

Abstract
Genetically engineered T cells expressing CD19-specific chimeric antigen receptors (CARs) have shown impressive activity against B cell malignancies, and preliminary results suggest that T cells expressing a first generation disialoganglioside (GD2)-specific CAR can also provide clinical benefit in patients with neuroblastoma. We sought to assess the potential of GD2-CAR therapies to treat pediatric sarcomas. We observed that 18/18 (100%) of osteosarcomas, 2/15 (13%) of rhabdomyosarcomas, and 7/35 (20%) of Ewing sarcomas expressed GD2. T cells engineered to express a third generation GD2-CAR incorporating the 14g2a-scFv with the CD28, OX40, and CD3ζ signaling domains (14g2a.CD28.OX40.ζ) mediated efficient and comparable lysis of both GD2+ sarcoma and neuroblastoma cell lines in vitro. However in xenograft models, GD2-CAR T cells had no antitumor effect against GD2+ sarcoma, despite effectively controlling GD2+ neuroblastoma. We observed that pediatric sarcoma xenografts, but not neuroblastoma xenografts, induced large populations of monocytic and granulocytic murine myeloid-derived suppressor cells (MDSCs) that inhibited human CAR T-cell responses in vitro. Treatment of sarcoma-bearing mice with all-trans retinoic acid (ATRA) largely eradicated monocytic MDSCs and diminished the suppressive capacity of granulocytic MDSCs. Combined therapy using GD2-CAR T cells plus ATRA significantly improved antitumor efficacy against sarcoma xenografts. We conclude that retinoids provide a clinically accessible class of agents capable of diminishing the suppressive effects of MDSCs, and that co-administration of retinoids may enhance the efficacy of CAR therapies targeting solid tumors.
Keywords: Chimeric antigen receptor (CAR), immunotherapy, myeloid-derived suppressor cells (MDSC), sarcoma, all-trans retinoic acid (ATRA)
Introduction
Chimeric antigen receptors (CARs) are synthetic immune receptors that link an antigen-binding domain, commonly a mAb-derived single chain variable fragment (scFv), to T-cell signaling domains (1,2). T cells genetically engineered to express CARs mediate non-MHC restricted, tumor-specific killing and provide new opportunities for the adoptive immunotherapy of cancer. This novel class of therapeutics has demonstrated impressive anti-leukemic activity against B cell hematologic malignancies (3-10), but has not yet shown clear evidence for efficacy against solid tumors. Studies using CARs targeting the α-folate receptor on ovarian cancer (11) or carboxy-anhydrase-IX on renal cell carcinoma (12) showed no objective responses in patients. CARs targeting the disialoganglioside GD2 on neuroblastoma showed modest antitumor activity in clinical trials, with 3 of 11 patients treated with active disease achieving a complete remission (13,14).
The basis for the limited clinical activity of CARs against solid tumors observed thus far is likely multifactorial. The GD2-specific CAR utilized in the study noted above incorporated only the CD3ζ signaling domain (i.e., a first generation CAR) and did not incorporate additional costimulatory domains that have since been demonstrated to enhance CAR expansion and persistence (8). Most current CARs under study incorporate one or two costimulatory domains in addition to CD3ζ. We have demonstrated that CAR potency can be diminished via T-cell exhaustion induced by tonic, antigen-independent CAR signaling (15). This phenomenon is present in CARs incorporating the 14g2a scFv, though the effects are diminished by anti-exhaustion signaling provided by TNFR family member costimulatory receptors. Finally, solid tumors evade immune responses via an immunosuppressive microenvironment, which may pose a key barrier to the efficacy of CAR therapies tested thus far (16). Myeloid-derived suppressor cells (MDSCs) have emerged as important contributors to solid tumor immune evasion. MDSCs are a heterogeneous population of immature myeloid cells that are expanded by and recruited to tumors. MDSCs dampen T-cell responses through numerous mechanisms, including depletion of essential nutrients via production of arginase I, iNOS, and indoleamine 2,3 dioxygenase, as well as production of suppressive reactive oxygen species, nitrosylation of chemokines, and expression of PD-L1 (17-20). The impact of MDSCs on CAR therapies has not been systemically studied to date.
GD2 is a validated cell surface antigen expressed ubiquitously in neuroblastoma. In addition to the promising early clinical results seen with first generation GD2-specific CAR T cells, clinical studies have also demonstrated improved survival in patients with high-risk neuroblastoma when mAbs to GD2 are administered in combination with standard therapy (21). GD2 expression has been reported in sarcomas (22-24). We sought to evaluate GD2 expression on the three most common pediatric sarcomas (osteosarcoma, rhabdomyosarcoma, and Ewing sarcoma) and to assess whether a third-generation GD2-CAR incorporating CD28 and OX40 costimulatory domains (GD2-CAR.OX40.28.ζ) could mediate antitumor activity in preclinical models of neuroblastoma and pediatric sarcoma. Here we report high GD2 expression on a majority of pediatric osteosarcomas and that a third generation GD2-CAR effectively lysed GD2+ osteosarcoma and neuroblastoma cell lines in vitro. In vivo, however, GD2-CAR T cells mediated potent antitumor activity against neuroblastoma xenografts, but only minimal activity against osteosarcoma xenografts. The lack of antitumor activity was associated with sizable expansions of CD11b+Ly6G+ murine MDSCs that suppress human T cells. Treatment of animals with all-trans retinoic acid (ATRA) diminished the number and suppressive functionality of MDSCs, and co-administration of ATRA with GD2-CAR T-cell therapy resulted in enhanced antitumor effects against sarcoma xenografts in vivo, compared to either agent alone.
Materials and Methods
Mice
Immunocompromised NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl /SzJ) were purchased from Jackson Laboratories. All studies were conducted according to NCI Animal Care and Use Committee approved protocols.
Cells and culture conditions
All cell lines used in this report were obtained between 2006 and 2014, authenticated by STR fingerprinting in 2014 (DDC Medical), and validated to be mycoplasma free by PCR. The 143b, G292, and MG63 lines were provided by C. Khanna (NCI, NIH); EW8, RMS559, and RH30 lines by L. Helman (NCI, NIH); Kelly and LAN5 lines by C. Thiele (NCI, NIH); 293GP line by the Surgery Branch, NCI, NIH; and a PG13 based stable producer line of the third generation GD2-CAR vector (SGF.iCasp9.2A.14g2a.CD28.OX40.ζ) by the Center for Cell and Gene Therapy, Baylor College of Medicine (25-28). The 293GP and the GD2-CAR vector producer lines were cultured in DMEM. All other cell lines were cultured in RPMI-1640. DMEM and RPMI-1640 media were supplemented with 10% heat inactivated fetal bovine serum (FBS, Gemini Bioproducts), 10 mM HEPES, 100 U/mL penicillin, 100 ug/mL streptomycin, and 2 mM L-glutamine (Invitrogen). Human PBMCs from healthy donors were obtained from the Department of Transfusion Medicine, NIH Clinical Center, under an NIH IRB approved protocol, after informed consent. PBMCs were depleted of monocytes by plastic adherence and cryopreserved. PBMCs were cultured in AIM-V (Invitrogen), supplemented with 5% FBS, 10 mM HEPES, 100 U/mL penicillin, 100 ug/mL streptomycin, 2 mM L-glutamine and r-human IL2 (aldesleukin, Prometheus).
CAR retroviral transduction of T cells
Retroviral supernatant for the third generation GD2-CAR (SGF.iCasp9.2A.14g2a.CD28.OX40.ζ) was harvested from the producer cell clone. Retroviral supernatants of the previously described GD2-specific CARs incorporating either CD28 or 4-1BB (MSGV.14g2a.CD28.ζ or MSGV.14g2a.4-1BB.ζ) were produced by transient transfection of the 293GP cell line using the RD114 envelope protein (15). Cryopreserved PBMCs were thawed and activated using αCD3/αCD28 beads (Invitrogen) at a 3:1 bead:T-cell ratio with 40 IU/mL rhIL2 for 3 days. Non-tissue coated 6-well plates were coated with 24 ug/well retronectin (Takara) for 2 hours at room temperature, and then blocked with 2.5% BSA for 30 min at room temperature. Plates were spin-coated with retroviral supernatant at 3050 rpm at 32°C for 3 hours. Activated human lymphocytes were added at 106 cells per well. Transduction was repeated the following day and cells were expanded in AIM-V media with 300 IU/mL rhIL2.
Lytic assay
The ability of CAR T cells to lyse target cell lines in vitro was evaluated using a 51Cr release assay. Target cells were labeled with 51Cr and incubated with GD2-CAR transduced effector T cells at varying effector-to-target (E:T) ratios. After 8 hours of co-incubation at 37°C, supernatant was removed and the amount of 51Cr released into the media was assessed using a Top Count reader. Lysis was calculated as follows: % lysis = [(experimental lysis-spontaneous lysis)/(maximum lysis-spontaneous lysis)] × 100%, where maximal lysis was induced by incubation in a 2% Triton X solution.
Chemokine production assay
G-CSF production by tumor cell lines was evaluated within supernatants by ELISA (R&D Systems). IL8 production by tumor cell lines was evaluated with the MesoScale Discovery Pro-Inflammatory multi-array 96 well system.
MDSC suppression assays
Where indicated, mice received surgically-implanted subcutaneous ATRA pellets (21 day timed release; 5mg; Innovative Research of America) or sham surgeries one day prior to tumor inoculation. MDSCs were then isolated via CD11b or Ly6G magnetic selection from spleens of tumor-bearing mice 15-20 days after tumor inoculation. Isolated MDSCs were titrated into human T-cell cultures at varying MDSC:T-cell ratios and incubated overnight. For flow-based proliferation assays, human T cells were pre-labeled with CellTrace Violet or CSFE (Invitrogen). Following overnight incubation, either human αCD3/αCD28 coated stimulatory beads (1:1) or the GD2-CAR anti-idiotype 1A7 antibody (0.5 μg/mL; Biological Research Branch of NCI) and a cross-linking anti-mouse F(ab’)2 (2.5 μg/mL, Jackson Immunoresearch) were added to the co-culture to serve as a proliferation stimulus. After 4-5 addition days of incubation, cells were harvested and proliferation evaluated by either flow cytometry (percent of divided calculated using FlowJo 9 software, Treestar) or direct cell count measurements. All MDSC suppression assays were conducted in custom RPMI 1640 containing physiologic levels of L-arginine (150 mM) supplemented with 10% FBS, 50 mM 2-mercaptoethanol, 10 mM HEPES, 100 U/mL penicillin, 100 ug/mL streptomycin, and 1 mM sodium pyruvate.
Flow cytometry and analysis
GD2 expression on tumor cell lines was performed using the anti-GD2 (clone 14g2a; BD Pharmingen) and compared to isotype controls. T-cell transduction was measured using the 14g2a anti-idiotype, clone 1A7 (Biological Research Branch of NCI) conjugated to Dylight 650 or 488 (Pierce Protein Biology Products). T-cell phenotypes were identified using the following antibodies to human CD4 (clone OKT4; eBioscience), human CD8 (clone RPA-T8; eBioscience), human CD45 (clone H130; eBioscience), human PD-1 (clone eBioJ105, eBioscience), human TIM-3 (clone F35-2E2, eBioscience), and human LAG-3 (clone 3DS223H, eBioscience). MDSCs were evaluated using antibodies to Ly6C (clone HK1.4; eBioscience), Ly6G (clone 1A8; eBioscience), CD11b (clone M1/70; eBioscience). All antibodies were used per manufacturers recommendations. Live/dead cells were distinguished with Fixable Viability Dye eFluor 780 or 506 (eBioscience). Flow cytometry was done using FACS Fortessa with FACS Diva software (BD Biosciences) and analyzed by FlowJo 9 software (Treestar).
Immunohistochemistry
Frozen tissue samples were obtained from surgical samples at the time of clinically indicated tumor resection or biopsy after informed consent. Sections were sliced at 5-micron thickness, fixed in cold acetone for 10 minutes, dried at room temperature for 5 minutes, and rehydrated in PBS for 10 minutes. Samples were quenched with endogenous peroxidase block (DAKO) for 10 minutes, washed for 5 minutes, and incubated for 60 minutes at 37°C with mouse anti-human GD2 (14G2a, Millipore) diluted in DAKO’s antibody diluent with background reducing components to a 1:100 concentration. After washing, sections were incubated for 30 minutes in anti-mouse (biotinylated goat anti-mouse IgG, Vector, Burlingame, CA) at a concentration of 2.5 μg/mL. Sections were washed with DAKO wash buffer, incubated in DAKO peroxidase substrate solution for 5 minutes, and washed in PBS. The reaction was developed by a 2-5 minute incubation with the of 3’-3’ diaminobenzidine chromogenic solution (Vector). Slides were then counterstained with hematoxylin, dehydrated with a series of alcohol solutions (50%-100%), followed by three changes of xylene and mounted with Cytoseal XYL (Thermo Scientific). Analysis was performed using standard microscopy.
Tumor models and treatment
Xenograft studies were performed using NSG mice 6-12 weeks of age. Mice were inoculated with 5 × 105 or 106 143b, EW8, or Kelly cells either periosteal to the tibia or subcutaneously on day 0. Where noted, tumors were injected with Matrigel (Corning) diluted 1:1 in PBS. Human GD2-CAR transduced or Mock T cells were adoptively transferred 3-5 days later, as indicated in individual experiments. Mice received cytokine support of 1 μg IL7 (Cytheris Inc.) and 5 μg M25 antibody (anti-IL7; Immunex) intraperitoneally three times per week following T-cell transfer (29). All-trans retinoic acid pellets (21 day timed release; 5mg; Innovative Research of America) were surgically implanted subcutaneously on day -1. Sham surgeries were performed on controls.
Graphs and statistical analysis
Graphs were generated using Graphpad Prism software. Graphs represent mean values ± standard error of the mean. P-values were calculated using Students t-test for comparing like groups, repeated-measures ANOVA for tumor growth curves, or Log-rank statistics for survival analyses. P<0.05 was considered statistically significant and is illustrated with an asterisk (*). Deaths of mice occurring before the establishment of tumor were censored from survival analysis.
Results
GD2 expressed on human pediatric sarcomas
To evaluate GD2 as a potential target for CAR T-cell therapies in pediatric sarcoma, we first analyzed GD2 expression on tissue samples. Frozen tissue sections from 68 pediatric sarcomas, obtained at the time of clinically indicated biopsy or resection, were analyzed via immunohistochemistry for GD2 expression (Fig. 1, Table 1, and (Supplementary Table S1). We observed that 100% of osteosarcoma samples expressed GD2 (n =18 samples from 17 patients). In 15/18 of the osteosarcoma samples (5/8 primary osteosarcomas and 10/10 metastatic osteosarcomas) expression was deemed high, based upon GD2 expression in > 60% of tumor cells. Among rhabdomyosarcoma (RMS) tissues studied (n =14 alveolar RMS and n=1 embryonal RMS), two samples were positive for GD2 and both demonstrated high-level expression. Among Ewing sarcoma tissues studied (n =35), seven were positive for GD2, but none showed high expression (Table 1). These results confirm that GD2 is a potential target for immunotherapy in patients with osteosarcoma, and suggest that GD2 could serve as a target in subsets of patients with rhabdomyosarcoma and Ewing sarcoma.
Figure 1. GD2 expression on sarcoma patient samples by immunohistochemistry.
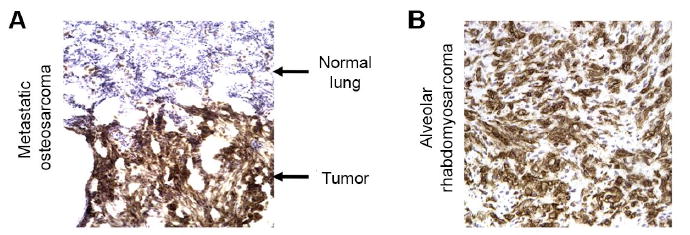
Representative immunohistochemical staining for GD2 on sarcoma patient samples, demonstrating high expression of GD2 on (A) metastatic osteosarcoma compared to adjacent lung tissue and on (B) alveolar rhabdomyosarcoma.
Table 1.
Summary of GD2 expression on pediatric sarcomas
| Samples analyzed | Number (%) of samples expressing GD2 | Number (%) of samples GD2hi |
|---|---|---|
| Osteosarcoma (n = 18) | 18 (100%) | 15 (83%) |
| Rhabdomyosarcoma (n = 15) | 2 (13%) | 2 (13%) |
| Ewing sarcoma family of tumors (n = 35) | 7 (20%) | 0 (0%) |
GD2hi = GD2 expression in >60% of tumor cells
GD2-CAR T cells efficiently lyse GD2+ sarcoma and neuroblastoma cell lines in vitro
We next sought to evaluate the capacity of T cells expressing a third generation GD2-CAR (14g2a.CD28.OX40.ζ) (25-28) to kill GD2+ human sarcoma cell lines in vitro, compared to killing of GD2+ neuroblastoma cell lines. GD2 was expressed on 2/2 neuroblastoma cell lines tested (Kelly, LAN5), 3/3 osteosarcoma cell lines tested (143b, MG63 and G292), 1/2 rhabdomyosarcoma cell lines tested (RMS559+, RH30−) and 1/1 Ewing sarcoma cell line tested (EW8; Fig. 2A). Surface expression was similar between the 143b osteosarcoma and Kelly neuroblastoma, and therefore these lines were chosen for further comparative studies aimed at assessing the relative efficacy of GD2-CAR T cells in targeting sarcoma versus neuroblastoma.
Figure 2. GD2-CAR T cells effectively lyse GD2+ sarcoma cell lines in vitro.
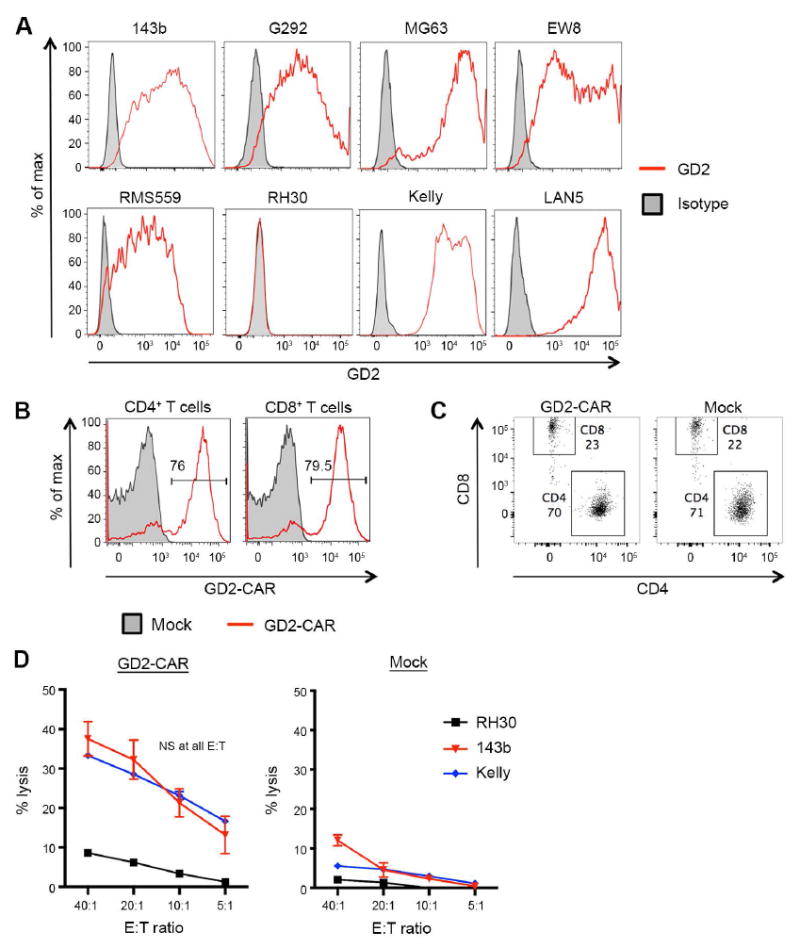
(A) Flow cytometric analysis of GD2 expression on human osteosarcoma (143b, G292, MG63), Ewing sarcoma (EW8), rhabdomyosarcoma (RMS559, RH30) and neuroblastoma (Kelly, LAN5) cell lines grown in vitro. Numerous sarcoma cell lines demonstrated strong GD2 surface expression. (B) Representative transduction efficiency of GD2-CAR T cells, evaluated on day 7 following initial activation. Transduction efficiency measured by staining with clone 1A7, an anti-idiotype antibody specific for the 14g2a antibody. (C) Representative CD4:CD8 ratios of GD2-CAR and Mock T-cell products, evaluated on day 7 following initial activation. (D) Lysis of target cells measured by 51Cr release. GD2-CAR T cells equivalently lyse the GD2+ 143b and Kelly cells lines, but do not lyse the GD2 negative cell line RH30.
Human T cells were transduced with the GD2-CAR, with consistently ~70% transduction efficiencies at day 7 post-activation for both CD4+ and CD8+ T cells (Fig. 2B). CD4:CD8 ratios within both the GD2-CAR and Mock T-cell products were typically ~3:1 (Fig. 2C). 51Cr release assays demonstrated equivalent lysis of the 143b osteosarcoma cell line and the Kelly neuroblastoma line (Fig. 2D), and GD2-CAR T cells were able to lyse all GD2+ cell lines tested (Supplementary Fig. S1). Very little lysis of the GD2 negative line RH30 was observed, indicating high specificity of T cells expressing the GD2-CAR. Thus, we conclude that T cells expressing the GD2-CAR have comparable efficacy against GD2+ sarcoma versus neuroblastoma tumor lines in vitro.
Poor in vivo efficacy associated with differential induction of MDSCs
We next compared the in vivo efficacy of GD2-CAR T cells against the 143b osteosarcoma line versus the Kelly neuroblastoma line in a xenograft model using immunodeficient NOD.SCID.γc−/− (NSG) mice. Despite lysing 143b tumor cells in vitro, GD2-CAR T cells showed minimal antitumor effect against 143b osteosarcomas tumors in vivo (Fig. 3A) and did not improve survival (Fig. 3B). Similar results were also seen against EW8 xenografts (Supplementary Fig. S2A-B). This poor in vivo efficacy could not be fully attributed to T-cell exhaustion induced by antigen-independent signaling of GD2-specific CARs (15), because the same GD2-CAR T cells provided a significant antitumor benefit against Kelly neuroblastoma tumors in vivo (Fig. 3C-D).
Figure 3. GD2-CAR has poor in vivo efficacy against the 143b osteosarcoma cell line in a xenograft model that is not fully attributable to T-cell exhaustion.
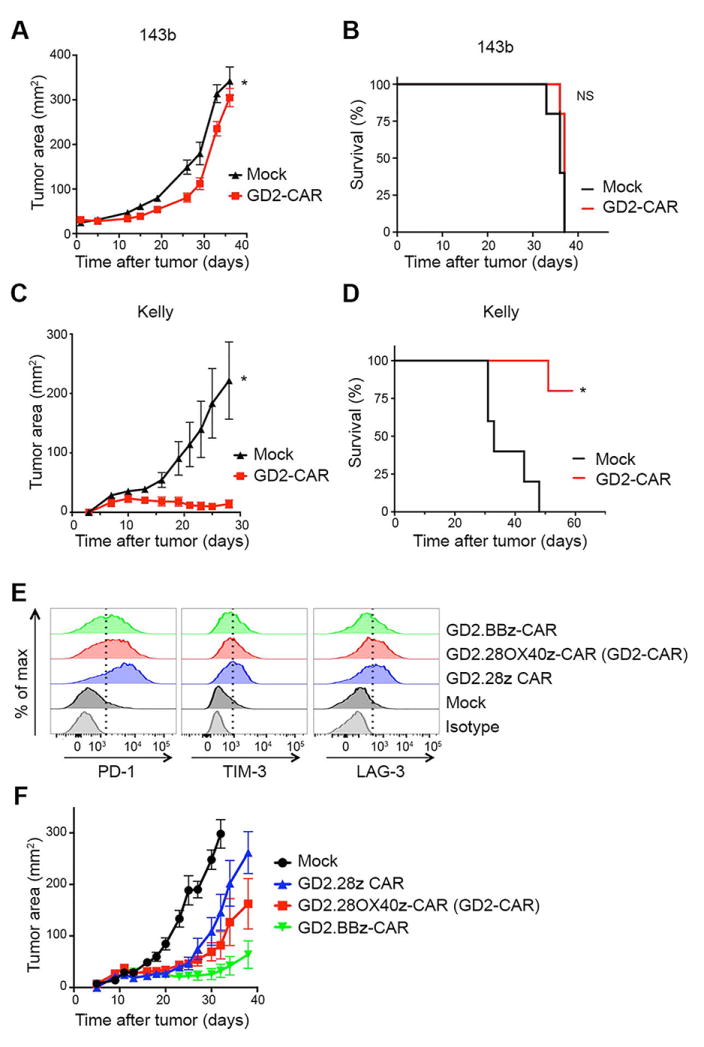
(A) Tumor growth curves and (B) survival of NSG mice inoculated with 106 143b cells periosteally on day 0, followed by adoptive transfer of 107 untransduced-Mock or GD2-CAR T cells on day 3. GD2-CAR T cells have no antitumor effect against 143b. n=5 mice/group. (C) Tumor growth curves and (D) survival of NSG mice inoculated with 106 Kelly cells subcutaneously with Matrigel on day 0, followed by adoptive transfer of 107 untransduced-Mock or GD2-CAR T cells on day 3. GD2-CAR T cells prevent outgrowth of Kelly tumors leading to enhanced overall survival. n=5 mice/group. (E) Exhaustion marker expression on T cells transduced with GD2-specific CARs incorporating different costimulatory domains (GD2-CAR, a third generation CAR with CD28 and OX40; a second generation CAR with CD28; and a second generation CAR with 4-1BB) 9 days following initial activation. The third generation GD2-CAR T cells have an intermediate expression of exhaustion markers compared to the second generation CD28 CAR and the 4-1BB CAR. (F) Tumor growth curves of NSG mice inoculated with 106 Kelly cells subcutaneously with Matrigel on day 0, followed by adoptive transfer of 107 untransduced-Mock or GD2-specific CAR T cells on day 3. The third generation GD2-CAR T cells had an intermediate efficacy compared to the second generation CD28 CAR and the 4-1BB CAR. n=10 mice/group.
All of the studies presented thus far were conducted with a third generation GD2-CAR incorporating both CD28 and OX40. The third generation CAR demonstrates intermediate exhaustion and functionality, with less exhaustion and improved in vivo activity compared to a second generation GD2-specific CAR incorporating only CD28 (Fig. 3E-F), but increased exhaustion and decreased functionality compared to a second generation CAR incorporating 4-1BB. These findings are consistent with previous reports that CD28 signaling contributes to exhaustion, while TNFR family member signaling ameliorates exhaustion (15).
The inability to fully attribute poor in vivo potency to T-cell exhaustion led us to question whether immunosuppressive properties of sarcomas that manifest in vivo, rather than CAR intrinsic properties, could be inhibiting GD2-CAR T-cell therapies. We have observed that granulocytic MDSCs are primary mediators of immune suppression in murine models of sarcoma (30). Therefore, we assessed whether tumor induced MDSCs could also be responsible for the poor efficacy of GD2-CAR T-cell therapies against sarcoma tumors.
We monitored the blood of NSG mice inoculated with sarcoma (143b and EW8) or neuroblastoma (Kelly) tumors for the expansion of myeloid populations, compared to non-tumor bearing mice. NSG mice bearing 143b and EW8 tumors showed an increase in CD11b+Ly6G+ and CD11b+Ly6C+ myeloid cell populations, compared to nontumor-bearing controls (Fig. 4A-B and (Supplementary Fig. S2C-E and S3A).We observed granulocytic CD11b+Ly6G+Ly6C− cells to be the major MDSC subset (>90% of CD11b+ cells) in the blood, spleen, and tumor compartments of sarcoma bearing mice. Despite bearing similarly sized tumors (Supplementary Fig. S3B), mice with Kelly tumors showed no significant expansion of similar myeloid cell populations (Fig. 4A-C and Supplementary Fig. S3A).
Figure 4. Human sarcoma implanted in NSG mice induces expansion of host MDSCs.
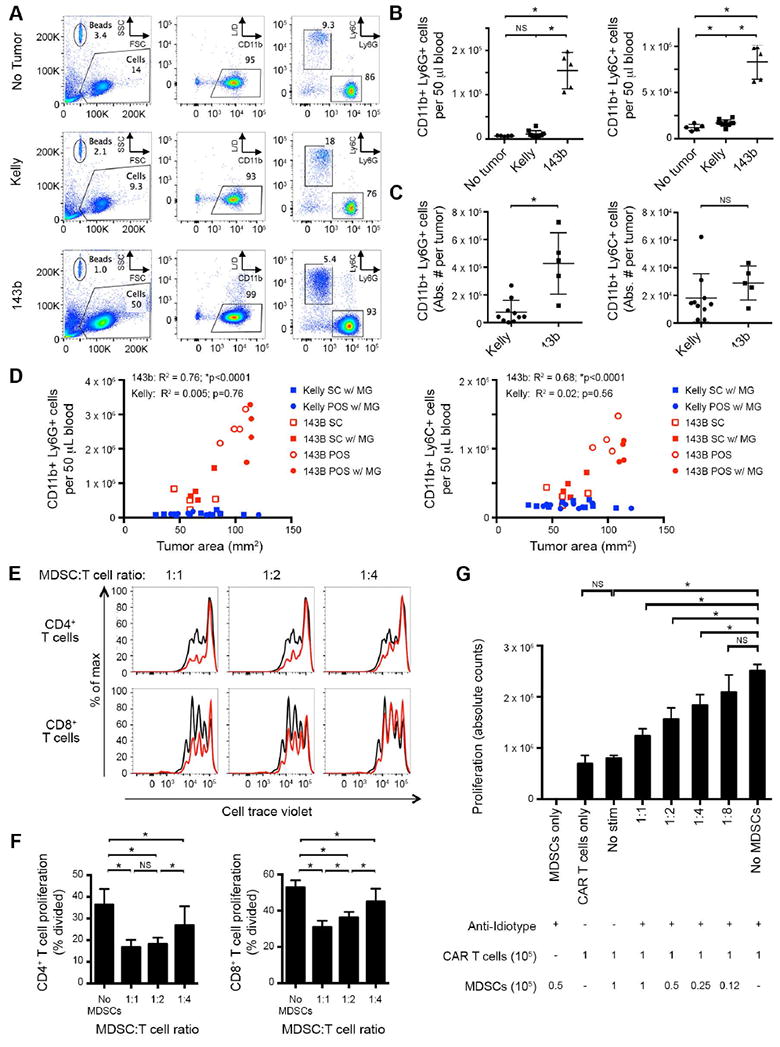
(A) Representative flow cytometry plots of peripheral blood, evaluating presence of CD11b+Ly6G+ or CD11b+Ly6C+ MDSCs in mice inoculated with 106 143b osteosarcoma periosteally or 106 Kelly cells subcutaneously with Matrigel. Evaluated day 28 after tumor inoculation. L/D = live/dead stain. (B) Cumulative data from A, quantifying absolute number of CD11b+Ly6G+ (left) and CD11b+Ly6C+ (right) cells per 50 μl of blood. 143b tumors induce significant expansion of both CD11b+Ly6C+ and CD11b+Ly6G+ populations, compared to non-tumor controls or Kelly tumor-bearing mice with similarly sized tumors. (C) Absolute number of CD11b+Ly6G+ (left) and CD11b+Ly6C+ (right) cells per tumor. 143b tumors contain significant CD11b+Ly6G+ populations, compared to Kelly tumors. (D) Impact of tumor location and Matrigel on induction of CD11b+Ly6G+ (left) and CD11b+Ly6C+ (right) cells in blood. NSG mice were inoculated with 106 143b (red) or Kelly (blue) tumor cells. MDSCs were quantified in the blood 20 days post tumor injection and plotted as a function of tumor size. Tumors were placed subcutaneously (SC, square) or periosteally (POS, circle), with (filled) or without (open) Matrigel (MG). R2 and p value statistics calculated from linear regression of pooled 143b and Kelly data. (E) Cell trace violet dilution of human T cells following co-incubation with murine CD11b+ MDSCs in vitro. Human αCD3/αCD28 beads were added as a proliferative stimulus. Flow cytometry was performed 4 days later. αCD3/αCD28 bead-induced proliferation without MDSCs in co-culture shown in black. CD11b+ MDSCs isolated from 143b tumor-bearing mice suppress human T-cell proliferation in a dose dependent manner (red). (F) Quantification of T-cell proliferation in E, measured as percentage of T cells divided. (G) Absolute cell counts of GD2-CAR T cells (9 days after initial activation) co-incubated with murine CD11b+ MDSCs derived from 143b tumor bearing mice. GD2-CAR T cells were stimulated via the CAR with the anti-idiotype 1A7 antibody (0.5 μg/mL) and a cross-linking anti-mouse F(ab’)2 (2.5 μg/mL). Cell counts from cultures were performed 5 days after CAR stimulation. CD11b+ MDSCs isolated from 143b tumor-bearing mice suppress human CAR T-cell proliferation in a dose dependent manner.
The difference in MDSC induction between 143b vs Kelly tumors appeared to be tumor cell intrinsic, rather than due to differences in tumor location or use of Matrigel in these tumor models. Independent of tumor location and use of Matrigel, 143b tumors induced significantly larger MDSC populations that correlated with tumor burden (Fig. 4D), as has been previously described (31,32). In contrast, Kelly tumors did not induce MDSCs, regardless of tumor burden or location. To identify potential mechanisms by which sarcoma may induce murine MDSCs in vivo, we evaluated 143b, EW8 and Kelly cell culture supernatants for the presence of growth factors and chemokines reported to influence MDSC expansion and migration. Associated with high level MDSC expansion in vivo, we observed that 143b cells produce greater quantities of G-CSF and IL8 compared to Kelly cells in vitro (Supplementary Fig. S3C). Such factors have been implicated in MDSC induction and expansion (30). However, IL8 was only modestly elevated in EW8 bearing mice, suggesting additional mechanisms may also be involved (17,33).
To assess whether the murine MDSC population could suppress the function of human T cells utilized in this xenograft model, we conducted coculture suppression assays with murine CD11b+ MDSCs and human T cells. Briefly, splenic CD11b+ cells were isolated from sarcoma tumor-bearing mice and were cultured at varying ratios with human T cells stimulated either through endogenous receptors with αCD3/αCD28 beads, or through the GD2-CAR with a crosslinking anti-idiotype antibody. After 4-5 days of in vitro culture, we assessed proliferation of human cells. Murine CD11b+ myeloid cells induced by human sarcoma xenografts inhibited human T-cell proliferative responses in a dose-dependent manner, when T cells were stimulated either through the native TCR (Fig. 4E-F and Supplementary Fig. S2D-E) or through the GD2-CAR (Fig. 4G and Supplementary Fig. S3D-E). Therefore, human sarcoma tumors implanted into immunodeficient mice expanded MDSCs that were capable of suppressing human CAR T cells.
ATRA reduces monocytic MDSCs and diminishes granulocytic MDSCs suppression
ATRA is a clinically approved drug that induces immature myeloid cells to differentiate into a nonsuppressive subtype (34-36). Therefore, we tested whether ATRA treatment altered the quantity or suppressive phenotype of 143b-induced MDSCs. NSG mice were implanted subcutaneously with a 21-day timed-release ATRA pellet prior to the injection of 143b tumor cells, and the peripheral blood MDSCs were monitored (Fig. 5A-B). We observed that ATRA treatment led to a significant reduction in the number of monocytic CD11b+Ly6C+ cells in the peripheral blood. Though the size of the granulocytic CD11b+Ly6G+ population was not consistently reduced, granulocytic MDSCs from ATRA-treated tumor-bearing mice had diminished suppressive potency compared to those from untreated tumor-bearing mice (Fig. 5C-E). Similar trends were also observed regarding the numbers of MDSCs present within tumors of mice treated with and without ATRA (Supplementary Fig. S3F). Thus, we observe that ATRA treatment of tumor-bearing mice dramatically reduces the quantity of monocytic MDSCs and diminishes the suppressive potency of granulocytic MDSCs in xenograft models of sarcoma.
Figure 5. ATRA treatment reduces number and suppressive capacity of murine MDSCs induced by sarcoma in NSG mice.
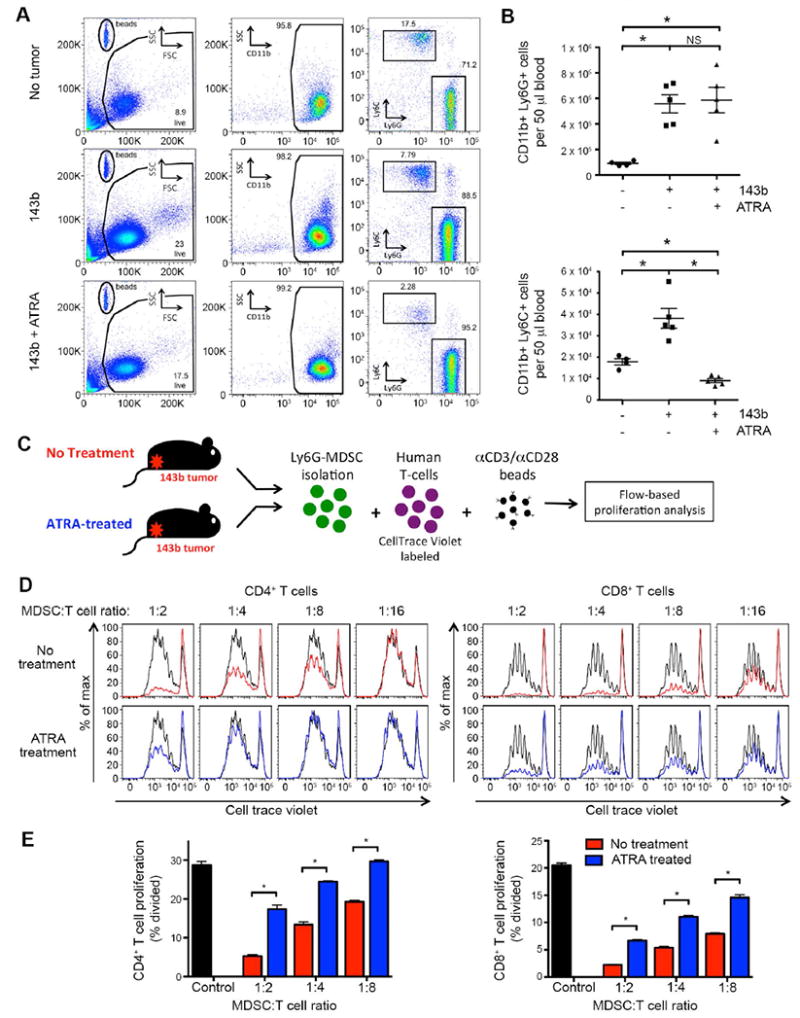
(A) Representative flow cytometry plots of peripheral blood, evaluating presence of CD11b+Ly6G+ or CD11b+Ly6C+ MDSCs in mice inoculated with 106 143b osteosarcoma periosteally on day 0, treated with or without ATRA sustained-release subcutaneous pellets on day -1. Evaluated day 15 after tumor inoculation. (B) Cumulative data from A, showing the absolute number of CD11b+Ly6C+ (top) and CD11b+Ly6C+ (bottom) cells per 50 μl of blood. ATRA treatment leads to a significant reduction in CD11b+Ly6C+ cells in 143b tumor-bearing mice. The number of CD11b+Ly6G+ MDSCs observed following ATRA treatment was not consistently decreased. (C) Schematic of MDSC T-cell suppression assay. Splenic Ly6G+ cells from 143b tumor-bearing mice (± ATRA treatment on day -1) were magnetically isolated and mixed at increasing ratios with CellTrace Violet-labeled human T cells. Flow cytometry was performed 4 days later. Human αCD3/αCD28 beads were added as a proliferative stimulus. (D) CellTrace Violet dilution of CD4+ and CD8+ human T cells following co-incubation with murine MDSCs in vitro. αCD3/αCD28 bead induced proliferation without MDSCs in co-culture shown in black. CD11b+Ly6G+ MDSCs isolated from 143b tumor-bearing mice not treated with ATRA suppress human T-cell proliferation in a dose dependent manner (red). CD11b+Ly6G+ MDSC isolated from 143b mice treated with ATRA have decreased ability to suppress T-cell proliferation (blue). (E) Quantification of T-cell proliferation in D, measured as percentage of CD4+ (left) or CD8+ (right) T cells dividing. n=6/group.
ATRA treatment enhances efficacy of GD2-CAR T cells against sarcoma in vivo
We next asked whether inhibiting the suppressive capabilities of MDSCs through ATRA treatment improved the efficacy of GD2-CAR T cells against tumors. Mice were implanted with sustained-release ATRA pellets day -1 prior to inoculation with sarcoma cells, and subsequently treated with GD2-CAR T cells. Treating tumor-bearing mice with both ATRA and GD2-CAR T cells led to an enhanced antitumor effect, with delayed tumor outgrowth and improved survival (Fig. 6A-B and Supplementary Fig. S2A-B) compared to animals receiving ATRA plus Mock T cells. We could detect no additional benefit of ATRA to GD2-CAR treatments in animals bearing neuroblastoma (Supplementary Fig. S4A).
Figure 6. Co-administration of ATRA with GD2-CAR T cells leads to enhanced antitumor efficacy against 143b osteosarcoma tumors in NSG mice.
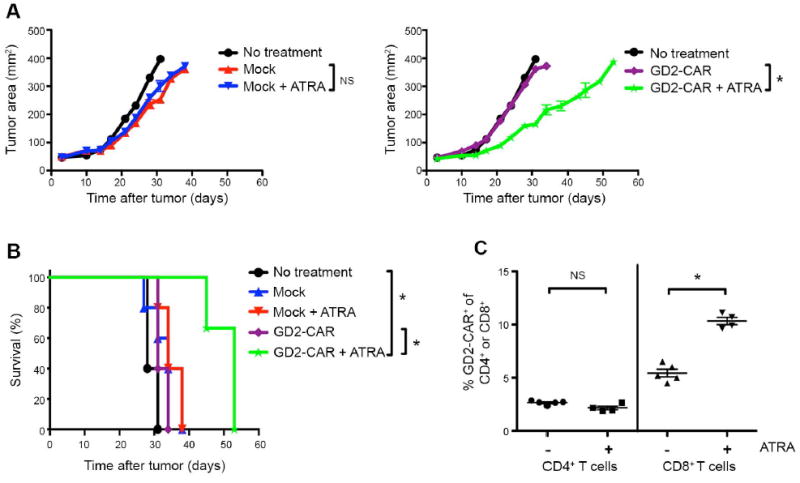
(A) Tumor growth curves and (B) survival of NSG mice inoculated with 5 × 105 143b cells on day 0, followed by adoptive transfer of 30 × 106 untransduced-Mock or GD2-CAR T cells on day 3 and 15 × 106 on day 5. Where indicated, mice received ATRA pellets subcutaneously or sham surgeries day -1 before tumor injection. GD2-CAR T cells have enhanced antitumor effect and prolonged survival against 143b tumors in the presence of ATRA. n=3-5 mice/group. (C) Frequency of GD2-CAR+ expression within CD4+ (left) and CD8+ (right) T cells populations in the peripheral blood 15 days after adoptive transfer, assessed by flow cytometry. Adoptively transferred T cells distinguished from host murine cells by hCD45+. CAR+ identified by 1A7 anti-idiotype.
Further suggesting that ATRA mediated modulation of MDSCs led to enhanced T-cell reactivity, we observed that 143b tumor-bearing mice treated with GD2-CAR T cells and ATRA had a higher frequency of CD8+ CAR+ T cells in the peripheral blood compared to animals which did not receive ATRA (Fig. 6C). Similar increases in GD2-CAR were not observed in Kelly tumor-bearing mice, which did not induce MDSC expansion and thus would not be expected to have enhanced T-cell reactivity following ATRA treatment (Supplementary Fig. S4B). The improved antitumor efficacy of GD2-CAR T cells in ATRA treated mice could not be attributed to changes in growth kinetics of the 143b tumor, because we saw no difference in the tumor growth between animals receiving Mock T cells alone versus those receiving Mock T cells + ATRA (Fig. 6A). Finally, we found no evidence that ATRA treatment increased GD2 antigen expression on tumors upon exposure either in vitro or in vivo (Supplementary Fig. S5). Together, these results suggest that ATRA may improve efficacy of the GD2-CAR against sarcoma tumors through its effects on MDSCs.
In addition to causing maturation of MDSCs, ATRA has also been reported to increase the frequency of T regulatory cells (37,38). Such a secondary effect of ATRA could theoretically inhibit the efficacy of CAR T-cell therapies. Therefore, we evaluated whether ATRA treatment led to an increased incidence of FOXP3+ T cells in vivo in this model system. Analysis of peripheral blood 21 days after T-cell injection demonstrated no detectable FOXP3 cells in either ATRA treated or non-treated mice (Supplementary Fig. S6). Taken together, we conclude that ATRA treatment enhances GD2-CAR T-cell efficacy against sarcoma tumors that induce MDSCs, without causing an increase in T regulatory CAR T cells.
Discussion
Recent clinical trials have demonstrated impressive activity of CAR T cells targeting CD19 on hematologic malignancies (3-10). However, similar results have not yet been observed using CARs to target antigens on other malignancies, including solid tumors (11-14). Despite this, we were encouraged by preliminary results suggesting clinical benefit of a first generation GD2-specific CAR in patients with neuroblastoma and sought to assess the efficacy of a costimulation-endowed CAR against pediatric sarcomas. We assessed GD2 expression on three common histologies and confirmed that it is expressed on essentially all osteosarcomas, with a sizable fraction of primary and metastatic tumors expressing a high level (22,39,40). Our results confirm those from Kailayangiri et al. demonstrating GD2 on Ewing sarcoma cell lines, although we did not observe significant expression of GD2 on Ewing sarcoma tissues (23). Information on GD2 expression in rhabdomyosarcoma is limited (41), and our data suggests that it may be a relevant target for a minority of patients. Together, these results indicated that GD2 is a rational target for the developing of CAR-based therapies for some pediatric sarcomas.
Using a third generation GD2-CAR that incorporated the CD28 and OX40 costimulatory domains, we observed equivalent in vitro killing of GD2-expressing cells, regardless of histology. However, we observed poor in vivo activity of GD2-CAR T cells against the 143b osteosarcoma cell line, despite strong in vivo activity against the Kelly neuroblastoma cell line. Granulocytic MDSCs are a primary mediator of immune evasion in murine models of sarcoma (30) so, we examined whether similar MDSCs were contributing to an immunosuppressive microenvironment that inhibits in vivo efficacy of GD2-CAR T cells against sarcomas. However, whether human tumors induce murine MDSCs in xenograft models of cancer, and whether such cells can suppress human T cells, has not been reported. Our data demonstrate that murine MDSCs can be expanded by human tumors in xenograft models of cancer, and that such murine MDSCs can suppress human T cells. The observation that MDSC-mediated T-cell suppression can cross species is consistent with a sizable literature that implicates nutrient depletion as a major mechanism of MDSC suppression (17-20). Our findings also suggest a possible role for G-CSF and IL8 in the recruitment of murine MDSCs, two factors known to influence MDSC expansion and migration. Interleukin-8 was only modestly elevated in EW8 bearing mice, consistent with previous evidence that such populations can be induced by a variety of cytokines and/or chemokines (17,33). The mechanism by which ATRA alters MDSC recruitment is likely complex and may differ depending on the model. Overall, the identification that murine MDSCs contribute to an immunosuppressive microenvironment within human xenograft tumors is an important finding that will likely impact future evaluation of immunotherapies within xenograft models.
Our results are also consistent with increasing evidence that MDSC populations are expanded in patients with sarcoma (30,42-44). We recently reported that human sarcoma patients have increased numbers of granulocytic MDSCs within unfractionated blood compared to healthy controls, and increased serum levels of cytokines responsible for MDSC expansion/migration (30). Fibrocyte MDSCs and hematopoietic stem/progenitor cells that develop into MDSCs are expanded in patients with pediatric sarcomas (42,43), and we reported that expanded populations of monocytic and granulocytic cells adversely affected the efficiency with which GD2-CAR T cells could be generated from apheresis products obtained from children and young adults with osteosarcoma and neuroblastoma (44). Though our present study highlights sarcoma-induced expansion of MDSCs and illustrates differential amounts of expansion between sarcoma cell lines and one neuroblastoma cell line, the absence of MDSCs may not be generalizable to other neuroblastoma models or neuroblastoma patients. Indeed, studies have suggested a clinical significance of tumor-associated myeloid cells in neuroblastoma in some settings (45,46). Thus, further work is needed to fully understand how pediatric tumors differentially induce MDSCs.
Sarcoma-induced MDSCs were associated with poor CAR T-cell efficacy against xenografts in vivo, implicating the immunosuppressive solid tumor microenvironment as a modulator of CAR T-cell efficacy. We found this by directly measuring MDSC inhibition of T-cell responses that had been triggered through CAR receptor signaling. Consistent with our findings, a reduction in MDSCs is associated with the enhanced efficacy of both VEGFR2-CAR T cells upon constitutive IL12 expression (47) and of ErbB2-CAR T cells upon anti-PD1 therapy (48). The CARS in both of these reports had the CD28 costimulatory domain in common with our GD2-CAR, but one was a third generation CAR that included 4-1BB (VEGFR2.CD28.4-1BB. ζ) and the other a second generation with only CD28 (ErbB2.CD28.ζ). . We chose to focus our evaluation on the third generation GD2-CAR with CD28 and OX40 costimulatory domains given its current use in clinical trials (NCT02107963). However, it is possible that different costimulatory domains may enhance susceptibility to MDSC suppression, and remains a focus of our ongoing work.
ATRA is one of the few clinically available drugs that modulates MDSCs in both animal models and in human patients (35,36). It mediates potent myeloid cell differentiating effects in the setting of acute promyelocytic leukemia and is well tolerated in this setting with cytotoxic chemotherapy (49), suggesting that it could be integrated into immunotherapeutic regimens for cancer. Use of our sarcoma xenograft models found that cotreatment with ATRA and GD2-CAR T cells led to enhanced antitumor effects compared to either single agent alone, which is likely mediated through a decreased number of monocytic MDSCs and potentially also via a reduction in the suppressive capacity of granulocytic MDSCs induced by sarcoma tumors. The diminishment in the suppressive capacity of granulocytic MDSC was approximately 50%, which may explain why we observed only a modest improvement in survival. It will be of interest to determine if this survival enhancement is improved further when treating subtypes of tumors that preferentially expand monocytic MDSC. Such effects of ATRA may be due to an increased expression of glutathione synthase in MDSCs (50), leading to higher glutathione and neutralization of reactive oxygen species (ROS), which play a role in T-cell suppression and prevent MDSC differentiation. Though ATRA can also induce FOXP3+ T regulatory cells in vivo, we found no evidence of such in our model system with adoptively transferred, activated human CAR T cells. Thus, retinoids provide a clinically accessible class of agents capable of modulating MDSCs that may enhance the efficacy of CAR therapies targeting solid tumors.
In conclusion, we identify GD2 as a promising target for CAR T-cell therapies against pediatric sarcoma. Though xenograft models suggest that sarcoma-induced MDSCs are capable of inhibiting CAR T-cell efficacy in vivo, we identify ATRA as an effective therapy in diminishing the suppressive capacity of MDSCs and enhancing the in vivo efficacy of GD2-CAR T-cell against sarcoma. Our findings demonstrate the need for enhanced understanding of the immunosuppressive mechanisms that may limit immunotherapies against solid tumors, and suggest that MDSCs may play a role in preventing CAR efficacy against solid tumors in patients.
Supplementary Material
Acknowledgments
We kindly thank M. Brenner (Baylor College of Medicine) for providing the retroviral producer line for the third generation GD2-CAR vector (SGF.iCasp9.2A.14g2a.CD28.OX40.ζ).
Financial Support: Research supported by the Intramural Research Program of the NIH, NCI (AHL, SLH, JPS, YC, AJW, SR, RE, SG, MT, RJO and CML) and an NIH T32 training grant awarded to Northwestern University (T32GM008152; AHL). Research supported by a Stand Up To Cancer - St. Baldrick’s Pediatric Dream Team Translational Research Grant (SU2C-AACR-DT1113. Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research. (AHL, JPS, YC, AJW, RJO, and CLM)
Footnotes
Disclosures: The authors report no conflict of interest exists.
References
- 1.Lee DW, Barrett DM, Mackall C, Orentas R, Grupp SA. The Future Is Now: Chimeric Antigen Receptors as New Targeted Therapies for Childhood Cancer. Clinical Cancer Research. 2012;18(10):2780–90. doi: 10.1158/1078-0432.CCR-11-1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sadelain M, Brentjens R, Rivière I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discovery. 2013;3(4):388–98. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. The Lancet. 2014 doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. New England Journal of Medicine. 2014;371(16):1507–17. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor–transduced T cells. Blood. 2012;119(12):2709–20. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor–modified T cells in chronic lymphoid leukemia. New England Journal of Medicine. 2011;365(8):725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. New England Journal of Medicine. 2013;368(16):1509–18. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor–modified T cells in lymphoma patients. The Journal of Clinical Investigation. 2011;121(5):1822–26. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Science Translational Medicine. 2013;5(177):177ra38–77ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Science Translational Medicine. 2014;6(224):224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clinical Cancer Research. 2006;12(20):6106–15. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lamers CHJ, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, et al. Treatment of Metastatic Renal Cell Carcinoma With CAIX CAR-engineered T cells: Clinical Evaluation and Management of On-target Toxicity. Mol Ther. 2013;21(4):904–12. doi: 10.1038/mt.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nature Medicine. 2008;14(11):1264–70. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park JR, DiGiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Molecular therapy. 2007;15(4):825–33. doi: 10.1038/sj.mt.6300104. [DOI] [PubMed] [Google Scholar]
- 15.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21(6):581–90. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gilham DE, Debets R, Pule M, Hawkins RE, Abken H. CAR–T cells and solid tumors: tuning T cells to challenge an inveterate foe. Trends in molecular medicine. 2012;18(7):377–84. doi: 10.1016/j.molmed.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 17.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–68. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arina A, Bronte V. Myeloid-derived suppressor cell impact on endogenous and adoptively transferred T cells. Current Opinion in Immunology. 2015;33(0):120–25. doi: 10.1016/j.coi.2015.02.006. [DOI] [PubMed] [Google Scholar]
- 19.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nature Reviews Immunology. 2009;9(3):162–74. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ostrand-Rosenberg S. Myeloid-derived suppressor cells: more mechanisms for inhibiting antitumor immunity. Cancer Immunology, Immunotherapy. 2010;59(10):1593–600. doi: 10.1007/s00262-010-0855-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Anti-GD2 Antibody with GM-CSF, Interleukin-2, and Isotretinoin for Neuroblastoma. New England Journal of Medicine. 2010;363(14):1324–34. doi: 10.1056/NEJMoa0911123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roth M, Linkowski M, Tarim J, Piperdi S, Sowers R, Geller D, et al. Ganglioside GD2 as a therapeutic target for antibody-mediated therapy in patients with osteosarcoma. Cancer. 2014;120(4):548–54. doi: 10.1002/cncr.28461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kailayangiri S, Altvater B, Meltzer J, Pscherer S, Luecke A, Dierkes C, et al. The ganglioside antigen GD2 is surface-expressed in Ewing sarcoma and allows for MHC-independent immune targeting. Br J Cancer. 2012;106(6):1123–33. doi: 10.1038/bjc.2012.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki M, Cheung N-KV. Disialoganglioside GD2 as a therapeutic target for human diseases. Expert Opinion on Therapeutic Targets. 2015;19(3):349–62. doi: 10.1517/14728222.2014.986459. [DOI] [PubMed] [Google Scholar]
- 25.Yvon E, Del Vecchio M, Savoldo B, Hoyos V, Dutour A, Anichini A, et al. Immunotherapy of Metastatic Melanoma Using Genetically Engineered GD2-Specific T cells. Clinical Cancer Research. 2009;15(18):5852–60. doi: 10.1158/1078-0432.CCR-08-3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gargett T, Brown MP. Different cytokine and stimulation conditions influence the expansion and immune phenotype of third-generation chimeric antigen receptor T cells specific for tumor antigen GD2. Cytotherapy. 2015 doi: 10.1016/j.jcyt.2014.12.002. [DOI] [PubMed] [Google Scholar]
- 27.Gargett T, Fraser CK, Dotti G, Yvon ES, Brown MP. BRAF and MEK Inhibition Variably Affect GD2-specific Chimeric Antigen Receptor (CAR) T-Cell Function In Vitro. Journal of Immunotherapy. 2015;38(1):12–23. doi: 10.1097/CJI.0000000000000061. [DOI] [PubMed] [Google Scholar]
- 28.Pule MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. 2005;12(5):933–41. doi: 10.1016/j.ymthe.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 29.Martin CE, van Leeuwen EMM, Im SJ, Roopenian DC, Sung Y-C, Surh CD. IL-7/anti IL-7 mAb complexes augment cytokine potency in mice through association with IgG-Fc and by competition with IL-7R. Blood. 2013;121(22):4484–92. doi: 10.1182/blood-2012-08-449215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Science translational medicine. 2014;6(237):237ra67–37ra67. doi: 10.1126/scitranslmed.3007974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bronte V, Apolloni E, Cabrelle A, Ronca R, Serafini P, Zamboni P, et al. Identification of a CD11b+/Gr-1+/CD31+ myeloid progenitor capable of activating or suppressing CD8+T cells. Blood. 2000;96(12):3838–46. [PMC free article] [PubMed] [Google Scholar]
- 32.Ostrand-Rosenberg S, Sinha P. Myeloid-Derived Suppressor Cells: Linking Inflammation and Cancer. The Journal of Immunology. 2009;182(8):4499–506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer. 2013;13(10):739–52. doi: 10.1038/nrc3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kusmartsev S, Cheng F, Yu B, Nefedova Y, Sotomayor E, Lush R, et al. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003;63(15):4441–9. [PubMed] [Google Scholar]
- 35.Mirza N, Fishman M, Fricke I, Dunn M, Neuger AM, Frost TJ, et al. All-trans-Retinoic Acid Improves Differentiation of Myeloid Cells and Immune Response in Cancer Patients. Cancer Research. 2006;66(18):9299–307. doi: 10.1158/0008-5472.CAN-06-1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iclozan C, Antonia S, Chiappori A, Chen D-T, Gabrilovich D. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunology, Immunotherapy. 2013;62(5):909–18. doi: 10.1007/s00262-013-1396-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu L, Lan Q, Li Z, Zhou X, Gu J, Li Q, et al. Critical role of all-trans retinoic acid in stabilizing human natural regulatory T cells under inflammatory conditions. Proceedings of the National Academy of Sciences. 2014;111(33):E3432–E40. doi: 10.1073/pnas.1408780111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Golovina TN, Mikheeva T, Brusko TM, Blazar BR, Bluestone JA, Riley JL. Retinoic Acid and Rapamycin Differentially Affect and Synergistically Promote the Ex Vivo Expansion of Natural Human T Regulatory Cells. PLoS ONE. 2011;6(1):e15868. doi: 10.1371/journal.pone.0015868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poon VI, Roth M, Piperdi S, Geller D, Gill J, Rudzinski ER, et al. Ganglioside GD2 expression is maintained upon recurrence in patients with osteosarcoma. Clinical sarcoma research. 2015;5(1):4. doi: 10.1186/s13569-014-0020-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heiner JP, Miraldi F, Kallick S, Makley J, Neely J, Smith-Mensah WH, et al. Localization of GD2-specific monoclonal antibody 3F8 in human osteosarcoma. Cancer Res. 1987;47(20):5377–81. [PubMed] [Google Scholar]
- 41.Chang HR, Cordon-Cardo C, Houghton AN, Cheung NK, Brennan MF. Expression of disialogangliosides GD2 and GD3 on human soft tissue sarcomas. Cancer. 1992;70(3):633–8. doi: 10.1002/1097-0142(19920801)70:3<633::aid-cncr2820700315>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 42.Zhang H, Maric I, DiPrima MJ, Khan J, Orentas RJ, Kaplan RN, et al. Fibrocytes represent a novel MDSC subset circulating in patients with metastatic cancer. 2013:1105–13. doi: 10.1182/blood-2012-08-449413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giles AJ, Reid CM, Evans JD, Murgai M, Vicioso Y, Highfill SL, et al. Activation of Hematopoietic Stem/Progenitor Cells Promotes Immunosuppression Within the Pre metastatic Niche. Cancer Research. 2016;76(6):1335–47. doi: 10.1158/0008-5472.CAN-15-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stroncek DF, Ren J, Lee DW, Tran M, Frodigh SE, Sabatino M, et al. Myeloid cells in Peripheral Blood Mononuclear Cell Concentrates Inhibit the Expansion of Chimeric Antigen Receptor T cells. Cytotherapy. 2016 doi: 10.1016/j.jcyt.2016.04.003. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Asgharzadeh S, Salo JA, Ji L, Oberthuer A, Fischer M, Berthold F, et al. Clinical Significance of Tumor-Associated Inflammatory Cells in Metastatic Neuroblastoma. Journal of Clinical Oncology. 2012;30(28):3525–32. doi: 10.1200/JCO.2011.40.9169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu D, Song L, Wei J, Courtney AN, Gao X, Marinova E, et al. IL-15 protects NKT cells from inhibition by tumor-associated macrophages and enhances antimetastatic activity. The Journal of Clinical Investigation. 2012;122(6):2221–33. doi: 10.1172/JCI59535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chinnasamy D, Yu Z, Kerkar SP, Zhang L, Morgan RA, Restifo NP, et al. Local delivery of lnterleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clinical Cancer Research. 2012;18(6):1672–83. doi: 10.1158/1078-0432.CCR-11-3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clinical Cancer Research. 2013;19(20):5636–46. doi: 10.1158/1078-0432.CCR-13-0458. [DOI] [PubMed] [Google Scholar]
- 49.Lo-Coco F, Cicconi L, Breccia M. Current standard treatment of adult acute promyelocytic leukaemia. British Journal of Haematology. 2016;172(6):841–54. doi: 10.1111/bjh.13890. [DOI] [PubMed] [Google Scholar]
- 50.Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of All-Trans Retinoic Acid Effect on Tumor-Associated Myeloid-Derived Suppressor Cells. Cancer Research. 2007;67(22):11021–28. doi: 10.1158/0008-5472.CAN-07-2593. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.