
Figure 2.
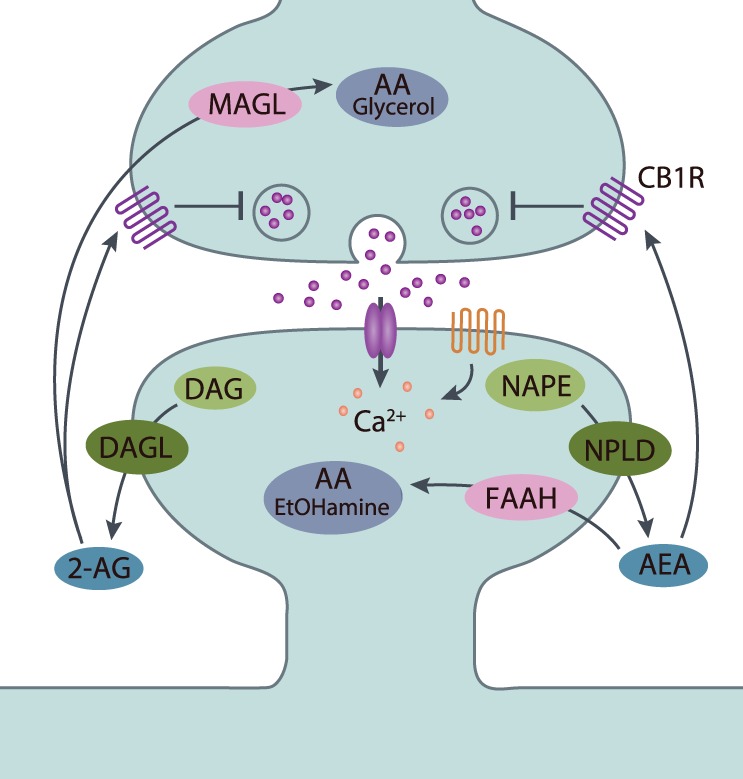
General mechanism of retrograde eCB signaling. Upon release of neurotransmitter (e.g., glutamate, GABA), postsynaptic depolarization results in elevations in intracellular calcium levels through activation of ionotropic receptors, Gq-coupled metabotropic receptors (e.g., Group I mGluRs, M1/M3 mAChRs, D2Rs), and/or voltage-gated calcium channels. The eCBs AEA and 2-AG are not stored in vesicles but instead are synthesized de novo from phospholipid precursors through calcium-dependent and -independent mechanisms. NAPE is hydrolyzed by N-arachidonoyl-phosphatidylethanolamine-specific phospholipase D (NPLD) to yield AEA, and DAG is converted to 2-AG by DAGL. Both eCB species traverse the synaptic cleft and activate presynaptic Gi/o-coupled CB1 receptors, thereby inhibiting adenylyl cyclase, regulating ion channels, and ultimately suppressing neurotransmitter release. eCB signaling is terminated following degradation by hydrolytic enzymes in the presynaptic and postsynaptic compartments. Primarily, AEA is converted to arachidonic acid (AA) and ethanolamine (EtOHamine) by fatty acid amide hydrolase (FAAH) localized to the postsynaptic cell, whereas 2-AG is hydrolyzed presynaptically into AA and glycerol by monacylglycerol lipase (MAGL).