fiber bundles (α-MHC) were subjected to various amplitude stretch/release perturbations (ΔL; ±0.1%, ±0.2%, ±0.3%, ±0.4%, ±0.5%, ±1.0%, ±1.5%, and ±2.0% of ML) and the corresponding steady-state force responses (ΔT) were recorded. The trace connecting the data (squares) represents the linear regression fit for the averaged ΔT-ΔL relationship from five muscle fiber bundles. The r2 value of the linear regression fit was 0.998, clearly indicating that the force scaled linearly with ML. Data are presented as mean ± SE. Standard error bars are smaller than symbols.
Measurement of rate of tension redevelopment (ktr)
The measurement of ktr was based upon the force response to a slightly modified version of the large slack-restretch ML maneuver, originally designed by Brenner and Eisenberg (1986). In brief, the muscle fiber in the steady-state of maximal Ca2+ activation (pCa 4.3) was first subjected to a rapid (1 ms) release by 10% of its ML using a high speed length-control device (322C, Aurora Scientific Inc., Ontario, Canada). After holding the fiber at the decreased length for 25 ms, it is quickly stretched past its ML by 10%, following which it was rapidly brought back to its ML and allowed to redevelop force. ktr was estimated by fitting the following mono-exponential function to the rising phase of the resulting force (F) response:
where Fss is steady-state force and Fres is residual force.
Data analysis
Normalized pCa-tension relationships were fitted to the Hill equation to derive pCa50 (an index of myofilament Ca2+ sensitivity) and nH (an index of myofilament cooperativity). We used a two-way ANOVA to analyze the contractile function parameters because our experimental model involved two factors, TnT (TnTWT and TnTR134W) and MHC (α-MHC and β-MHC). First, we assessed if the MHC-TnT interaction effect on a given contractile parameter was significant. A significant MHC-TnT interaction effect does not suggest a direct interaction between MHC and TnT but it demonstrates that the effects of TnTR134W on a parameter are dissimilar in α- and β-MHC fiber bundles. When the MHC-TnT interaction effect was not significant, we interpreted the main effect of TnT. To probe the cause for a significant MHC-TnT interaction effect or a main effect of TnT, multiple post-hoc t-tests were carried out using uncorrected Fisher's Least Significant Difference (LSD) method. Statistical significance was set at P < 0.05. Data are expressed as mean ± standard error of the mean (SEM).
Results
Incorporation levels of recombinant TnT in α- and β-MHC fiber bundles
We have previously demonstrated that the expression level of β-MHC in TG mouse hearts was ~70% of the total MHC (Gollapudi et al., 2015). This overexpression of β-MHC had no impact on either the stoichiometry of other sarcomeric proteins or the phosphorylation levels of contractile regulatory proteins (Gollapudi et al., 2015). We used the Western blot to quantify the extent of recombinant Tn incorporation into muscle fiber bundles. The addition of an 11-amino acid c-myc tag at the N-terminus of recombinant TnT proteins (TnTWT or TnTR134W) allowed us to separate the recombinant and endogenous TnT on an SDS gel, and to assess the extent of recombinant Tn incorporation in muscle fiber bundles. The inclusion of c-myc epitope had no impact on the TnT-mediated function in cardiac muscle (Tardiff et al., 1998; Montgomery et al., 2001). A representative Western blot showing the incorporation levels of recombinant TnT in α- and β-MHC fiber bundles is presented in Figure 2. Densitometric analysis revealed that the incorporation levels of TnTWT and TnTR134W in α-MHC fiber bundles were 93% and 74%, while those in β-MHC fiber bundles were 90% and 72%, respectively. Similar incorporation levels of TnTR134W in both α- and β-MHC fiber bundles provided a good model to probe the interplay between TnTR134W- and MHC-mediated effects on various contractile parameters.
Figure 2.

Western blot showing the incorporation levels of recombinant TnT in α- and β-MHC fiber bundles. Reconstituted fiber bundles were solubilized in 2.5% SDS solution and their final concentrations were adjusted to 2 mg/ml. Equal amounts (5 μg) of standardized protein samples were loaded and run on an 8% small SDS-gel for optimal separation of recombinant and endogenous TnT. Proteins were then transferred onto a PVDF membrane and TnT was probed using an anti-TnT primary, followed by an anti-TnT secondary antibody.
TnTR134W-mediated impact on Ca2+-activated maximal tension and ED in α- and β-MHC fiber bundles
We assessed whether TnTR134W altered maximal activation in an MHC-dependent manner by analyzing the steady-state tension measurements at pCa 4.3. Two-way ANOVA of maximal tension did not reveal a significant MHC-TnT interaction effect (P = 0.35) or a main effect of TnT (P = 0.50). This is because TnTR134W showed no impact on maximal tension in either α- or β-MHC fiber bundles. The mean ± SEM values of maximal tension (in mN·mm−2) in α-MHC+TnTWT and α-MHC+TnTR134W fiber bundles were 46.82 ± 1.03 (n = 13) and 46.11 ± 1.60 (n = 12), while those in β-MHC+TnTWT and β-MHC+TnTR134W fiber bundles were 45.50 ± 1.16 (n = 14) and 49.58 ± 1.24 (n = 14), respectively.
Previously, we have demonstrated that maximal tension is correlated to ED (Campbell et al., 2004; Mamidi et al., 2013; Chandra et al., 2015). Therefore, to support our observations in maximal tension, we assessed ED. ED is an approximate measure of the number of force-bearing XBs in the isometric steady-state prior to ML change (Campbell et al., 2004; Ford et al., 2010). Two-way ANOVA did not show a significant MHC-TnT interaction effect (P = 0.37) on ED or a main effect of TnT (P = 0.14). Thus, TnTR134W did not alter ED in either α- or β-MHC fiber bundles. The mean ± SEM values of ED (in mN·mm−3) in α-MHC+TnTWT and α-MHC+TnTR134W fiber bundles were 1041 ± 42 (n = 13) and 895 ± 49 (n = 12), while those in β-MHC+TnTWT and β-MHC+TnTR134W fiber bundles were 980 ± 59 (n = 14) and 951 ± 29 (n = 14), respectively. Similar observations in both maximal tension and ED substantiate that TnTR134W did not affect maximal activation regardless of the MHC isoform.
TnTR134W-mediated impact on the pCa-tension relationship in α- and β-MHC fiber bundles
A comparison of pCa-tension relationships showed that TnTR134W induced a larger rightward shift in the pCa-tension relationship in α-MHC fiber bundles (Figure 3A) than in β-MHC fiber bundles (Figure 3B). A closer examination of the pCa-tension relationships also revealed that TnTR134W did not alter the steepness of the pCa-tension relationship in α-MHC fiber bundles (Figure 3A) but it decreased the steepness in β-MHC fiber bundles (Figure 3B). To quantify the magnitude of such effects in α- and β-MHC fiber bundles, we analyzed the Hill model-derived parameters, pCa50 (myofilament Ca2+ sensitivity) and nH (myofilament cooperativity). Two-way ANOVA of pCa50 did not show a significant MHC-TnT interaction effect (P = 0.17) but showed a significant main effect of TnT (P < 0.001). Post-hoc analysis revealed that TnTR134W significantly decreased pCa50 in both α- and β-MHC fiber bundles; however, the magnitude of attenuation was different (Figure 3C). For example, TnTR134W significantly attenuated pCa50 by 0.14 pCa units (P < 0.001) in α-MHC fiber bundles and by 0.08 pCa units (P < 0.001) in β-MHC fiber bundles. These observations suggest that TnTR134W decreases myofilament Ca2+ sensitivity to a greater extent in the presence of α-MHC than in the presence of β-MHC. To quantify such changes in pCa50 in terms of tension, we also compared the steady-state tension data at submaximal Ca2+ activation (pCa 5.5) among groups. Our analysis showed that TnTR134W significantly attenuated tension at pCa 5.5 by 46% (P < 0.001) in α-MHC fiber bundles and by 26% (P < 0.01) in β-MHC fiber bundles. These observations substantiate that, at submaximal Ca2+ levels, the attenuating effect of TnTR134W on thin filament activation is stronger in α-MHC than in β-MHC fiber bundles.
Figure 3.

Effect of TnTR134W on pCa-tension relationships in α- and β-MHC fiber bundles. Steady-state tensions, normalized to the value at pCa 4.3, were plotted against pCa to construct the pCa-tension relationship. Hill model was fitted to these pCa-tension relationships to estimate pCa50 (myofilament Ca2+ sensitivity) and nH (myofilament cooperativity). TnTR134W-mediated impact on pCa-tension relationship in (A) α-MHC and (B) β-MHC fiber bundles. Traces connecting the data (empty or filled circles) are the Hill model fits. TnTR134W-mediated impact on (C) pCa50 and (D) nH in α- and β-MHC fiber bundles. Statistical differences were analyzed by two-way ANOVA and subsequent post-hoc t-tests using Fishers LSD method. ***P < 0.001 indicate a significant result compared to TnTWT (NS, not significant). The number of fiber bundles measured is as follows: 13 for α-MHC+TnTWT, 12 for α-MHC+TnTR134W, 14 for β-MHC+TnTWT, and 14 for β-MHC+TnTR134W. Data are presented as mean ± SE. Standard error bars are smaller than symbols in some cases.
Two-way ANOVA of nH showed a significant MHC-TnT interaction effect (P < 0.05), suggesting that the TnTR134W-mediated impact on nH was dissimilar in α- and β-MHC fiber bundles. Post-hoc analysis revealed that TnTR134W showed no effect (P = 0.30; Figure 3D) on nH in α-MHC fiber bundles, but it significantly decreased nH by 26% (P < 0.001; Figure 3D) in β-MHC fiber bundles. These observations suggest that TnTR134W does not affect myofilament cooperativity in the presence of α-MHC, but attenuates myofilament cooperativity in the presence of β-MHC. Collectively, these observations demonstrate that α- and β-MHC isoforms differently modulate the TnTR134W-mediated impact on thin filaments at submaximal Ca2+ activation.
TnTR134W-mediated impact on XB detachment kinetics in α- and β-MHC fiber bundles
To determine whether TnTR134W affected the XB detachment rate (g) in an MHC-dependent manner, we assessed tension cost and c. Tension cost was estimated as the slope of the linear relationship between tension and ATPase data at various pCa (de Tombe and Stienen, 1995; Stienen et al., 1995; Ford and Chandra, 2013). Within the context of a two-state XB model (Huxley, 1957), the ratio of steady-state ATPase activity (fg/(f + g)) and tension (f/(f + g)) is proportional to g; thus, tension cost is an approximate measure of g. c, which is the rate constant of the immediate force decay, following a sudden change in ML (Ford et al., 2010), is also a measure of g because it is positively correlated to tension cost (Campbell et al., 2004).
A comparison showed that TnTR134W induced a downward shift in the tension-ATPase plot in α-MHC fiber bundles (Figure 4A), which suggested a decrease in the slope of this relationship. On the other hand, TnTR134W showed no effect on the tension-ATPase plot in β-MHC fiber bundles (Figure 4B). These disimilar effects of TnTR134W on tension cost in α- and β-MHC fiber bundles gave rise to a significant MHC-TnT interaction effect (P < 0.001). Post-hoc t-tests showed that TnTR134W significantly decreased tension cost by 17% (P < 0.001; Figure 4C) in α-MHC fiber bundles, while it showed no effect (P = 0.39; Figure 4D) in β-MHC fiber bundles. Observed effects in tension cost were also validated by our findings in c. A comparison of force responses to 2% stretch showed that TnTR134W induced a rightward shift in the immediate force decay phase in α-MHC fiber bundles (Figure 5A), which suggested a slower c. However, TnTR134W showed no effect on the immediate force response in β-MHC fiber bundles (Figure 5B). Two-way ANOVA showed a significant MHC-TnT interaction effect (P < 0.01) on c, which suggested that the effect of TnTR134W on c was different in α- and β-MHC fiber bundles. Post-hoc t-tests showed that TnTR134W significantly decreased c by 15% (P < 0.01; Figure 5C) in α-MHC fiber bundles, while it showed no effect (P = 0.21; Figure 5D) in β-MHC fiber bundles. Similar effects in tension cost and c suggest that TnTR134W-induced changes in thin filaments interact differently with those induced by α- and β-MHC isoforms to differently modulate the effect on g.
Figure 4.

Effect of TnTR134W on tension cost in α- and β-MHC fiber bundles. Simultaneous measurements of steady-state tension and ATPase activity were made in muscle fiber bundles at various pCa, as described in Materials and Methods. Tension cost was estimated as the slope of the linear regression fit to the tension-ATPase plot. TnTR134W-mediated impact on tension-ATPase relation in (A) α-MHC and (B) β-MHC fiber bundles. Traces connecting the data (empty or filled circles) are the linear regression fits. TnTR134W-mediated impact on tension cost in (C) α-MHC and (D) β-MHC fiber bundles. Statistical differences were analyzed by two-way ANOVA and subsequent post-hoc t-tests using Fishers LSD method. ***P < 0.001 indicate a significant result compared to TnTWT (NS, not significant). The number of fiber bundles measured is as follows: 13 for α-MHC+TnTWT, 12 for α-MHC+TnTR134W, 14 for β-MHC+TnTWT, and 14 for β-MHC+TnTR134W. Data are presented as mean ± SE.
Figure 5.

Effect of TnTR134W on c in α- and β-MHC fiber bundles. TnTR134W-mediated impact on force response to a 2% stretch in muscle length (ML) in (A) α-MHC and (B) β-MHC fiber bundles. Force data were normalized by the isometric steady-state value, Fss, prior to stretch. c describes the rate of force decay to a minimum force point (nadir), b governs the rate of delayed force rise following an increase in ML, and Fnss represents the new steady-state force following an increase in ML. TnTR134W-mediated impact on c in (C) α-MHC and (D) β-MHC fiber bundles. Statistical differences were analyzed by two-way ANOVA and subsequent post-hoc t-tests using Fishers LSD method. **P < 0.01 indicate a significant result compared to TnTWT (NS, not significant). The number of fiber bundles measured is as follows: 13 for α-MHC+TnTWT, 12 for α-MHC+TnTR134W, 14 for β-MHC+TnTWT, and 14 for β-MHC+TnTR134W. Data are presented as mean ± SE.
TnTR134W-mediated impact on XB turnover rate in α- and β-MHC fiber bundles
To determine whether TnTR134W differently altered XB turnover rate in α- and β-MHC fiber bundles, we assessed two independent rate parameters, ktr and b. While ktr represents the rate of force redevelopment following a large release-restretch length maneuver (Brenner and Eisenberg, 1986), b describes the rate of delayed force rise following a sudden stretch in ML (Ford et al., 2010). Both ktr and b have been previously shown to be approximate measures of XB turnover rate (Ford et al., 2010; Gollapudi et al., 2013, 2015; Chandra et al., 2015).
A representative comparison of force responses to a large release-restretch length meneuver showed that, in α-MHC fiber bundles (Figure 6A), TnTR134W induced a rightward shift in the rising force phase, which suggested a slower rate of force rise. On the contrary, TnTR134W showed no effect on the force response in β-MHC fiber bundles (Figure 6B). Therefore, two-way ANOVA showed a significant MHC-TnT interaction effect on ktr (P < 0.05), which suggested that the effects of TnTR134W on ktr against α- and β-MHC were dissimilar. Post-hoc analysis confirmed that TnTR134W attenuated ktr by 14% (P < 0.01; Figure 6C) in α-MHC fiber bundles, while it showed no effect (P = 0.57; Figure 6C) in β-MHC fiber bundles. Our analysis of b also revealed similar findings. A comparison of force responses to a 2% stretch showed that TnTR134W induced a rightward shift in the delayed force rise phase in α-MHC fiber bundles (Figure 5A), which suggested attenuation of b. However, TnTR134W displayed no effect on the delayed force rise phase in β-MHC fiber bundles (Figure 5B). These differential effects of TnTR134W on b in α- and β-MHC fiber bundles gave rise to a significant MHC-TnT interaction effect (P < 0.01). Post-hoc analysis confirmed that TnTR134W attenuated b by 17% (P < 0.001; Figure 6D) in α-MHC fiber bundles, while it showed no effect (P = 0.86; Figure 6D) in β-MHC fiber bundles. Therefore, similar effects in ktr and b demonstrate that the XB turnover rate is attenuated by TnTR134W only in the presence of α-MHC.
Figure 6.

Effect of TnTR134W on ktr and b in α- and β-MHC fiber bundles. ktr was estimated by fitting a mono-exponential function to the rising phase of the force response following a large release-restretch length maneuver (Brenner and Eisenberg, 1986). b was estimated by fitting the NLRD model to a family of force responses to various amplitude ML perturbations (Ford et al., 2010). TnTR134W-mediated effect on the force response to a large release-restretch maneuver in (A) α-MHC and (B) β-MHC fiber bundles. Force data were normalized by the isometric steady-state value following the length perturbation. TnTR134W-mediated effect on (C) ktr and (D) b in α- and β-MHC fiber bundles. Statistical differences were analyzed by two-way ANOVA and subsequent post-hoc t-tests using Fishers LSD method. **P < 0.01 and ***P < 0.001 indicate significant results compared to TnTWT (NS, not significant). The number of fiber bundles measured is as follows: 13 for α-MHC+TnTWT, 12 for α-MHC+TnTR134W, 14 for β-MHC+TnTWT, and 14 for β-MHC+TnTR134W. Data are presented as mean ± SE.
TnTR134W-mediated impact on ER in α- and β-MHC fiber bundles
To investigate whether TnTR134W differentially altered the magnitude of stretch activation in α- and β-MHC fiber bundles, we assessed estimates of ER at maximal Ca2+ activation (pCa 4.3). ER represents the magnitude of muscle length-mediated recruitment of new force-bearing XBs (ER) and is equivalent to the magnitude of stretch activation (Campbell and Chandra, 2006; Stelzer et al., 2006b, 2007; Ford et al., 2010). ER is derived as the slope of the linear regression between (Fnss − Fss) and ΔL (see Figures 5A,B), where Fnss is the force corresponding to the new-steady state attained after the change in ML, Fss is the steady-state isometric force prior to the change in ML, and ΔL is the imposed ML change. Thus, ER increases when Fnss increases and vice versa. Comparison of force responses to a 2% stretch showed that TnTR134W attenuated Fnss in α-MHC fiber bundles (Figure 5A), which suggested a decrease in ER. On the other hand, TnTR134W showed no effect on Fnss in β-MHC fiber bundles (Figure 5B). These different effects of TnTR134W on ER in α- and β-MHC fiber bundles gave rise to a significant MHC-TnT interaction effect (P < 0.05). Post-hoc analysis showed that TnTR134W significantly decreased ER by 24% (P < 0.01; Figure 7) in α-MHC fiber bundles, while it showed no effect (P = 0.92; Figure 7) in β-MHC fiber bundles. These observations demonstrate that the magnitude of stretch activation mediated by TnTR134W is differently altered by α- and β-MHC.
Figure 7.
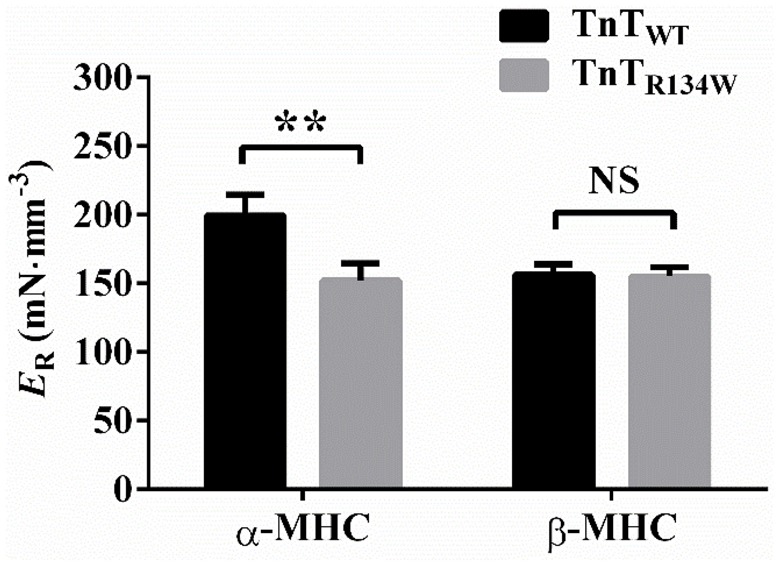
Effect of TnTR134W on ER in α- and β-MHC fiber bundles. ER was estimated as the slope of a linear relationship between (Fnss − Fss) elicited by muscle fiber bundles to imposed ML changes, ΔL (see Figures 5A,B for details on Fnss and Fss). Statistical differences were analyzed by two-way ANOVA and subsequent post-hoc t-tests using Fishers LSD method. **P < 0.01 indicate a significant result compared to TnTWT fibers (NS, not significant). The number of fiber bundles measured is as follows: 13 for α-MHC+TnTWT, 12 for α-MHC+TnTR134W, 14 for β-MHC+TnTWT, and 14 for β-MHC+TnTR134W. Data are presented as mean ± SE.
Discussion
The severity of DCM phenotypes in humans varies so widely that a commonly attributed change in a steady-state contractile parameter, such as a modest decrease in myofilament Ca2+ sensitivity, precludes us from explaining disparate cardiac phenotypes. Because dynamic aspects of cardiac contraction dominate under conditions in which the heart muscle normally operates, dynamic contractile indices may provide more meaningful clues to link disparate phenotypes to different mutations. Our extensive steady-state and dynamic contractile data demonstrate that alterations in contractile dynamics (both rate and magnitude), in addition to the differential impact of α- and β-MHC on myofilament Ca2+ sensitivity, allow us to expand our view on how some mutations in TnT affect heart function and cardiac phenotypes.
TnTR134W attenuates myofilament Ca2+ sensitivity to a greater extent in α-MHC than in β-MHC fiber bundles
A greater magnitude of decrease in myofilament Ca2+ sensitivity in α-MHC+TnTR134W than in β-MHC+TnTR134W fiber bundles (Figure 3C) raises two questions: (1) how does TnTR134W attenuate thin filament activation?; and (2) why is the effect of TnTR134W on thin filaments minimized in the presence of β-MHC? Previous studies have associated residues 112–136 of human TnT in the strong interaction of CR of TnT with Tm at the Tm-Tm overlap junction (Hinkle and Tobacman, 2003). This CR-Tm interaction acts as a gateway not only for the activation of regulatory units (RU; Tn-Tm complex) but also for cooperative interactions between near-neighbor RUs and between near-neighbor RUs and XBs (Schaertl et al., 1995; Razumova et al., 2000; Tobacman et al., 2002; Moss et al., 2004). There is evidence to suggest that the R131W mutation in TnT decreases RU activation by increasing the rate of dissociation of Ca2+ from Tn (Liu et al., 2012). While such attenuation of RU activation may primarily involve altered CR-Tm interaction, another study suggests that the R131W-induced structural changes in the CR may also spread to the T2 region of TnT to modify Ca2+-sensitive interactions between TnT and TnI/TnC (Mogensen et al., 2004). Based on these findings, we posit that TnTR134W alters allosteric/cooperative mechanisms that underlie RU activation. At submaximal [Ca2+], such actions of TnTR134W increase the amount of Ca2+ required to attain the magnitude of RU activation that is normally observed in TnTWT, leading to attenuation of pCa50 in both α- and β-MHC fiber bundles. However, the magnitude of the impact on pCa50 is lower in β-MHC+TnTR134W than in α-MHC+TnTR134W fiber bundles, suggesting that β-MHC partially counters the effect of TnTR134W on RU activation. Given that enhanced XB-RU, but not XB-XB, cooperativity increases pCa50 (Razumova et al., 2000), the ability of β-MHC to counter the influence of TnTR134W on pCa50 appears to arise from greater XB-RU cooperativity.
How differences in XB cycling kinetics permit α- and β-MHC (Rundell et al., 2005; Stelzer et al., 2007; Ford and Chandra, 2013) to differently modify XB-RU cooperativity may be gleaned by considering the initial conditions of thin filaments. At submaximal [Ca2+]free, the TnTR134W-induced attenuation of RU activation leaves behind a larger than normal pool of RUs in the off state, thereby increasing the scope for strong XBs to cooperatively influence XB-RU interactions. Therefore, the slow cycling β-MHC may exert a positive effect on thin filaments by amplifying XB-RU cooperativity. This enhanced XB-RU cooperativity by β-MHC is expected to facilitate the transition of RUs from the off to the on state, thereby resulting in an increase in RU activation and a subsequent increase in the number of force-bearing XBs. Indeed, tension is augmented at submaximal activation in β-MHC+TnTR134W fiber bundles when compared to α-MHC+TnTR134W fiber bundles. This explains why β-MHC is able to partially counter the negative influence of TnTR134W on pCa50. Such β-MHC-mediated increase in XB-RU cooperativity may have exhausted the pool of RUs from which RU-RU cooperativity could recruit (Razumova et al., 2000), thereby decreasing the contributions of RU-RU cooperativity to nH in β-MHC+TnTR134W fiber bundles. Because RU-RU cooperativity has the greatest influence on nH (Razumova et al., 2000), a decrease in RU-RU cooperativity may likely explain the decrease in nH in β-MHC+TnTR134W fiber bundles.
β-MHC neutralizes the attenuating effect of TnTR134W on XB turnover rate and XB detachment rate
Our observations on two contractile rate parameters, ktr and b (Figure 6), confirm that TnTR134W attenuates XB turnover rate in α-MHC fiber bundles, but shows no effect in β-MHC fiber bundles. In our previous studies (Campbell, 1997; Campbell et al., 2004; Ford et al., 2010; Gollapudi and Chandra, 2012), we have shown that attenuation of b may be brought about by the following: (1) attenuation of RU on/off kinetics; (2) attenuation of XB cycling kinetics, f and g; (3) augmentation of XB-based cooperativity; or (4) a combinatorial effect of 1, 2, and 3. However, unaltered maximal tension and ED in α-MHC+TnTR134W fiber bundles suggest that the impact of TnTR134W on RU activation is minimized. Under these conditions, the available pool of RUs and XBs from which XB-based cooperativity may recruit is expected to be similar in both α-MHC+TnTWT and α-MHC+TnTR134W fiber bundles. Thus, it is unlikely that enhanced XB-based cooperativity is responsible for slower b in α-MHC+TnTR134W fiber bundles. Therefore, our observations suggest that attenuation of b may result from the slowing effect of TnTR134W on XB cycling kinetics. Because ktr = f + g, as per the two-state XB model (Huxley, 1957; Brenner, 1988; de Tombe and Stienen, 2007), a significant attenuation of ktr in α-MHC+TnTR134W fiber bundles also substantiates our assertion that attenuation of b is due to a slowing effect on f and/or g.
Evidence to substantiate that attenuation of g may underlie the slowed XB turnover rate (ktr and b; Figure 6) in α-MHC+TnTR134W fiber bundles comes from our observations on tension cost (Figure 4) and c (Figure 5). Furthermore, other observations suggest that the attenuation of XB turnover rate in α-MHC+TnTR134W fiber bundles may also involve a slowing effect on f; this is because steady-state isometric force is proportional to f/(f + g) (Huxley, 1957). Therefore, a decrease in g alone should increase force produced in α-MHC+TnTR134W fiber bundles. However, both maximal tension and ED are unaltered in α-MHC+TnTR134W fiber bundles, which suggests that TnTR134W does not impact f/(f + g) in the presence of α-MHC at maximal activation. Thus, this conjectural evidence may indicate that f decreases in proportion to g in α-MHC+TnTR134W fiber bundles. In contrast, a lack of effect on g in β-MHC+TnTR134W fiber bundles (Figures 4, 5) suggests that β-MHC negates the attenuating effect of TnTR134W on g. Our observation on g, in conjunction with unaltered ktr, b, and maximal tension in β-MHC+TnTR134W fiber bundles, suggests that TnTR134W does not alter f in the presence of β-MHC. These observations demonstrate that the interplay between the TnT- and MHC-mediated effects on the thin filament modulate XB cycling kinetics.
β-MHC neutralizes the attenuating effect of TnTR134W on the magnitude of stretch activation
Another notable finding from our study is that TnTR134W attenuates the magnitude of stretch activation (ER) in α-MHC but shows no effect in β-MHC fiber bundles (Figure 7). The magnitude of ER is dependent on the ML-related XB recruitment mechanisms that operate within thin filaments. For example, XB-based (XB-RU/XB-XB) cooperativity, which is mediated through thin filaments, strongly influences ER such that a decrease in XB-based cooperativity decreases ER and vice versa (Campbell et al., 2004; Campbell and Chandra, 2006; Stelzer et al., 2006b). Our data suggest that different outcomes on ER in α-MHC+TnTR134W and β-MHC+TnTR134W fiber bundles may be closely linked to differential effects on XB-RU cooperativity. To clarify, a decrease in XB-RU cooperativity may be responsible for the attenuation of ER in TnTR134W+α-MHC fiber bundles, while unaltered XB-RU cooperativity explains why ER is unaffected in β-MHC+TnTR134W fiber bundles. One source of this difference between α- and β-MHC fiber bundles may be related to our earlier assertion that the negative effect of TnTR134W on RU activation remains more prominent in α-MHC than in β-MHC fiber bundles. Therefore, ER is attenuated in α- but not in β-MHC fiber bundles.
Implications of our findings for heart function in mice and humans
TnTR134W attenuated XB turnover and detachment rates in α-MHC fiber bundles, but not in β-MHC fiber bundles. When extrapolated to the whole heart level, these observations suggest slower rise and slower fall of ventricular pressure in α-MHC containing hearts but not in β-MHC containing hearts. In addition, slower rates of XB turnover and detachment may also slow dynamics of ejection in α-MHC-expressing hearts. Previous studies also implicate mechanisms such as stretch activation in maintaining the ventricular force output during the late phase of ejection (Stelzer et al., 2006a,c). Thus, attenuation of ER in α-MHC+TnTR134W fiber bundles, in conjunction with slowed XB turnover and detachment rates, suggests that the ejection phase may be prematurely terminated in α-MHC-expressing hearts. Inferences drawn from dynamic studies demonstrate that the magnitude of cardiac contractile impairment, induced by TnTR134W, differ significantly in α- and β-MHC expressing fiber bundles. The effect on pCa50 also shows that the severity of contractile deficits induced by TnTR134W is different in α- and β-MHC fiber bundles; for instance, TnTR134W decreases pCa50 to a greater extent in α-MHC fiber bundles (Figure 3C). Although the attenuation of pCa50 alone may suggest DCM in both α- and β-MHC background, the severity of cardiac phenotype is expected to be greater in mouse hearts because various indices of contractile dynamics were attenuated only in α-MHC fiber bundles.
Author contributions
Contribution of SG: Conception and design, acquisition of data, analysis and interpretation of data, drafting and revising the manuscript. Contribution of MC: Conception and design, interpretation of data, drafting and revising the manuscript.
Funding
This work was supported, in part, by National Institutes of Health Grant No. HL-075643 (to MC) and a Poncin grant supported by the Autzen foundation.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Sherif M. Reda and Alexis V. Mickelson for proof reading the manuscript.
Glossary
Abbreviations
- ANOVA
analysis of variance
- CR
central region
- DCM
dilated cardiomyopathy
- HR
high relaxing
- LSD
least significant differences
- MHC
myosin heavy chain
- ML
muscle length
- NLRD
nonlinear recruitment distortion
- NTG
non-transgenic
- RU
regulatory unit
- SL
sarcomere length
- TG
transgenic
- Tm
tropomyosin
- Tn
troponin
- TnC
troponin C
- TnI
troponin I
- TnT
troponin T
- WT
wild-type
- XB
crossbridge.
References
- Brenner B. (1988). Effect of Ca2+ on cross-bridge turnover kinetics in skinned single rabbit psoas fibers: implications for regulation of muscle contraction. Proc. Natl. Acad. Sci. U.S.A. 85, 3265–3269. 10.1073/pnas.85.9.3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner B., Eisenberg E. (1986). Rate of force generation in muscle: correlation with actomyosin ATPase activity in solution. Proc. Natl. Acad. Sci. U.S.A. 83, 3542–3546. 10.1073/pnas.83.10.3542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell K. (1997). Rate constant of muscle force redevelopment reflects cooperative activation as well as cross-bridge kinetics. Biophys. J. 72, 254–262. 10.1016/S0006-3495(97)78664-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell K. B., Chandra M. (2006). Functions of stretch activation in heart muscle. J. Gen. Physiol. 127, 89–94. 10.1085/jgp.200509483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell K. B., Chandra M., Kirkpatrick R. D., Slinker B. K., Hunter W. C. (2004). Interpreting cardiac muscle force-length dynamics using a novel functional model. Am. J. Physiol. Heart Circ. Physiol. 286, H1535–H1545. 10.1152/ajpheart.01029.2003 [DOI] [PubMed] [Google Scholar]
- Chandra M., Montgomery D. E., Kim J. J., Solaro R. J. (1999). The N-terminal region of troponin T is essential for the maximal activation of rat cardiac myofilaments. J. Mol. Cell. Cardiol. 31, 867–880. 10.1006/jmcc.1999.0928 [DOI] [PubMed] [Google Scholar]
- Chandra M., Tschirgi M. L., Ford S. J., Slinker B. K., Campbell K. B. (2007). Interaction between myosin heavy chain and troponin isoforms modulate cardiac myofiber contractile dynamics. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, R1595–R1607. 10.1152/ajpregu.00157.2007 [DOI] [PubMed] [Google Scholar]
- Chandra M., Tschirgi M. L., Rajapakse I., Campbell K. B. (2006). Troponin T modulates sarcomere length-dependent recruitment of cross-bridges in cardiac muscle. Biophys. J. 90, 2867–2876. 10.1529/biophysj.105.076950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra V., Gollapudi S. K., Chandra M. (2015). Rat cardiac troponin T mutation (F72L)-mediated impact on thin filament cooperativity is divergently modulated by alpha- and beta-myosin heavy chain isoforms. Am. J. Physiol. Heart Circ. Physiol. 309, H1260–H1270. 10.1152/ajpheart.00519.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Tombe P. P., Stienen G. J. (1995). Protein kinase A does not alter economy of force maintenance in skinned rat cardiac trabeculae. Circ. Res. 76, 734–741. 10.1161/01.RES.76.5.734 [DOI] [PubMed] [Google Scholar]
- de Tombe P. P., Stienen G. J. (2007). Impact of temperature on cross-bridge cycling kinetics in rat myocardium. J. Physiol. 584(Pt 2), 591–600. 10.1113/jphysiol.2007.138693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiato A., Fabiato F. (1979). Calculator programs for computing the composition of the solutions containing multiple metals and ligands used for experiments in skinned muscle cells. J. Physiol. (Paris). 75, 463–505. [PubMed] [Google Scholar]
- Ford S. J., Chandra M. (2013). Length-dependent effects on cardiac contractile dynamics are different in cardiac muscle containing alpha- or beta-myosin heavy chain. Arch. Biochem. Biophys. 535, 3–13. 10.1016/j.abb.2012.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford S. J., Chandra M., Mamidi R., Dong W., Campbell K. B. (2010). Model representation of the nonlinear step response in cardiac muscle. J. Gen. Physiol. 136, 159–177. 10.1085/jgp.201010467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford S. J., Mamidi R., Jimenez J., Tardiff J. C., Chandra M. (2012). Effects of R92 mutations in mouse cardiac troponin T are influenced by changes in myosin heavy chain isoform. J. Mol. Cell. Cardiol. 53, 542–551. 10.1016/j.yjmcc.2012.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollapudi S. K., Chandra M. (2012). Cardiomyopathy-related mutations in cardiac troponin C, L29Q and G159D, have divergent effects on rat cardiac myofiber contractile dynamics. Biochem. Res. Int. 2012:824068. 10.1155/2012/824068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollapudi S. K., Chandra M. (2016). The effect of cardiomyopathy mutation (R97L) in mouse cardiac troponin T on the muscle length-mediated recruitment of crossbridges is modified divergently by α- and β-myosin heavy chain. Arch. Biochem. Biophys. 601, 105–112. 10.1016/j.abb.2016.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollapudi S. K., Gallon C. E., Chandra M. (2013). The tropomyosin binding region of cardiac troponin T modulates crossbridge recruitment dynamics in rat cardiac muscle fibers. J. Mol. Biol. 425, 1565–1581. 10.1016/j.jmb.2013.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollapudi S. K., Mamidi R., Mallampalli S. L., Chandra M. (2012). The N-terminal extension of cardiac troponin T stabilizes the blocked state of cardiac thin filament. Biophys. J. 103, 940–948. 10.1016/j.bpj.2012.07.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollapudi S. K., Tardiff J. C., Chandra M. (2015). The functional effect of dilated cardiomyopathy mutation (R144W) in mouse cardiac troponin T is differently affected by alpha- and beta-myosin heavy chain isoforms. Am. J. Physiol. Heart Circ. Physiol. 308, H884–H893. 10.1152/ajpheart.00528.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X., Wattanapermpool J., Palmiter K. A., Murphy A. M., Solaro R. J. (1994). Mutagenesis of cardiac troponin I. Role of the unique NH2-terminal peptide in myofilament activation. J. Biol. Chem. 269, 15210–15216. [PubMed] [Google Scholar]
- Hershberger R. E., Pinto J. R., Parks S. B., Kushner J. D., Li D., Ludwigsen S., et al. (2009). Clinical and functional characterization of TNNT2 mutations identified in patients with dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2, 306–313. 10.1161/CIRCGENETICS.108.846733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinkle A., Tobacman L. S. (2003). Folding and function of the troponin tail domain. Effects of cardiomyopathic troponin T mutations. J. Biol. Chem. 278, 506–513. 10.1074/jbc.M209194200 [DOI] [PubMed] [Google Scholar]
- Huxley A. F. (1957). Muscle structure and theories of contraction. Prog. Biophys. Biophys. Chem. 7, 255–318. [PubMed] [Google Scholar]
- Jackson P., Amphlett G. W., Perry S. V. (1975). The primary structure of troponin T and the interaction with tropomyosin. Biochem. J. 151, 85–97. 10.1042/bj1510085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenz M., Robbins J. (2004). Impact of beta-myosin heavy chain expression on cardiac function during stress. J. Am. Coll. Cardiol. 44, 2390–2397. 10.1016/j.jacc.2004.09.044 [DOI] [PubMed] [Google Scholar]
- Krenz M., Sadayappan S., Osinska H. E., Henry J. A., Beck S., Warshaw D. M., et al. (2007). Distribution and structure-function relationship of myosin heavy chain isoforms in the adult mouse heart. J. Biol. Chem. 282, 24057–24064. 10.1074/jbc.M704574200 [DOI] [PubMed] [Google Scholar]
- Krenz M., Sanbe A., Bouyer-Dalloz F., Gulick J., Klevitsky R., Hewett T. E., et al. (2003). Analysis of myosin heavy chain functionality in the heart. J. Biol. Chem. 278, 17466–17474. 10.1074/jbc.M210804200 [DOI] [PubMed] [Google Scholar]
- Kushner J. D., Nauman D., Burgess D., Ludwigsen S., Parks S. B., Pantely G., et al. (2006). Clinical characteristics of 304 kindreds evaluated for familial dilated cardiomyopathy. J. Card. Fail. 12, 422–429. 10.1016/j.cardfail.2006.03.009 [DOI] [PubMed] [Google Scholar]
- Liu B., Tikunova S. B., Kline K. P., Siddiqui J. K., Davis J. P. (2012). Disease-related cardiac troponins alter thin filament Ca2+ association and dissociation rates. PLoS ONE 7:e38259. 10.1371/journal.pone.0038259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q. W., Wu X. Y., Morimoto S. (2013). Inherited cardiomyopathies caused by troponin mutations. J. Geriatr. Cardiol. 10, 91–101. 10.3969/j.issn.1671-5411.2013.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamidi R., Mallampalli S. L., Wieczorek D. F., Chandra M. (2013). Identification of two new regions in the N-terminus of cardiac troponin T that have divergent effects on cardiac contractile function. J. Physiol. 591(Pt 5), 1217–1234. 10.1113/jphysiol.2012.243394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marston S. B. (2011). How do mutations in contractile proteins cause the primary familial cardiomyopathies? J. Cardiovasc. Transl. Res. 4, 245–255. 10.1007/s12265-011-9266-2 [DOI] [PubMed] [Google Scholar]
- Mirza M., Marston S., Willott R., Ashley C., Mogensen J., McKenna W., et al. (2005). Dilated cardiomyopathy mutations in three thin filament regulatory proteins result in a common functional phenotype. J. Biol. Chem. 280, 28498–28506. 10.1074/jbc.M412281200 [DOI] [PubMed] [Google Scholar]
- Mogensen J., Murphy R. T., Shaw T., Bahl A., Redwood C., Watkins H., et al. (2004). Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 44, 2033–2040. 10.1016/j.jacc.2004.08.027 [DOI] [PubMed] [Google Scholar]
- Montgomery D. E., Tardiff J. C., Chandra M. (2001). Cardiac troponin T mutations: correlation between the type of mutation and the nature of myofilament dysfunction in transgenic mice. J. Physiol. 536(Pt 2), 583–592. 10.1111/j.1469-7793.2001.0583c.xd [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss R. L., Razumova M., Fitzsimons D. P. (2004). Myosin crossbridge activation of cardiac thin filaments: implications for myocardial function in health and disease. Circ. Res. 94, 1290–1300. 10.1161/01.RES.0000127125.61647.4F [DOI] [PubMed] [Google Scholar]
- Palm T., Graboski S., Hitchcock-DeGregori S. E., Greenfield N. J. (2001). Disease-causing mutations in cardiac troponin T: identification of a critical tropomyosin-binding region. Biophys. J. 81, 2827–2837. 10.1016/S0006-3495(01)75924-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B. S., Johnson R. G., Jr. (1996). Interaction of cardiotonic thiadiazinone derivatives with cardiac troponin C. J. Biol. Chem. 271, 817–823. 10.1074/jbc.271.2.817 [DOI] [PubMed] [Google Scholar]
- Pearlstone J. R., Smillie L. B. (1977). The binding site of skeletal alpha-tropomyosin on troponin-T. Can. J. Biochem. 55, 1032–1038. 10.1139/o77-154 [DOI] [PubMed] [Google Scholar]
- Razumova M. V., Bukatina A. E., Campbell K. B. (2000). Different myofilament nearest-neighbor interactions have distinctive effects on contractile behavior. Biophys. J. 78, 3120–3137. 10.1016/S0006-3495(00)76849-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rundell V. L., Manaves V., Martin A. F., de Tombe P. P. (2005). Impact of beta-myosin heavy chain isoform expression on cross-bridge cycling kinetics. Am. J. Physiol. Heart Circ. Physiol. 288, H896–H903. 10.1152/ajpheart.00407.2004 [DOI] [PubMed] [Google Scholar]
- Schaertl S., Lehrer S. S., Geeves M. A. (1995). Separation and characterization of the two functional regions of troponin involved in muscle thin filament regulation. Biochemistry 34, 15890–15894. 10.1021/bi00049a003 [DOI] [PubMed] [Google Scholar]
- Stelzer J. E., Brickson S. L., Locher M. R., Moss R. L. (2007). Role of myosin heavy chain composition in the stretch activation response of rat myocardium. J. Physiol. 579(Pt 1), 161–173. 10.1113/jphysiol.2006.119719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer J. E., Dunning S. B., Moss R. L. (2006a). Ablation of cardiac myosin-binding protein-C accelerates stretch activation in murine skinned myocardium. Circ. Res. 98, 1212–1218. 10.1161/01.RES.0000219863.94390.ce [DOI] [PubMed] [Google Scholar]
- Stelzer J. E., Larsson L., Fitzsimons D. P., Moss R. L. (2006b). Activation dependence of stretch activation in mouse skinned myocardium: implications for ventricular function. J. Gen. Physiol. 127, 95–107. 10.1085/jgp.200509432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer J. E., Patel J. R., Moss R. L. (2006c). Acceleration of stretch activation in murine myocardium due to phosphorylation of myosin regulatory light chain. J. Gen. Physiol. 128, 261–272. 10.1085/jgp.200609547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stienen G. J., Zaremba R., Elzinga G. (1995). ATP utilization for calcium uptake and force production in skinned muscle fibres of Xenopus laevis. J. Physiol. 482 (Pt 1), 109–122. 10.1113/jphysiol.1995.sp020503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardiff J. C., Factor S. M., Tompkins B. D., Hewett T. E., Palmer B. M., Moore R. L., et al. (1998). A truncated cardiac troponin T molecule in transgenic mice suggests multiple cellular mechanisms for familial hypertrophic cardiomyopathy. J. Clin. Invest. 101, 2800–2811. 10.1172/JCI2389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobacman L. S., Nihli M., Butters C., Heller M., Hatch V., Craig R., et al. (2002). The troponin tail domain promotes a conformational state of the thin filament that suppresses myosin activity. J. Biol. Chem. 277, 27636–27642. 10.1074/jbc.M201768200 [DOI] [PubMed] [Google Scholar]
- Willott R. H., Gomes A. V., Chang A. N., Parvatiyar M. S., Pinto J. R., Potter J. D. (2010). Mutations in Troponin that cause HCM, DCM AND RCM: what can we learn about thin filament function? J. Mol. Cell. Cardiol. 48, 882–892. 10.1016/j.yjmcc.2009.10.031 [DOI] [PubMed] [Google Scholar]