

Abstract
The traditional structure-function paradigm has provided significant insights for well-folded proteins in which structures can be easily and rapidly revealed by X-ray crystallography beamlines. However approximately one third of the human proteome are comprised of intrinsically disordered proteins and regions (IDPs/IDRs) that do not adopt a dominant well-folded structure, and therefore remain “unseen” by traditional structural biology methods. This Perspective article considers the challenges raised by the “Dark Proteome”, in which determining the diverse conformational substates of IDPs in their free states, in encounter complexes of bound states, and in complexes retaining significant disorder, requires an unprecedented level of integration of multiple and complementary solution-based experiments that are analyzed with state-of-the art molecular simulation, Bayesian probabilistic models, and high throughput computation. We envision how these diverse experimental and computational tools can work together through formation of a “computational beamline” that will allow key functional features to be identified in IDP structural ensembles.
INTRODUCTION
Intrinsic protein disorder can refer to either local, disordered regions of a protein containing one or more folded domains (IDRs), or global protein disorder without any stable structure along the entire sequence (IDPs). IDPs/IDRs, which are estimated to make up approximately one-third of the human genome1–4, pose new challenges for the structure-function paradigm since they take advantage of their disordered state to interact with numerous partners for signaling, regulation and transcription5–8. At the same time disease-related proteins are highly enriched in IDRs, including those that are central to neurodegenerative disorders such as Parkinson’s disease, Huntington’s disease, prion diseases, and Alzheimer’s disease (AD), as well as cancer-associated proteins that have a primary function in regulatory protein interactions.9
The greater biological challenge posed by the IDP class of proteins relative to the singular folded counterpart is that the disorder in their free and bound complex states is integral to their function. In their monomeric or unbound forms, IDPs adopt neither a single nor a small number of stable folded conformations, and their energy landscape of the free IDP lacks a deep minimum, unlike that of a folded globular protein.10,11. Nonetheless, the structural characterization of an IDP in its free state is paramount to understanding the biology of the static or dynamic complexes that it forms with other ordered or disordered proteins. Depending on the dissociation constant, KD, there can be a significant population of the unbound form even in the crowded cell for some IDPs.12 A high degree of disorder and rapid interconversion between states is necessary for different IDRs to become accessible or inaccessible to binding and/or post-translational modifications important for regulation and signaling in the protein complex13. In describing the disorder-to-order transitions that can occur when disordered proteins fold upon binding to their targets, Arai and co-workers have found that the sub-populations of the unbound protein ensemble influence the mechanism of complex formation14, a conclusion that needs to be examined in the context of a range of different IDP systems. In regards the recent discovery of a small molecule drug that in cellular models of α–synuclein-mediated dysfunction points to a potential strategy for treating Parkinson’s disease15, the accurate determination of the conformational ensemble of the free protein might aid in a corresponding molecular interpretation of how such a drug works and therefore how best to target other disease-related IDPs.
The biological activities of IDPs are typically identified by the protein complexes that they form: an IDP can make both “static” (well-ordered) and/or “dynamic” (disordered) interactions with different sites on the target protein surface. Many intrinsically disordered proteins contain short amphipathic motifs, termed molecular recognition elements (MoRFs)16, which fold into regular secondary structures such as α-helix or β-strand or adopt irregular structures upon binding to their targets. An important feature of the recognition elements in many IDPs is that they exhibit conformational plasticity, i.e. they can fold into different structures on binding to different targets. Additional dynamic interactions involving adjacent regions to the MoRFs of the IDP frequently enhance the binding affinity, enabling them to interact with multiple targets and to create accessibility of sites for post-translational modification.
IDPs can bind with high specificity but modest affinity6,7, an attribute that enables spontaneous dissociation or displacement after signaling is complete.6,7,17–19 This is supported by the fact that when comparing IDPs to folded proteins, their average affinity is indeed lower than for folded proteins20, and while the distribution of KD shows considerable overlap between IDPs and folded proteins20, the definition of high specificity needs to be put in perspective. In particular, in order for a folded protein to obtain the degree of specificity given by a disordered sequence, the latter which can wrap around a target and provide extremely large contact area, in a fully-folded protein the degree of specificity would have to be even larger. The dynamics within a complex with multiple exchanging interacting element that can also facilitate displacement, particularly for cases with higher affinity interactions. One such example is the low nM affinity complex of the eukaryotic translation initiation factor 4E and the disordered 4E-binding protein 2 (4E-BP2).21 This complex involves two adjacent elements of 4E-BP2, a canonical 4E-binding helical motif and a less regular C-terminal region, that appear to dynamically exchange on the millisecond-microsecond timescale, facilitating phosphorylation at the interface required to break the complex22 and enable translation to proceed. Another functionally important dynamic complex involves the disordered Sic1 cyclin-dependent kinase inhibitor and the Cdc4 subunit of an SCF ubiquitin ligase. Phosphorylation of ~6 or more of the 9 sites on Sic1 enables low micromolar affinity binding. Dynamic exchange of each site on and off of Cdc4 facilitates ultrasensitive binding leading to controlled degradation and a sharp cell-cycle switch, as well as efficient multi-site ubiquitination.23–25 A further illustration of the dynamic complexes that IDPs can form is found with the disordered regulatory (R) region of the cystic fibrosis transmembrane conductance regulator (CFTR), that acts as an interaction hub with both intramolecular and intermolecular partners to integrate input for controlling CFTR channel activity.26,27 One of these partners, 14-3-3β, involved in CFTR processing, has two binding sites within its dimer into which 9 phosphorylated segments of the CFTR R region dynamically exchange.28 Thus, structural characterization of IDPs/IDRs in both free and bound forms, with a range of dynamics and disorder, is paramount to understanding their biology, and expansion of the structure-function paradigm to a structural ensemble is a necessary consideration for this class of protein.
Structures of biomolecules have driven functional insight into molecular biology and biochemistry ever since Watson and Crick advanced the structural model of DNA, and computational models are integral to rendering structural and dynamical information relevant to structure and function. The idea of ensemble structure modeling is, of course, also relevant for folded proteins and their unfolded states, exemplified by studies starting 25 years ago by Kuriyan et al.29 and continued more recently by the groups of Vendruscolo and Dobson30,31 and De Groot and Grubmuller 32. Starting with the seminal work of Nilges and co-workers33 which introduced Bayesian inference to derive a probability distribution for the folded structure using NMR, other research groups have made important theoretical contributions to the problem of ensemble structure determination using probabilistic frameworks.34–39 The quantitative challenge in understanding IDPs is how to build models of even more diverse structural ensembles that allow researchers to gain insight into their nature, to form hypotheses about their functional roles and to target them for small drug therapeutics. For folded proteins, the first structures determined by X-ray40, NMR41 and cryoEM42 helped propel their continued development into robust techniques for providing a concrete, predictive and conceptually straightforward model for the structure-function relationship. For example, scientists at protein crystallography beamlines have defined an increasingly automated workflow of tasks needed to solve the 3D structures of folded globular and membrane proteins and complexes: determining crystallization conditions; X-ray data collection from protein crystals; model building, refinement, and validation.43,44 Computational methods have also advanced to become critical partners to experiment in providing further insight through study of protein dynamics, folding kinetics, lead optimization in drug design, and the transition state energetics for enzymatic mechanisms.
However, IDPs are not amenable to static structural determination methods such as X-ray and electron crystallography and microscopy40,42,45,46, necessitating an adjustment in the core methodology of protein structure determination for the so-called “dark proteome” that can capture their dynamics and disorder. Nuclear magnetic resonance (NMR) and Small Angle X-ray Scattering (SAXS) are the experimental tools of choice for characterizing the solution structure and dynamics of IDPs in an aqueous environment23,46,47. Even so, since IDPs typically interconvert between conformations on the ~ns-ms timescale, most solution based experimental observables are highly averaged and thus obscure the characterization of the conformational sub-states of an IDP that are tied to biological function. Structural descriptions of IDPs/IDRs are highly underdetermined, that is, their number of degrees of freedom will far exceed the number of experimental restraints. Since experiments alone will likely be unable to provide a detailed structural ensemble, it is important to build the connection between the averaged experimental observables over the IDP structural ensemble to the conformational sub-populations within the ensemble6,48–54 using state-of-the-art computational methods and models31,55.
In analogy to crystallographic beamlines and their role in streamlining protein crystallography, we propose that the IDP community could develop a “computational beamline” to build up the requisite experimental and computational tools to model structural ensembles for a broad class of IDPs and IDRs and their complexes. The computational beamline would serve in several roles including as a repository for information from the best experimental techniques such as NMR and SAXS, to examine whether other techniques such as circular dichroism (CD), electron paramagnetic resonance (EPR) and double electron-electron resonance (DEER), fluorescence resonance energy transfer (FRET), and mass spectrometry can add valuable information, and to combine relevant experimental data with the best theoretical tools such as de novo molecular dynamics54–58, Markov State models59,60, Monte Carlo methods to sample side chain ensembles61, Bayesian probabilistic analysis10,34,62, and quantum mechanical methods to predict NMR observables63. In this Perspective we examine the current state of experimental approaches and computational methods applied to the IDP problem, what future directions can be usefully advanced within each area, and finally, how the two approaches can be combined into a powerful new resource that would culminate in an “analysis end-station” that could develop and apply new correlative methods to yield quantitative insight into key structural aspects that define the free and complexed IDPs to their functionally relevant states.
EXPERIMENTAL METHODS AND FUTURE INNOVATIONS
The current experimental solution methods for characterizing protein intrinsic disorder include infrared spectroscopy (IR), ultraviolet (UV) spectroscopy, circular dichroism (CD) spectroscopy, single molecule fluorescence spectroscopy, mass spectroscopy (MS), CD, Wide Angle X-ray scattering (WAXS), and the primary techniques of NMR and SAXS7,50. CD and IR spectroscopy report on the amount of secondary structure and hydrodynamic techniques such as SAXS, gel filtration, and dynamic light scattering report on the radius of gyration or hydrodynamic radius. Lack of a cooperative folding transition and proteolytic sensitivity are also attributes of IDPs and some of their complexes that are useful in forming a complete picture of a certain level of disorder.
NMR observables that can be used to restrain the IDP structural ensemble include chemical shifts of backbone and side chain nuclei, which aid in structural assignments and probe conformational information through their surrounding environment, spin-spin couplings (J-couplings) which independently report on dihedral angles, and residual dipolar couplings (RDCs) which have been used to describe the relative orientation of spatially separated regions of a disordered protein64–70. These types of measurements are highly useful in describing local and/or short-range interactions of the bound and free IDP state, while global descriptors of order/disorder are usefully defined through SAXS or SANS experiments for categorization of a free IDP into collapsed semi-ordered ensembles, collapsed disordered ensembles, or extended disordered ensembles1,2,4,50, based on the distribution of heavy atom distances. Additional NMR and ESR experiments such as through-space dipole-dipole interactions that give rise to the Nuclear Overhauser Effect (NOE), and paramagnetic relaxation enhancements (PRE) from an attached spin label71, as well as the more recent EPR72 and DEER73 experiments, are in principle information-rich since they report on both local and non-local tertiary structure contacts that would be valuable in restraining the IDP ensemble.
The joint application of SAXS and NMR to study the structural ensemble of IDPs has been pioneered based on a number of developments in North America and Europe. The SAXS program Ensemble Optimization Method (EOM) by Bernado, Svergun, Blackledge and collaborators was designed to work with NMR observables for IDPs74–76, and ENSEMBLE from the Forman-Kay group works with multiple different types of data including NMR and SAXS for IDPs77,78. The SIBYLS group in the U.S. has developed SAXS analysis for the characterization of large IDRs.79–81, including the analysis programs BilboMD and MES82, which have provided novel and disease relevant insights into IDRs. Data deposited into the SIBYLS based SAXS data repository83 bioisis.net was mined for the development of quantitative measures of flexibility84 and the extraction of mass81 even when flexibility is present.
However in all cases we are still faced with new challenges in applying these solution-based techniques to IDPs. For example, the data for optimal characterization of IDPs lie at the extremes of typical SAXS data for folded proteins since for their molecular weight, IDPs can be extremely extended, thereby requiring very low-Q data from SAXS for the characterization of maximum dimensions. For example while typical SAXS data collection standards occur in the Q range of 0.01 to 0.32Å−1 with exposure times of 1 second, for IDPs SAXS data is taken down to a low Q value of 0.005 Å−1 and with exposure times rising to tens of seconds. IDPs can also benefit from WAXS since localized structural features generally occur over short length scalesin the high Q region.
For NMR the generally poor chemical shift dispersion (particularly in the 1H dimension) can be mitigated with optimized pulse sequences for IDPs85. The pronounced motional averaging of NOEs has limited the use of NOEs, and yet the information about intermediate range NOEs may be particularly important in defining conformational ensembles. For example, NOE data collected for the Aβ40 and Aβ42 peptides contained ~1100 and ~700 crosspeaks, respectively, but only ~20% of these can be uniquely determined from experimental information alone, due to chemical shift ambiguity, and most of these are due to short-range intraresidue, sequential, and i to i+2 contacts57. With the addition of 13C/15N labeling for resolution in 3D or 4D experiments it becomes possible to assign many more NOE cross-peaks, adding new experimental constraints for ensemble generation; for example particular combinations of hydrophobic amino acids labeled with 13C for the unfolded state N-terminal SH3 domain of Drkproved to be valuable in these studies86–88.
Further progress can be made with techniques like PRE which has been valuable in detecting transient interactions, particularly intermolecular interactions89. The power of this approach comes from the fact that close proximity to an unpaired electron (on a metal or nitroxide group covalently bound to the protein) causes a large increase in relaxation rate that is dependent on the inverse sixth power of the distance from the spin label. By labeling one binding partner with the relaxation agent, and the other with isotopes such as 15N for selective NMR detection, characterization of even transient complexes is possible.90 This approach can also be applied to investigate short-lived intramolecular contacts in IDPs, since the transient folding leads to proximity of the electron and nuclei, and is again observed through changes in relaxation. But there are some caveats to this approach that requires careful calibration when applied to IDPs. First, because of the covalent attachment of the relaxation agent it can never get very far from the region of the peptide where it is attached, so there is always locally enhanced relaxation, although more interesting are the sites of enhanced relaxation that are far, in the sense of covalent structure, from the attachment site. The relaxation agents are invariably larger than an amino acid sidechain, and interactions of the agent with other amino acids may affect the transient folding of the IDP, requiring additional controls to assess whether this occurs using chemical shifts and NOE spectra of the modified peptide, or even better a diamagnetic version using a reduced spin label, or diamagnetic metal substituting for the paramagnetic one in a chelator, and comparing with the unmodified IDP. Nonetheless when carefully calibrated, a number of high quality PRE experiments have been successfully carried out on IDPs71,91, and thus these experimental methods are particularly promising – a conclusion which has been nicely reviewed elsewhere92.
DEER gives the distance distribution between two nitroxides93 or metal (Gd+3)94 labeled sites, complementing PRE data where it can be difficult to unambiguously separate spatial and temporal components of the distance distribution function. However, DEER measurements have challenges when applied to IDPs. For DEER measurements the electron relaxation rates must be reduced by rapid freezing to form a glass at liquid N2 temperatures, and a concern is whether freezing as well as the addition of the two labels, which may also involve mutations to create the labeling sites, perturbs the distribution of conformers present in the unlabeled IDP in solution. A second challenge in analysis of DEER data on IDPs is extraction of distance from the dipolar echo modulation pattern, although existing analysis tools are available such as the DeerAnalysis2013 software95. Nevertheless, IDPs inherently will give rise to a broad distance distribution for the pairwise electron-electron interactions, so the number of parameters to be extracted from the data is inherently larger than for folded proteins, where in favorable cases a single, narrow distance distribution applies. A priori, it remains unclear how robust such an analysis will be when taking into account the limited signal-to-noise, and possible systematic sources of error resulting from finite pulse widths and orientational selection at high magnetic fields. Therefore, substantial experimental work is needed to fully explore the potential of DEER in the analysis of IDPs. We note the significant analogy of EPR DEER to optical FRET experiments using attached fluorescent dyes96–98 rather than nitroxides or metals to IDPs; thus FRET can also be useful for restraining the IDP ensemblesif the similar challenges described for the DE ER measurements can be overcome.
The integration of multiple solution-based experimental techniques on IDPs requires optimization from both a data acquisition and analysis perspective. For example, while each individual SAXS measurement does not contain as much information as a high resolution NMR measurement, NMR and EPR are “low” throughput techniques whereas SAXS and dynamic light scattering (DLS) data can be collected and analyzed much more quickly. To illustrate, ideal IDP concentrations for NMR can be rapidly identified through the analysis of high throughput solution results generated by SAXS for oligomerization and DLS to determine the maximum concentrations allowed before signals of aggregation are apparent. Part of the growing resurgence of SAXS as a technique is that many measurements allow for relative comparisons on how structure changes with sequence and conditions – typical of many IDP projects. These ideas have been advanced by Hura and co-workers through formulation of a general heatmap based method79 for comprehensively viewing several SAXS data sets and application to the human DNA mismatch repair protein MutSβ which contains 300 flexible amino-acids (Figure 1).
Figure 1. Heatmap comparing pairs of profiles from multiple experimental techniques including SAXS, DLS and CD.

Each cell is a pair wise comparison between a condition or construct. Similarity between SAXS curves is measured by the metric Vr, and displayed as a gradient color where red indicates similarity (low Vr score) and white indicates dissimilarity. The black square diagonals are self-comparisons.
Although there have been many NMR studies on IDPs, and some have included other data such as SAXS, there have not been extensive and systematic evaluations of the value of different types and combinations of data in defining the IDP conformational ensemble. However a few approaches are starting to emerge that deal with this important issue. One is a study99 that performed ENSEMBLE calculations for three IDPs based on a variety of experimental inputs, including chemical shifts, RDCs, PREs, and SAXS. Comparison of ensembles calculated with subsets of the experimental data missing defined types were used to quantify which measurements most affected secondary structure, tertiary contacts and molecular size distribution, and hence are high priorities for data acquisition to restrain IDP structural ensembles. It was found that secondary structure was most strongly restrained using 13Cα chemical shifts and to some degree using 3JHNHα couplings, whereas the accuracy of calculated tertiary structure is dependent on the number of PRE distance restraints used.99 RDCs were found to provide a small but significant probe of short- to medium-range tertiary structure whereas as SAXS was important for restraining the size distribution.99
At the same time, if the chemical shifts are not highly dispersed along the sequence of a particular IDP, such as is found for the amyloid-β peptides57, then the chemical shifts have more limited value as a experimental refinement input or as a validation measure.55 In addition, the optimization phase of methods such as ENSEMBLE77,78 and ASTEROIDS47 rely on heuristic back-calculation methods such as SHIFTX2100 for chemical shifts, PALES101 for RDCs, and mere structural approximations to NMR observables such as NOEs, even though the dynamical origins of NOE intensities can be better used for determining the IDP ensemble55. Alternative approaches based on Bayesian probabilistic modeling can offer a more solid foundation for examining the usefulness of experimental data types10,62 as described in the n ext section.
COMPUTATIONAL METHODS AND FUTURE INNOVATIONS
An important area of IDP research is to build robust all-atom models of IDP ensembles that can successfully interface with experimental data to provide predictions of the structural ensembles of IDP monomers and their complexes. One approach is to identify sets of conformers that in aggregate agree with experimental data, to derive the IDP structural ensemble. Such “experimental data knowledge” methods are the foundation of NMR structure determination of folded proteins using experimentally derived constraints based on NOE data, RDCs, J-couplings and chemical shifts, embodied in software packages such as CANDID102, CYANA103, X-Plor-NIH104,105, and TALOS106. Most often, experimental data knowledge approaches for IDPs start with an extensive set of statistical coil conformations derived from software platforms such as Flexible-Meccano107 and TraDES108, which can generate both coil- an/or various structure-biased conformers. This basis set of structures is then culled for the subset of conformations and their populations that are in best agreement with experimental data to create the IDP ensemble. Examples are the energy-minima mapping and weighting method109,110, ENSEMBLE77,78, and ASTEROIDS47. ASTEROIDS uses a genetic search algorithm to select structures that together best match experimental chemical shifts, PREs, or RDCs47,68 while the ENSEMBLE method77,78,87,88 selects structures from the starting pool using a Monte Carlo algorithm with an energy-weighting scheme for each type of experimental input. These programs contain modules for several different experimental data types. For example, ENSEMBLE is able to accommodate data from a very wide range of sources including chemical shifts, RDCs, PREs, J-couplings, NOEs and relaxation rate-derived contact densities, as well as hydrodynamic radii (Rh) and SAXS77 and hydrodynamic radii (Rh) from NMR, size exclusion chromatograph or dynamic light scattering.
In addition to the ENSEMBLE and ASTEROIDS approaches that use experimental data for conformational selection, a number of researchers have advanced the combination of applying knowledge from NMR to restrain the IDP ensemble generated during an MD trajectory30,111–114. For example, MD simulations have been combined with RDC restraint data for folded proteins, which then allows for the analysis of other features of the ensemble, such as conformational fluctuations. NMR restrained MD has also been applied to IDPs such as α–synuclein by incorporating distance restraints derived from PRE experiments to guide the MD so that the radius of gyration distribution of the ensembleis in good agreement with the experimental value 111.
While conformer creation and selection methods such as TraDES108, ENSEMBLE77,78, Flexible-Meccano107 and ASTEROIDS47 have proven very useful to the IDP community, qualitative changes are required in the theoretical approach to IDP structure solvers. First, IDP ensemble construction has typically relied on low-complexity statistical coil descriptions that are not Boltzmann weighted and do not contain any important dynamical information55 that can be compared to NMR observables such as relaxation rates and NOEs. Second, our ability to back-calculate NMR or SAXS data from structures is actually very poor, thus losing an important discriminator for selecting an IDP structural ensemble that is most consistent with the abundant availability of chemical shift and scalar coupling experimental data.55,62 Third, characterizing both the free and bound IDP ensembles is important to understanding their biology, and the computational techniques must be able to accurately describe the range of environments from solvent-exposed disordered monomer ensembles through to protein-protein complexes where the IDP folds or remains partially or fully disordered. Thus we require higher accuracy in IDP ensemble generation using robust force fields and advanced sampling methods, including appropriate accounting of timescales, and increased sensitivity of back-calculations with high quality NMR spectral simulation tools. Finally, due to the under-determined nature of the IDP problem, we need to utilize statistical approaches, such as Bayesian analysis115,116, to rank alternate IDP conformational ensembles to determine the most probable one based on agreement with the experimental data.
To illustrate this confluence of issues, Brookes and Head-Gordon developed a Bayesian approach to determine the most probable IDP structural ensemble model that takes full advantage of experimental data, their known errors and variances, and the quality of the theoretical back-calculation from structure to experimental observables.62 The experimental inferential structure determination (EISD) method is formulated to determine the most probable structure (for folded proteins) or structural ensemble (for IDPs) by decomposing the posterior probability distribution p(X, ξ|D, I) using Bayes’ Theorem:
| (1) |
where P(D|X, ξ, I) is the conditional probability that relates , a structural ensemble containing N structures, to a set of M experimental data observations . The parameters of the Bayesian model are the set of so-called “nuisance” parameters, ξ, which are uncertain values that cannot be determined directly from the data, such as the uncertainties in the experimental measurements or back-calculation equations. I represents any prior information about the system, such as experimental information embodied in p(ξ|I), or structural information via p(X|I) in which the latter is typically modeled as either a uniform (uninformative) prior or using Boltzmann weighting that requires a robust energy function.
One of the primary advances of the EISD model is that we use all of the available information about the separate distributions of different experimental data types. We utilize the variable quality with which we can back-calculate these observables, o, from structure, by optimizing within the experimental and back -calculation “nuisance” parameters that are treated as random variables with known Gaussian distributions, p(ξ(exp)i) and p(ξ(back)i), respectively. More specifically, the posterior probability can be modeled as follows
| (2) |
where
| (3) |
otherwise and < > denotes an average over the candidate IDP ensemble of structures used to back-calculate experimental observables, since all that is known for a given NMR measurement on an IDP is that it corresponds to an average of that measurement over every structure in the ensemble.
We applied the EISD Bayesian method to evaluate the relative probabilities of 7 qualitatively different structural ensembles for the Aβ42 IDP monomer: one random coil ensemble generated from TraDES108, one ensemble generated from a replica exchange simulation (de novo MD)55, one statistical coil ensemble that incorporates bioinformatics knowledge about independent local secondary structure at each residue (Pred-SS)55, and four ensembles generated by adding experimental restraints from NMR (RDCs, NOEs, scalar couplings, and chemical shifts) operating on the de novo MD and Pred-SS ensembles using ENSEMBLE (MD-ENS1, MD-ENS2, MD-ENS4, and Pred-SS-ENS)77,88,99. We used only two NMR data types: chemical shifts and J-couplings55,57,117,118, and an uninformative uniform structural prior was used to generate the results shown in Figure 2.
Figure 2.

log p(X, ξ|D, I) evaluated for X equal to the following qualitatively different ensembles for the Aβ42 monomer: random coil (RC), statistical secondary structure (Pred-SS), de novo MD, and ENSEMBLE optimized ensembles (MD-ENS1, MD-ENS2, MD-ENS4, and Pred-SS-ENS) using (a) chemical shift data only, (b) J-coupling data only, and (c) J-coupling and chemical shift data together. Adapted with permission from [62], copyright 2016 American Chemical Society.
Figure 2 indicates that the rankings of the Aβ structural ensembles primarily depend on our ability to quantitatively back-calculate from structure to observable, in this case using SHIFTX2100 for chemical shifts and the Karplus equation for 3-bond couplings119
| (4) |
where ϕ represents a dihedral angle of interest, and A, B, C are typically parameterized on folded proteins120–122. A comparison of Figures 2a and 2b show that using chemical shifts and J-couplings on their own result in a large difference in the structural ensemble rank order. When they are used together in Eq. (2), the relative rankings among ensembles are qualitatively unchanged from using J-couplings alone (Figure 2c). However, whether using J-couplings alone or together with chemical shifts in the Bayesian model, it still is not possible to differentiate between the extended RC ensemble, equivalent to a protein under high denaturant conditions, and the collapsed and structured MD ensembles that would be representative of low denaturant conditions.
While adding additional data types such as SAXS or PREs to the EISD model would certainly help to overcome this problem, the abundant availability of chemical shift and scalar coupling data would be better used if back-calculations were more robust. For example, although improvements realized by SHIFTX2100 over SHIFTX123 were significant for folded proteins with the introduction of structural homology information, the level of difference between the SHIFTX2 and SHIFTX calculators is negligible as we have shown for the Aβ42 example62, since structural homology plays no role for IDPs. Even for scalar couplings, Karplus anticipated that further refinements of Eq. (4) were necessary for quantitative prediction, such as the inclusion of electron orbital and dipolar electron spin terms, a more careful choice of underlying electronic structure methods, consideration of chemical substitutions when applied to other molecules, the number of bonds separating spins, and dependence on additional geometric features such as bond angles or other dihedral angles119. Although some studies have adopted some of these suggestions, there is still a primary focus on use of the original Karplus equation to predict J-couplings of folded and flexible peptides and proteins, where the constants A, B, and C must capture the large variations in dihedral fluctuations across many peptide and protein data sets122,124.
Accordingly, quantitative back-calculations of the NMR and SAXS observables are clearly a necessary objective to make better use of the experimental data in order to generate tighter spatial restraints for discriminating among alternative structural ensemble models. To be more specific, computational predictions of chemical shifts in proteins, such as ShiftX2100 and Sparta125, rely on knowledge-based algorithms that employ sequence and/or structural information together with experimental NMR data for folded proteins. Their residual error is typically only about 3-fold smaller than the typical range of such shifts, and orders of magnitude larger than the experimental uncertainty in the measured chemical shift values. As such their applicability to partially or fully extended IDP conformations is clearly limited, and improvements are highly desirable and may well be absolutely necessary. As Case recently commented126: “Although quantum calculations of chemical shifts in proteins have not yet reached the level of accuracy obtained by empirical models, there are good reasons to push forward. Quantum models allow study of unusual conformations, including fibrils (and) partially disordered systems”.
Therefore a future challenge is to deploy (and further develop) high accuracy QM-based methods for scalar couplings, and environment-dependent 13C, 1H, and 15N NMR chemical shifts, accounting for more than intervening dihedral angles and backbone ϕ and ψ torsion angles, respectively, at tractable computational cost for characterizing the structural diversity of IDP ensembles. However, there are three distinct challenges to improving chemical shift and scalar coupling back-calculation accuracy. At the basic level, common-place density functional theory (DFT) suffers from inadequate accuracy127 for chemical shifts128 because of fundamental limitations: rigororously, the functional must depend not just upon the density, but also on the paramagnetic current density129, or the field itself130, which is an unsolved challenge at present. Another way to push forwards is via wave function methods131, which are far superior to present-day DFT for chemical shift calculations (~10X higher accuracy132) while advances in QM treatment of spin-spin couplings63 would need to occur simultaneously.
To illustrate we have compared the chemical shift prediction capabilities of DFT (using the B3LYP functional) to that possible with second order Møller-Plesset perturbation theory (MP2) when benchmarked against a highly accurate CCSD(T) calculation (Table 1). Using a small blocked dipeptide whose conformational space is spanned by its backbone ϕ and ψ dihedral angles, the chemical shifts are computed for every atom of four diverse conformers (β sheet, extended, α helix, γ turn) using all three levels of theory with the cc-pVTZ basis set. The RMSD in the chemical shifts is calculated relative to small molecule primary references (CH4, NH3, H2O, and H2 for C, N, O, and H, respectively) and relative to a secondary reference which is the planar conformer.
Table 1. RMSD in ppm of B3LYP DFT and MP2 with respect to CCSD(T) when using small molecules as the reference (primary) vs. a planar peptide reference (secondary).
Four different conformers of glycine dipeptide are used to explore chemical shift accuracies with respect to geometrical changes in ϕ and ψ dihedral angles. All geometries were optimized at the MP2 level of theory with the φ and ψ dihedral angles constrained to the values shown
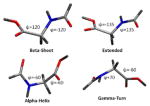
|
Atom | DFT | MP2 | ||
|
| |||||
| Primary | Secondary | Primary | Secondary | ||
|
| |||||
| C | 7.63 | 0.50 | 1.08 | 0.07 | |
|
| |||||
| N | 17.65 | 0.40 | 5.08 | 0.15 | |
|
| |||||
| O | 32.77 | 2.73 | 6.23 | 1.93 | |
|
| |||||
| H | 0.26 | 0.06 | 0.08 | 0.03 | |
|
| |||||
Focusing on 13C NMR, the results in Table 1 show that MP2 yields RMS errors that are over 7 times smaller than B3LYP. This gap is preserved when using the secondary reference, showing that MP2 yields greater fidelity to the reference for the biologically relevant shifts in each atom relative to its value in the reference planar environment. Therefore, given a bonding motif, the MP2 method does very well (less than 0.1 ppm relative RMSD for 13C) at predicting how the chemical shift will change due to changes in bond lengths, angles, and dihedral angles. Given databases of environment-dependent calculated NMR chemical shifts of this quality or better, there is clearly scope for building more accurate software tools for NMR chemical shift prediction from structure in the future. This will require incorporation of other environmental effects, including hydrogen bonding.
These theoretical and methodological advances all operate on a three-dimensional conformation, and thus they will be dependent on the generation of representative and complete IDP structural ensembles. While methods such as Flexible-Meccano107 and TraDES108 are valuable for generation of random coil conformers, molecular dynamics simulations are capable of generating a true Boltzmann weighted ensemble if the underlying energy surface is accurate and if sampling on this surface is complete. However, presently available energy functions that work well for folded proteins are imperfect when applied to IDPs. Because there are many degrees of freedom to sample over ns-ms timescales, MD simulations rely on computationally cheap fixed-charge force fields that allow for adequate sampling of the conformational space of an IDP. However, given the range of IDP environments including extreme solvent exposure in the monomer state through to buried residue interactions at the IDP-protein complex interface, a number of new protein and water fixed charge force fields offer better balance for the energetics of relative conformational energies and peptide-water interactions.133–138 Alternatively, polarizable force field offer the best future hope for more accuracy139,140 since they have the necessary physics to respond to a range of IDP environments experienced by the free as well as bound states of an IDP. However, polarizable models come with an increase in computational expense that in turn limits needed sampling. Nonetheless, recent efforts to reduce the computational expense of polarizable models are starting to take hold141, and thus will be an important future direction in the simulation of robust IDP structural ensembles.
There is always tension between potential energy surface accuracy and adequate sampling of conformational space of an IDP due to its heterogeneous nature, as both increase computational cost. This requires that we develop sampling methods that converge faster to the Boltzmann weighted ensemble for an IDP. Generalized Ensemble (GE) Methods that use temperature, ionic strength, dielectric constant, and protonation states as the scaling variable142–153 will be important for IDPs. Markov State Model (MSM) approaches combined with adaptive sampling (AS) such as the MinCounts154 algorithm can be tailored to sample the heterogeneous states of IDPs, including those lacking preferential structure and those with partial folding59,154,155.
The MinCounts method is a means for pushing sampling to slow, orthogonal degrees of freedom – even those that haven’t been discovered yet. Mincounts looks at the counts of transitions seen in MD simulations that started in state i and ended up in state j after some lag time Δt, and runs more simulations at states with few counts. This has been shown to be the most effective scheme for adaptive sampling as shown in Figure 3.154, and has been applied to numerous systems, including simple models (where sampling can be tested exactly as a “gold standard” is known) as well as in MD simulations of protein folding, protein unfolded states, and protein conformational change. Using these sampling methods and the Folding@home distributed computing project, the Pande lab has simulated the conformational change of kinases156 and GPCRs156 on the submillisecond timescale.60
Figure 3. Convergence time for Fs (capped 21 alanine) peptide transition matrix generated with various hybrid sampling schemes.
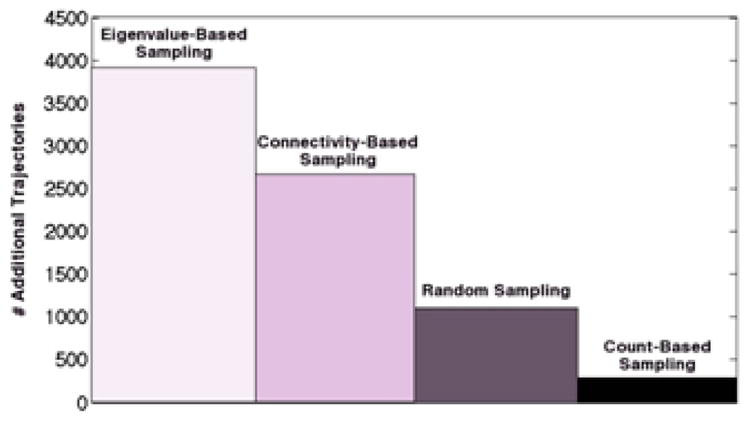
Time is measured in the number of eigenvalue-based trajectories needed to converge to an absolute error of 2.00 after 1000 initial trajectories are run from a chosen sampling method. Absolute error is defined as the sum of absolute deviations in transition matrix elements. Convergence times for each method were, averaged over 10 simulations, 1) 3913 for pure eigenvalue-based sampling, 2) 2669 for connectivity-based hybrid sampling, 3) 1107 for even sampling hybrid sampling, and 4) 286 for min count-based hybrid sampling. Reprinted with permission from [154]; copyright 2011 American Chemical Society.
In addition to backbone degrees of freedom, the generation of side chain ensembles for folding upon binding intermediates and IDP complexes will be necessary. However theoretical approaches for sampling the low energy alternative side chain arrangements of a protein is a difficult problem, and while molecular dynamics (MD) simulations give a good description of side chain conformational change on the nanosecond to microsecond level157, the experimental estimates indicate that the timescales are much longer. Therefore to circumvent the sampling issues imposed by MD, many groups have resorted to advanced Monte Carlo (MC) schemes158–160 which are designed to more exhaustively sample the Boltzmann weighted populations of side chain repackings, especially in the interior of the protein that may undergo low-probability rotamer transitions61,159 and and have been shown to extend into the microsecond to millisecond time scale.161
We have recently introduced a new Monte Carlo Side Chain Ensemble (MC-SCE)61 approach for calculating side chain ensembles, entropy, and mutual information that is more quantitative compared to past efforts, by using a better convergent Rosenbluth sampling scheme, an augmented Dunbrack library162,163, a robust physics-based energy function using an implicit solvent model, and side chain rotamer sampling on a ensemble of backbone structures using backrub sampling159. We have now used our MC-SCE algorithm to generate tens of thousands different side chain packings for hundreds of different protein backbones, including protein-protein complexes for 60 different protein systems. These include cryogenically cooled and room temperature X-ray crystallographic structures for CypA and H-Ras164,165 as well as NMR J-coupling data for CypA and Eglin-C, and DHFR binary complexes of E:THF and E:FOL, in which we found overall excellent agreement across the full range of X-ray and NMR data 164,166,167. (see Figure 4). Although the MSM/AS and MC-SCE methods have been primarily validated and then used for prediction on folded proteins, their extension to IDPs is clearly the next frontier for generation of more complete structural ensembles.
Figure 4. J-coupling constants (a) 3JCγN and (b) 3JCγCO for the DHFR binary product complex E:THF and (c) 3JCγN and (d) 3JCγCO for the DHFR binary product complex E:FOL.

The red symbols are the experimental data from 167. The blue symbols are calculated from the MC-SCE ensemble using backbones from molecular dynamics and the Karplus parameterization from 167. Reprinted with permission from [61]; copyright 2015 Elsevier.
CREATING A COMPUTATIONAL BEAMLINE FOR IDPS
The traditional structural biology approach of crystal structure visualization and analysis operations done on a single set of coordinates ultimately fails when applied to IDP ensembles. The goal of an IDP computational beamline is to better connect observed structural or dynamical motifs derived from the interplay of experiment and computation into functional relevance for free IDPs and their bound complexes. We envision a central resource that will integrate experimental and simulation data, simulation codes, and analysis tools across the IDP research community (Figure 5). The computational beamline will run workflows of software to create structural ensembles, store and index the variety of data types that help create, restrain, and/or validate ensembles, and collect analyses into an analysis end station that would formulate hypotheses about the relevance of structural ensembles to biological function.
Figure 5.

Conceptualization of a Computational Beamline for the IDP community.
To scale up the execution of software tools from a handful of manually managed runs to thousands of runs continuously executing on parallel computing resources, the computational beamline will use proven scientific workflow software. For example, The Materials Project has developed a high-throughput workflow software called FireWorks53 that has run millions of materials science calculations on supercomputers at national laboratories and cloud resources such as in the NSF XSEDE project. Scientific workflow software like FireWorks automatically manages multi-step, branching, and iterative calculations, and can continuously launch and monitor new calculations from a queue spanning months of time and many millions of compute hours.
While more sophistication in workflow software will dramatically increase the capability to perform experiments and gather new data, this scale of computation will also introduce new challenges: re-creating and debugging runs is no longer within the capacity of a single person’s memory. In order to know exactly which code produced which result, there must be a disciplined curation of all the relevant software tools used and developed in the IDP community, including methodology such as ENSEMBLE, QM back-calculations, Bayesian analysis, and advanced methods to derive the detailed structural ensembles of IDPs and their complexes using new force fields and backbone and side chain sampling methods. We envision that workflow software like FireWorks could be used to take an IDP of interest and combine multiple methodologies into a single workflow, then run that workflow in parallel and for a large number of timesteps on a cluster or supercomputer, automatically collecting all the results generated during the run.
The computational beamline workflows will enable automated and systematic collection of the results of the calculations into a central data repository, and a second type of “data-intensive” workflow will perform the same function to continuously collect and normalize the available experimental data. Consequently, the computational beamline data repository will integrate all available experimental data, all spectral simulations, and finally calculated ensemble data into an integrated data resource. The data repository would develop methods to ingest and organize a wide variety of experimental data (i.e. SAXS, NMR, FRET, DEER-EPR, etc) and conformer ensembles, with data categorized and coded according to a common vocabulary. Use of industry-standard database technologies will provide powerful and flexible search capabilities. Data sharing to external databases such as BioIsis (bioisis.net)83, pE-DB168, and BMRB (www.bmrb.wisc.edu)169 could be performed from this repository and made accessible to the IDP community.
Just as in a cyclotron or synchrotron, the scientific impact of a computational beamline aso depends on the capabilities of the “analysis end-station”, where IDP researchers can derive knowledge from the integrated experimental and simulation data with new correlative methods that would enable collaborative and reproducible analyses via flexible interfaces and web-enabled analysis environments, such as the Jupyter170 notebook. The end-station could provide a set of transformations to extract selected result sets into the formats and parameters that are needed to feed into other existing or new analysis tools. The analysis end-station could first build upon existing analysis tools such as DSSP171, k-means clustering or Principal Components Analysis, and combine them with new analysis protocols such as hydrophobic and electrostatic clustering, identification of regions of compaction or extension, probabilistic contact maps to define short and long-range interactions.
We can also envision even more novel analysis tools for IDPs based on kinetic clustering from Markov State Models (MSMs)59,60,154. Kinetic clustering is a natural outcome of MSMs, and would bring to the IDP community a much more physical, natural, and biologically relevant means to conceptualize IDPs, by clustering not due to geometric similarity (which may or may not be relevant for function), but due to kinetic similarity, i.e. grouping structures together in a cluster if they rapidly interconvert kinetically, which is a natural and very physical definition for a “state”, versus structures that interconvert more slowly and thus can be classified as distinct sub-populations. Finally, data mining and machine learning methods will play a particularly important role in IDPs, especially in determining and understanding key structural or dynamical motifs that are difficult to identify just by visual inspection, including repeated transient structure and more sophisticated correlative motions.
SUMMARY
One of the central primary objectives of IDP research is to provide atomic level structural and dynamical information on the free IDP conformational ensembles and their relationships to the ensembles of IDP complexes exhibiting a broad range of order to disorder that is important for understanding their function. Given the fact that IDPs/IDRs are underdetermined systems, and thus a unique structural ensemble cannot be defined by experiment alone, three important areas for future progress have been identified. First is large-scale experimental data acquisition, including defining the most information-rich experimental techniques to provide for discriminatory information between competing ensemble definitions for IDPs. Second is improving the accuracy of IDP ensemble model generation (free and bound forms) with (i) advanced molecular simulation approaches, (ii) new QM/MM spectral simulation tools that enhance the discriminatory power of the solution-based experimental techniques, and (iii) probabilistic models such as recent Bayesian formulations to provide measures of uncertainty quantification in the experimental and simulation data generated. Third is centralizing the experimental data and computational tasks into an automated workflow, including development of a comprehensive set of analysis tools that can connect observed structural or dynamical motifs with functional relevance to the biological questions being addressed in a wide range of IDP projects. The need for collaborative teamwork to create this infrastructure is obvious, as are the ultimate benefits to the IDP and general protein structural communities.
Acknowledgments
THG thanks the National Science Foundation grant CHE-1363320 for support of this work. This work was supported by grants GM113251 (HJD) and CA96865 (PEW) from the National Institutes of Health. JDF-K is a Tier 1 Canada Research Chair in Intrinsically Disordered Proteins and acknowledges support from the Natural Sciences and Engineering Research Council of Canada (NSERC, RGPIN-2016-06718). We thank the two reviewers for careful reading of the manuscript and for their many excellent suggestions for clarifications.
References
- 1.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z. J Mol Graph Model. 2001;19:26. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 2.Dunker AK, Silman I, Uversky VN, Sussman JL. Curr Opin Struct Bio. 2008;18:756. doi: 10.1016/j.sbi.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 3.Uversky V, Gillespie J, Fink A. Proteins. 2000;41:415. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 4.Uversky VN, Dunker AK. Biochim Biophys Acta. 2010;1804:1231. doi: 10.1016/j.bbapap.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Csizmok V, Follis AV, Kriwacki RW, Forman-Kay JD. Chem Rev. 2016;116:6424. doi: 10.1021/acs.chemrev.5b00548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dyson HJ, Wright PE. Nature Reviews: Mol Cell Biol. 2005;6:197. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 7.Wright PE, Dyson HJ. J Mol Biol. 1999;293:321. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 8.Wright PE, Dyson HJ. Nature Reviews Molecular Cell Biology. 2015;16:18. doi: 10.1038/nrm3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uversky VN. Front Biosci. 2009;14:5188. doi: 10.2741/3594. [DOI] [PubMed] [Google Scholar]
- 10.Fisher CK, Stultz CM. J Am Chem Soc. 2011;133:10022. doi: 10.1021/ja203075p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dunker AK, Gough J. Curr Opin Struct Bio. 2011;21:379. doi: 10.1016/j.sbi.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 12.Theillet FX, Binolfi A, Frembgen-Kesner T, Hingorani K, Sarkar M, Kyne C, Li C, Crowley PB, Gierasch L, Pielak GJ, Elcock AH, Gershenson A, Selenko P. Chem Rev. 2014;114:6661. doi: 10.1021/cr400695p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Binolfi A, Limatola A, Verzini S, Kosten J, Theillet F-X, Rose HM, Bekei B, Stuiver M, Rossum Mv, Selenko P. Nature Comm. 2016;7:10251. doi: 10.1038/ncomms10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arai M, Sugase K, Dyson HJ, Wright PE. Proc Natl Acad Sci USA. 2015;112:9614. doi: 10.1073/pnas.1512799112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Toth G, Gardai SJ, Zago W, Bertoncini CW, Cremades N, Roy SL, Tambe MA, Rochet JC, Galvagnion C, Skibinski G, Finkbeiner S, Bova M, Regnstrom K, Chiou SS, Johnston J, Callaway K, Anderson JP, Jobling MF, Buell AK, Yednock TA, Knowles TPJ, Vendruscolo M, Christodoulou J, Dobson CM, Schenk D, McConlogue L. PloS One. 2014;9:e87133. doi: 10.1371/journal.pone.0087133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mohan A, Oldfield CJ, Radivojac P, Vacic V, Cortese MS, Dunker AK, Uversky VN. J Mol Biol. 2006;362:1043. doi: 10.1016/j.jmb.2006.07.087. [DOI] [PubMed] [Google Scholar]
- 17.Kriwacki RW, Hengst L, Tennant L, Reed SI, Wright PE. Proc Natl Acad Sci USA. 1996;93:11504. doi: 10.1073/pnas.93.21.11504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oldfield CJ, Cheng Y, Cortese MS, Romero P, Uversky VN, Dunker AK. Biochem. 2005;44:12454. doi: 10.1021/bi050736e. [DOI] [PubMed] [Google Scholar]
- 19.Tompa P. FEBS Lett. 2005;579:3346. doi: 10.1016/j.febslet.2005.03.072. [DOI] [PubMed] [Google Scholar]
- 20.Shammas SL, Rogers JM, Hill SA, Clarke J. Biophysical journal. 2012;103:2203. doi: 10.1016/j.bpj.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lukhele S, Bah A, Lin H, Sonenberg N, Forman-Kay JD. Structure. 2013;21:186. doi: 10.1016/j.str.2013.08.030. [DOI] [PubMed] [Google Scholar]
- 22.Bah A, Vernon RM, Siddiqui Z, Krzeminski M, Muhandiram R, Zhao C, Sonenberg N, Kay LE, Forman-Kay JD. Nature. 2015;519:106. doi: 10.1038/nature13999. [DOI] [PubMed] [Google Scholar]
- 23.Mittag T, Orlicky S, Choy WY, Tang X, Lin H, Sicheri F. Proc Natl Acad Sci USA. 2008;105:17772. doi: 10.1073/pnas.0809222105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Borg M, Mittag T, Pawson T, Tyers M, Forman-Kay JD, Chan HS. Proc Natl Acad Sci USA. 2007;104:9650. doi: 10.1073/pnas.0702580104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mittag T, Marsh J, Grishaev A, Orlicky S, Lin H, Sicheri F, Tyers M, Forman-Kay JD. Structure. 2010;18:494. doi: 10.1016/j.str.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bozoky Z, Krzeminski M, Muhandiram R, Birtley JR, Al-Zahrani A, Thomas PJ, Frizzell RA, Ford RC, Forman-Kay JD. Proc Natl Acad Sci USA. 2013;110:4427. doi: 10.1073/pnas.1315104110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bozoky Z, Krzeminski M, Chong PA, Forman-Kay JD. FEBS J. 2013;280:4407. doi: 10.1111/febs.12422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang X, Paula ACD, Bozóky Z, Zhang H, Bertrand CA, Peters KW, Forman-Kay JD, Frizzella RA. Mol Biol Cell. 2012;23:996. doi: 10.1091/mbc.E11-08-0662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuriyan J, Ösapay K, Burley SK, Brünger AT, Hendrickson WA, Karplus M. Proteins. 1991;10:340. doi: 10.1002/prot.340100407. [DOI] [PubMed] [Google Scholar]
- 30.Lindorff-Larsen K, Kristjansdottir S, Teilum K, Fieber W, Dobson CM, Poulsen FM, Vendruscolo M. J Am Chem Soc. 2004;126:3291. doi: 10.1021/ja039250g. [DOI] [PubMed] [Google Scholar]
- 31.Vendruscolo M. Curr Opin Struct Bio. 2007;17:15. doi: 10.1016/j.sbi.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 32.Lange OF, Lakomek N-A, Fares C, Schröder GF, Walter KFA, Becker S, Meiler J, Grubmüller H, Griesinger C, Groot BLd. Science. 2008;320:1471. doi: 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- 33.Rieping W, Habeck M, Nilges M. Science. 2005;309:303. doi: 10.1126/science.1110428. [DOI] [PubMed] [Google Scholar]
- 34.Hummer G, Kofinger J. J Chem Phys. 2015;143:243150. doi: 10.1063/1.4937786. [DOI] [PubMed] [Google Scholar]
- 35.Roux B, Weare J. J Chem Phys. 2013;138:084107. doi: 10.1063/1.4792208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boomsma W, Ferkinghoff-Borg J, Lindorff-Larsen K. PLoS Comput Biol. 2014;10:e1003406. doi: 10.1371/journal.pcbi.1003406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Antonov LD, Olsson S, Boomsma W, Hamelryck T. Phys Chem Chem Phys. 2016 doi: 10.1039/c5cp04886a. [DOI] [PubMed] [Google Scholar]
- 38.Olsson S, Vogeli BR, Cavalli A, Boomsma W, Ferkinghoff-Borg J, Lindorff-Larsen K, Hamelryck T. J Chem Theory Comput. 2014;10:3484. doi: 10.1021/ct5001236. [DOI] [PubMed] [Google Scholar]
- 39.Cavalli A, Camilloni C, Vendruscolo M. J Chem Phys. 2013;138:094112. doi: 10.1063/1.4793625. [DOI] [PubMed] [Google Scholar]
- 40.Kendrew JC, Bodo G, Dintzis HM, Parrish RG, Wyckoff H, Phillips DC. Nature. 1958;181:662. doi: 10.1038/181662a0. [DOI] [PubMed] [Google Scholar]
- 41.Wüthrich K. NMR of Proteins and Nucleic Acids. John Wiley & Sons; 1986. [Google Scholar]
- 42.Henderson R, Baldwin JM, Ceska TA, Zemlin F, Beckmann E, Downing KH. J Mol Biol. 1990;213:899. doi: 10.1016/S0022-2836(05)80271-2. [DOI] [PubMed] [Google Scholar]
- 43.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS. Acta Crystallographica Section D: Biological Crystallography. 1998;54:905. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 44.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW. Acta Crystallographica Section D: Biological Crystallography. 2010;66:213. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lange OF, Schäfer LV, Grubmüller H. J Comp Chem. 2006;27:1693. doi: 10.1002/jcc.20473. [DOI] [PubMed] [Google Scholar]
- 46.Wright PE, Dyson HJ. Curr Opin Struct Bio. 2009;19:31. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schneider R, Huang JR, Yao M, Communie G, Ozenne V, Mollica L, Salmon L, Jensen MR, Blackledge M. Mol BioSys. 2012;8:56. doi: 10.1039/c1mb05291h. [DOI] [PubMed] [Google Scholar]
- 48.Dunker AK, Brown CJ, Lawson JD. Biochem. 2002 [Google Scholar]
- 49.Iakoucheva LM, Brown CJ, Lawson JD, Obradović Z, Dunker AK. J Mol Biol. 2002;323:573. doi: 10.1016/s0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- 50.Tompa P. Trends in Biochem Sci. 2002;27:527. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 51.Uversky VN, Oldfield CJ, Dunker AK. J Mol Recognition. 2005;18:343. doi: 10.1002/jmr.747. [DOI] [PubMed] [Google Scholar]
- 52.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Uversky VN, Obradović Z. J Proteome Res. 2007;6:1882. doi: 10.1021/pr060392u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oldfield CJ, Meng J, Yang JY, Yang MQ, Uversky VN, Dunker AK. BMC genomics. 2008;S1 doi: 10.1186/1471-2164-9-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ball KA, Phillips AH, Wemmer DE, Head-Gordon T. Biophys J. 2013;104:2714. doi: 10.1016/j.bpj.2013.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ball KA, Wemmer DE, Head-Gordon T. J Phys Chem B. 2014;118:6405. doi: 10.1021/jp410275y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fawzi NL, Phillips AH, Ruscio JZ, Doucleff M, Wemmer DE, Head-Gordon T. J Am Chem Soc. 2008;130:6145. doi: 10.1021/ja710366c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ball KA, Phillips AH, Nerenberg PS, Fawzi NL, Wemmer DE, Head-Gordon TL. Biochem. 2011;50:7612. doi: 10.1021/bi200732x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sgourakis NG, Merced-Serrano M, Boutsidis C, Drineas P, Du Z, Wang C, Garcia A. J Mol Biol. 2011;405:570. doi: 10.1016/j.jmb.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bowman GR, Ensign DL, Pande VS. J Chem Theory Comput. 2010;6:787. doi: 10.1021/ct900620b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lane TJ, Shukla D, Beauchamp KA, Pande VS. Current opinion in structural biology. 2013;23:58. doi: 10.1016/j.sbi.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bhowmick A, Head-Gordon T. Structure. 2015;23:44. doi: 10.1016/j.str.2014.10.011. [DOI] [PubMed] [Google Scholar]
- 62.Brookes DH, Head-Gordon T. Journal of the American Chemical Society. 2016;138:4530. doi: 10.1021/jacs.6b00351. [DOI] [PubMed] [Google Scholar]
- 63.Helgaker T, Jaszunski M, Pecul M. Prog NMR Spect. 2008;53:249. [Google Scholar]
- 64.Esteban-Martín S, Fenwick RB, Salvatella X. J Am Chem Soc. 2010;132:4626. doi: 10.1021/ja906995x. [DOI] [PubMed] [Google Scholar]
- 65.Marsh JA, Baker JMR, Tollinger M, Forman-Kay JD. J Am Chem Soc. 2008;130:7804. doi: 10.1021/ja802220c. [DOI] [PubMed] [Google Scholar]
- 66.Montalvao RW, Simone AD, Vendruscolo M. J Biomol NMR. 2012;53:281. doi: 10.1007/s10858-012-9644-3. [DOI] [PubMed] [Google Scholar]
- 67.Showalter SA, Brüschweiler R. J Am Chem Soc. 2007;129:4158. doi: 10.1021/ja070658d. [DOI] [PubMed] [Google Scholar]
- 68.Salmon L, Nodet G, Ozenne V, Yin G, Jensen MR, Zweckstetter M, Blackledge M. J Am Chem Soc. 2010;132:8407. doi: 10.1021/ja101645g. [DOI] [PubMed] [Google Scholar]
- 69.Huang JR, Grzesiek S. Journal of the American Chemical Society. 2009;132:694. doi: 10.1021/ja907974m. [DOI] [PubMed] [Google Scholar]
- 70.Mukrasch MD, Markwick P, Biernat J, von Bergen M, Bernado P, Griesinger C, Mandelkow E, Zweckstetter M, Blackledge M. Journal of the American Chemical Society. 2007;129:5235. doi: 10.1021/ja0690159. [DOI] [PubMed] [Google Scholar]
- 71.Bibow S, Ozenne V, Biernat J, Blackledge M, Mandelkow E, Zweckstetter M. Journal of the American Chemical Society. 2011;133:15842. doi: 10.1021/ja205836j. [DOI] [PubMed] [Google Scholar]
- 72.Drescher M. In: EPR Spectroscopy: Applications in Chemistry and Biology. Drescher M, Jeschke G, editors. Springer Berlin Heidelberg; Berlin, Heidelberg: 2012. p. 91. [Google Scholar]
- 73.Theillet F-X, Binolfi A, Bekei B, Martorana A, Rose HM, Stuiver M, Verzini S, Lorenz D, Rossum Mv, Goldfarb D, Selenko P. Nature. 2016;530:45. doi: 10.1038/nature16531. [DOI] [PubMed] [Google Scholar]
- 74.Mylonas E, Hascher A, Bernado P, Blackledge M, Mandelkow E, Svergun DI. Biochemistry-Us. 2008;47:10345. doi: 10.1021/bi800900d. [DOI] [PubMed] [Google Scholar]
- 75.Bernado P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. Journal of the American Chemical Society. 2007;129:5656. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- 76.Bernado P, Svergun DI. Mol Biosyst. 2012;8:151. doi: 10.1039/c1mb05275f. [DOI] [PubMed] [Google Scholar]
- 77.Krzeminski M, Marsh JA, Neale C, Choy WY, Forman-Kay JD. Bioinformatics. 2013;29:398. doi: 10.1093/bioinformatics/bts701. [DOI] [PubMed] [Google Scholar]
- 78.Choy WY, Forman-Kay JD. J Mol Biol. 2001;308:1011. doi: 10.1006/jmbi.2001.4750. [DOI] [PubMed] [Google Scholar]
- 79..
- 80.Schneidman-Duhovny D, Hammel M, Sali A. Nucleic Acids Research. 2010;38:W540. doi: 10.1093/nar/gkq461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rambo RP, Tainer JA. Nature. 2013;496:477. doi: 10.1038/nature12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pelikan M, Hura GL, Hammel M. General physiology and biophysics. 2009;28:174. doi: 10.4149/gpb_2009_02_174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, 2nd, Tsutakawa SE, Jenney FE, Jr, Classen S, Frankel KA, Hopkins RC, Yang SJ, Scott JW, Dillard BD, Adams MW, Tainer JA. Nat Methods. 2009;6:606. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rambo RP, Tainer JA. Biopolymers. 2011;95:559. doi: 10.1002/bip.21638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zawadzka-Kazimierczuk A, KoŸmiński W, Šanderová H, Krásný L. Journal of Biomolecular NMR. 2012;52:329. doi: 10.1007/s10858-012-9613-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Crowhurst KA, Forman-Kay JD. Biochem. 2003;42:8687. doi: 10.1021/bi034601p. [DOI] [PubMed] [Google Scholar]
- 87.Marsh JA, Neale C, Jack FE, Choy WY, Lee AY, Crowhurst KA, Forman-Kay JD. J Mol Biol. 2007;367:1494. doi: 10.1016/j.jmb.2007.01.038. [DOI] [PubMed] [Google Scholar]
- 88.Marsh JA, Forman-Kay JD. J Mol Biol. 2009;391:359. doi: 10.1016/j.jmb.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 89.Clore GM. Biochemical Society transactions. 2013;41:1343. doi: 10.1042/BST20130232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Anthis N, Clore GM. Quart Rev Biophys. 2015;48:35. doi: 10.1017/S0033583514000122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bertoncini C, Jung Y, Fernandez C, Hoyer W, Griesinger C, Jovin T, Zweckstetter M. Proc Natl Acad Sci USA. 2005;102:1430. doi: 10.1073/pnas.0407146102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jensen M, Zweckstetter M, Huang J, Blackledge M. Chem Rev. 2014;114:6632. doi: 10.1021/cr400688u. [DOI] [PubMed] [Google Scholar]
- 93.de Vera IM, Blackburn ME, Galiano L, Fanucci GE. In: Current protocols in protein science/editorial board. Unit 17. Coligan John E, et al., editors. Vol. 74. 2013. p. 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Goldfarb D. Physical chemistry chemical physics: PCCP. 2014;16:9685. doi: 10.1039/c3cp53822b. [DOI] [PubMed] [Google Scholar]
- 95.Jeschke G, Chechik V, Ionita P, Godt A, Zimmermann H, Banham J, Timmel CR, Hilger D, Jung H. Appl Magn Reson. 2006;30:473. [Google Scholar]
- 96.Haas E. Methods in molecular biology. 2012;895:467. doi: 10.1007/978-1-61779-927-3_28. [DOI] [PubMed] [Google Scholar]
- 97.Schuler B, Muller-Spath S, Soranno A, Nettels D. Methods in molecular biology. 2012;896:21. doi: 10.1007/978-1-4614-3704-8_2. [DOI] [PubMed] [Google Scholar]
- 98.Zerze GH, Best RB, Mittal J. Biophysical journal. 2014;107:1654. doi: 10.1016/j.bpj.2014.07.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Marsh JA, Forman-Kay JD. Proteins: Struct, Func, Bioinform. 2012;80:556. doi: 10.1002/prot.23220. [DOI] [PubMed] [Google Scholar]
- 100.Han B, Liu YF, Ginzinger SW, Wishart DS. Journal of Biomolecular NMR. 2011;50:43. doi: 10.1007/s10858-011-9478-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zweckstetter M, Bax A. J Am Chem Soc. 2000;122:3791. [Google Scholar]
- 102.Herrmann T, Güntert P, Wüthrich K. J Mol Biol. 2002;319:209. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- 103.López-Méndez B, Güntert P. J Am Chem Soc. 2006;128:13112. doi: 10.1021/ja061136l. [DOI] [PubMed] [Google Scholar]
- 104.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. J Magn Res. 2003;160:66. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 105.Schwieters CD, Kuszewski JJ, Clore GM. Progr NMR Spect. 2006;48:47. [Google Scholar]
- 106.Shen Y, Delaglio F, Cornilescu G, Bax A. J Biomol NMR. 2009;44:213. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ozenne V, Bauer F, Salmon L, Huang JR, Jensen MR, Segard S, Bernado P, Charavay C, Blackledge M. Bioinformatics. 2012;28:1463. doi: 10.1093/bioinformatics/bts172. [DOI] [PubMed] [Google Scholar]
- 108.Feldman HJ, Hogue CW. Proteins: Struct, Func, Bioinform. 2000;39:112. [PubMed] [Google Scholar]
- 109.Huang A, Stultz CM. PLoS Comp Bio. 2008;4:e1000155. doi: 10.1371/journal.pcbi.1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yoon M, Venkatachalam V, Huang A, Choi B, Stultz C, Chou J. Prot Sci. 2008 doi: 10.1002/pro.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dedmon MM, Lindorff-Larsen K, Christodoulou J, Vendruscolo M, Dobson CM. J Am Chem Soc. 2005;127:476. doi: 10.1021/ja044834j. [DOI] [PubMed] [Google Scholar]
- 112.Richter B, Gsponer J, Várnai P, Salvatella X, Vendruscolo M. J Biomol NMR. 2007;37:117. doi: 10.1007/s10858-006-9117-7. [DOI] [PubMed] [Google Scholar]
- 113.Allison JR, Várnai P, Dobson CM, Vendruscolo M. J Am Chem Soc. 2009;131:18314. doi: 10.1021/ja904716h. [DOI] [PubMed] [Google Scholar]
- 114.Vise P, Baral B, Stancik A, Lowry DF, Daughdrill GW. Proteins: Struct, Func, Bioinform. 2007;67:526. doi: 10.1002/prot.21364. [DOI] [PubMed] [Google Scholar]
- 115.Fisher CK, Huang A, Stultz CM. Journal of the American Chemical Society. 2010;132:14919. doi: 10.1021/ja105832g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fisher CK, Stultz CM. Curr Opin Struct Bio. 2011;21:426. doi: 10.1016/j.sbi.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hou L, Shao H, Zhang Y, Li H, Menon NK, Neuhaus EB, Brewer JM, Byeon IJ, Ray DG, Vitek MP, Iwashita T, Makula RA, Przybyla AB, Zagorski MG. J Am Chem Soc. 2004;126:1992. doi: 10.1021/ja036813f. [DOI] [PubMed] [Google Scholar]
- 118.Sgourakis NG, Yan Y, McCallum SA, Wang C, Garcia AE. J Mol Biol. 2007;368:1448. doi: 10.1016/j.jmb.2007.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Karplus M, Grant DM. Proc Natl Acad Sci U S A. 1959;45:1269. doi: 10.1073/pnas.45.8.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Vuister GW, Bax A. J Am Chem Soc. 1993;115:7772. [Google Scholar]
- 121.Case DA. Acc Chem Res. 2002;35:325. doi: 10.1021/ar010020l. [DOI] [PubMed] [Google Scholar]
- 122.Case DA, Scheurer C, Bruschweiler R. J Am Chem Soc. 2000;122:10390. [Google Scholar]
- 123.Neal S, Nip AM, Zhang H, Wishart DS. J Biomol NMR. 2003;26:215. doi: 10.1023/a:1023812930288. [DOI] [PubMed] [Google Scholar]
- 124.Hennig M, Bermel W, Schwalbe H, Griesinger C. J Am Chem Soc. 2000;122:6268. [Google Scholar]
- 125.Shen Y, Bax A. J Biomol NMR. 2010;48:13. doi: 10.1007/s10858-010-9433-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Case DA. Curr Opin Struct Bio. 2013;23:172. doi: 10.1016/j.sbi.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Auer A, Gauss J, Stanton J. J Chem Phys. 2003;118:10407. [Google Scholar]
- 128.Lodewyk MW, Siebert MR, Tantillo DJ. Chem Rev. 2012;112:1839. doi: 10.1021/cr200106v. [DOI] [PubMed] [Google Scholar]
- 129.Tellgren E, Kvaal S, Sagvolden E, Ekstrom U, Teale A, Helgaker T. Phys Rev A. 2012;86:062506. [Google Scholar]
- 130.Grayce CJ, Harris RA. Phys Rev A. 1994;50:3089. doi: 10.1103/physreva.50.3089. [DOI] [PubMed] [Google Scholar]
- 131.Helgaker T, Coriani S, Jorgensen P, Kristensen K, Olsen J, Ruud K. Chem Rev. 2012;112:543. doi: 10.1021/cr2002239. [DOI] [PubMed] [Google Scholar]
- 132.Teale AM, Lutnaes OB, Helgaker T, Tozer DJ, Gauss J. J Chem Phys. 2013;138:024111. doi: 10.1063/1.4773016. [DOI] [PubMed] [Google Scholar]
- 133.Piana S, Donchev AG, Robustelli P, Shaw DE. J Phys Chem B. 2015;119:5113. doi: 10.1021/jp508971m. [DOI] [PubMed] [Google Scholar]
- 134.Best R, Hummer G. J Phys Chem B. 2009;113:9004. doi: 10.1021/jp901540t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Best RB, Mittal J. Journal of Physical Chemistry B. 2010;114:14916. doi: 10.1021/jp108618d. [DOI] [PubMed] [Google Scholar]
- 136.Nerenberg PS, Head-Gordon T. J Chem Theory Comput. 2011;7:1220. doi: 10.1021/ct2000183. [DOI] [PubMed] [Google Scholar]
- 137.Nerenberg P, Jo B, So C, Tripathy A, Head-Gordon T. Journal of physical Chemistry B. 2012;116:4524. doi: 10.1021/jp2118373. [DOI] [PubMed] [Google Scholar]
- 138.Best RB, Zheng W, Mittal J. J Chem Theory Comp. 2014;10:5113. doi: 10.1021/ct500569b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang L-P, Head-Gordon T, Ponder JW, Ren P, Chodera JD, Eastman PK, Martinez TJ, Pande VS. J Phys Chem B. 2013 doi: 10.1021/jp403802c. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Demerdash ON, Yap E-H, Head-Gordon T. Ann Rev Phys Chem. 2014;65:149. doi: 10.1146/annurev-physchem-040412-110040. [DOI] [PubMed] [Google Scholar]
- 141.Albaugh A, Boateng HA, Bradshaw RT, Demerdash O, Dziedzic J, Mao Y, Margul DT, Swails J, Zeng Q, Case DA, Eastman P, Essex JW, Head-Gordon M, Pande VS, Ponder JW, Shao Y, Skylaris C-K, Todorov IT, Tuckerman ME, Head-Gordon T. J Phys Chem B (Feature article) 2016 doi: 10.1021/acs.jpcb.6b06414. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nymeyer H. J Chem Phys. 2010;133:114113. doi: 10.1063/1.3480013. [DOI] [PubMed] [Google Scholar]
- 143.Huang X, Bowman GR, Pande VS. J Chem Phys. 2008;128:205106. doi: 10.1063/1.2908251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Park S. Phys Rev E Stat Nonlin Soft Matter Phys. 2008;77:016709. doi: 10.1103/PhysRevE.77.016709. [DOI] [PubMed] [Google Scholar]
- 145.Hagen M, Kim B, Liu P, Friesner RA, Berne BJ. The journal of physical chemistry B. 2007;111:1416. doi: 10.1021/jp064479e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sorin EJ, Rhee YM, Nakatani BJ, Pande VS. Biophysical journal. 2003;85:790. doi: 10.1016/S0006-3495(03)74520-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Rhee YM, Pande VS. Biophysical journal. 2003;84:775. doi: 10.1016/S0006-3495(03)74897-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sugita Y, Okamoto Y. Chemical Physics Letters. 1999;314:141. [Google Scholar]
- 149.Mitsutake A, Sugita Y, Okamoto Y. Peptide Science. 2001;60:96. doi: 10.1002/1097-0282(2001)60:2<96::AID-BIP1007>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 150.Brown S, Head-Gordon T. Journal of Computational Chemistry. 2003;24:68. doi: 10.1002/jcc.10181. [DOI] [PubMed] [Google Scholar]
- 151.Zuckerman DM, Lyman E. J Chem Theory Comput. 2006;2:1200. doi: 10.1021/ct600297q. [DOI] [PubMed] [Google Scholar]
- 152.Itoh SG, Okumura H. Journal of Computational Chemistry. 2013;34:622. doi: 10.1002/jcc.23167. [DOI] [PubMed] [Google Scholar]
- 153.Flores-Canales JC, Kurnikova M. J Chem Theory Comput. 2015;11:2550. doi: 10.1021/ct501090y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Weber JK, Pande VS. J Chem Theory Comput. 2011;7:3405. doi: 10.1021/ct2004484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Huang X, Bowman GR, Bacallado S, Pande VS. Proc Natl Acad Sci U S A. 2009;106:19765. doi: 10.1073/pnas.0909088106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kohlhoff KJ, Shukla D, Lawrenz M, Bowman GR, Konerding DE, Belov D, Altman RB, Pande VS. Nature chemistry. 2014;6:15. doi: 10.1038/nchem.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Li D-W, Brüschweiler R. Journal of the American Chemical Society. 2009;131:7226. doi: 10.1021/ja902477s. [DOI] [PubMed] [Google Scholar]
- 158.DuBay KH, Geissler PL. Journal of molecular biology. 2009;391:484. doi: 10.1016/j.jmb.2009.05.068. [DOI] [PubMed] [Google Scholar]
- 159.Friedland GD, Linares AJ, Smith Ca, Kortemme T. Journal of molecular biology. 2008;380:757. doi: 10.1016/j.jmb.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zhang J, Liu JS. PLoS computational biology. 2006;2:e168. doi: 10.1371/journal.pcbi.0020168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hattori M, Li H, Yamada H, Akasaka K, Hengstenberg W, Gronwald W, Kalbitzer HR. Prot Sci. 2004;13:3104. doi: 10.1110/ps.04877104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Dunbrack RL. Current opinion in structural biology. 2002;12:431. doi: 10.1016/s0959-440x(02)00344-5. [DOI] [PubMed] [Google Scholar]
- 163.Shapovalov MV, Dunbrack RL. Structure (London, England: 1993) 2011;19:844. doi: 10.1016/j.str.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Fraser JS, Clarkson MW, Degnan SC, Erion R, Kern D, Alber T. Nature. 2009;462:669. doi: 10.1038/nature08615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Fraser JS, van den Bedem H, Samelson AJ, Lang PT, Holton JM, Echols N, Alber T. Proc Natl Acad Sci U S A. 2011;108:16247. doi: 10.1073/pnas.1111325108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Clarkson MW, Gilmore SA, Edgell MH, Lee AL. Biochemistry-Us. 2006;45:7693. doi: 10.1021/bi060652l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tuttle LM, Dyson HJ, Wright PE. Biochemistry-Us. 2013;52:3464. doi: 10.1021/bi400322e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Varadi M, Kosol S, Lebrun P, Valentini E, Blackledge M, Dunker AK, Felli IC, Forman-Kay JD, Kriwacki RW, Pierattelli R, Sussman J, Svergun DI, Uversky VN, Vendruscolo M, Wishart D, Wright PE, Tompa P. Nuc Acids Res. 2014;42:D326. doi: 10.1093/nar/gkt960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ulrich EL, Akutsu H, Doreleijers JF, Harano Y, Ioannidis YE, Lin J, Livny M, Mading S, Maziuk D, Miller Z, Nakatani E, Schulte CF, Tolmie DE, Wenger RK, Yao H, Markley JL. Nucleic Acids Research. 2008;36:D402. doi: 10.1093/nar/gkm957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Pérez F, Granger BE. Computing in Science & Engineering. 2007;9:21. [Google Scholar]
- 171.Kabsch W, Sander C. Biopoly. 1983;22:2577. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]