

Abstract
Glycosylation plays an important role in cell-cell adhesion, ligand-binding and subcellular recognition. Current approaches for predicting protein glycosylation are primarily based on sequence-derived features, while little work has been done to systematically assess the importance of structural features to glycosylation prediction. Here, we propose a novel bioinformatics method called GlycoMinestruct(http://glycomine.erc.monash.edu/Lab/GlycoMine_Struct/) for improved prediction of human N- and O-linked glycosylation sites by combining sequence and structural features in an integrated computational framework with a two-step feature-selection strategy. Experiments indicated that GlycoMinestruct outperformed NGlycPred, the only predictor that incorporated both sequence and structure features, achieving AUC values of 0.941 and 0.922 for N- and O-linked glycosylation, respectively, on an independent test dataset. We applied GlycoMinestruct to screen the human structural proteome and obtained high-confidence predictions for N- and O-linked glycosylation sites. GlycoMinestruct can be used as a powerful tool to expedite the discovery of glycosylation events and substrates to facilitate hypothesis-driven experimental studies.
Glycosylation is a major type of protein post-translational modification (PTM) through which a carbohydrate (i.e., a glycosyl donor) is attached to specific functional groups on target proteins (i.e., glycosyl acceptors). It is among the most complicated of PTMs occurring in protein biosynthesis1 and is ubiquitous across different species and cell types1. Glycosylation plays an important role in a myriad of biological processes involving protein folding, sorting, trafficking, degradation, and immune response2,3,4,5. Due to its fundamental importance in cell biology, protein glycosylation has also been implicated in a number of human diseases, including congenital muscular dystrophies6, alcoholism7, Alzheimer’s disease8, and cancer6.
The three major types of glycosylation, N-, O-, and C-linked glycosylation, are distinguished in the functional groups in the protein side chain being modified with the carbohydrate moiety. While little is known about the factors contributing to C-linked glycosylation, asparagine residues can be modified by N-linked glycosylation when located within a consensus sequence motif (Asn-X-Ser/Thr, where X denotes any amino acid except Pro9). Oligosaccharyltransferase is the central enzyme of protein N-glycosylation in eukaryotes, catalyzing the formation of an N-glycosidic linkage of oligosaccharides to the side-chain amide of target asparagine residues. This catalysis occurs selectively on consensus sequons Asn-X-Ser/Thr in substrate proteins10. This pathway occurs co-translationally (i.e. as unfolded substrate polypeptides enter the endoplasmic reticulum) or post-translationally (i.e. after substrate polypeptides have folded in the lumen of the endoplasmic reticulum). Since cell surface and extracellular proteins are first translocated into the endoplasmic reticulum, protein N-glycosylation is responsible for much of the glycan modification of these extracellular proteins. O-linked glycosylation involves glycan attachment to serine or threonine residues. There exists at least five classes of O-glycosyl modifications, including O-N-acetylgalactosamine (O-galNAc), O-fucose, O-glucose, O-N-acetylglucosamine (O-GlcNAc) and O-mannose11. These reactions can occur in the cytosol, to proteins that will remain in the cytosol or enter into the nucleus12,13, or in the cis-, medial- and trans-Golgi compartments after secretory proteins traffic from the endoplasmic reticulum14,15. To date, no biologically significant sequons have been identified for any class of O-linked glycosylation16. Whether it occurs in the cytosol or the Golgi compartment, O-linked glycosylation occurs post-translationally so that only some potential glycosylation sites would be available to the glycosyltransferases that mediate this PTM11. Likewise, only a sub-set of all Asn-X-Ser/Thr sequences will be accessed by the glycosyltransferases that catalyze N-linked glycosylation17. In addition to the accessibility criterion limiting O- and N-glycosylation11, it was suggested that sequences surrounding a potential glycosylation site and/or distances to the next glycosylation site can impact whether an acceptor Asn-X-Ser/Thr sequence is actually N-glycosylated18,19.
Mass spectrometry is perhaps now the predominant experimental method to detect protein glycosylation sites20,21. In recent years, other techniques that can perform medium and high-throughput identification and quantification of glycosylation sites (including glycan structures and glycan occupancy) have been developed and applied22,23,24, including flow cytometry25, solid-phase extraction26 and lectin-based methods27,28. All of these experimental approaches require considerable time and effort. This hinders their ability to keep pace with data generated from high-throughput sequencing endeavors, given the enormous volume of proteomic data generated by these and other new technologies. Compared with other important types of PTM, such as phosphorylation, acetylation, and ubiquitination, bioinformatics-based prediction of glycosylation has lagged behind29.
In light of this, computational approaches to address this issue are attractive options, particularly with advances in data-mining and machine-learning algorithms. Current computational tools for protein-glycosylation prediction (e.g., GlycoMine30, NetNGlyc31, NetOGlyc32, EnsembleGly33, and GPP34) were constructed based on sequence features, which have been widely used as a basic feature for the construction of computational models. Sequence-based features generally include physical/chemical properties (e.g., hydrophobicity and AAindex), statistical features (e.g., position-specific scoring matrices (PSSMs)), predicted features by third-party computational methods (e.g., protein secondary structure), and functional annotations from publicly available databases.
Protein structural features have not been systematically examined or incorporated for glycosylation prediction. An interplay between N-linked glycosylation sites and secondary structures was revealed, suggesting that secondary structure features are important for distinguishing glycosylation sites from non-glycosylation sites19,35. To the best of our knowledge, NGlycPred35 is the only tool that has incorporated protein structural features for N-linked glycosylation prediction. The balanced predictive performance of NGlycPred based on 10-fold cross-validation in terms of accuracy (ACC) was 68.7%. Furthermore, NGlycPred is limited to N-linked glycosylation-site prediction. These underline the necessity for developing an improved approach considering both sequence-derived and three-dimensional protein-structure information.
Here, we proposed a novel computational framework, GlycoMinestruct, for N- and O-linked glycosylation-site prediction that integrates both protein-sequence and protein-structural features. This is the only computational framework to date that assembles protein-sequence and protein-structural features for both N- and O-glycosylation-site prediction. Effective feature-selection methods, combining linear support vector machine (SVM)-based feature selection and incremental feature selection, were applied to extract the most informative sequence-based and structural features for N- and O-linked glycosylation prediction. In empirical studies, our proposed method achieved outstanding predictive performance in terms of area under the curve (AUC; 0.948 and 0.923) for N- and O-linked glycosylation sites, respectively, using a benchmark dataset and outperformed NGlycPred on an independent test dataset. Additionally, we applied GlycoMinestruct to scan the entire human structural proteome to identify N- and O-glycosylation sites, thereby providing a comprehensive dataset to the community for further in-depth glycosylation studies and experimental investigations.
Results
Methodology overview
A flowchart describing GlycoMinestruct is illustrated in Fig. 1, with the four major steps denoted by different colors: dataset collection and preprocessing (blue), feature extraction (yellow), feature analysis and selection (red), and model evaluation (green). The first step involves data collection and extraction from publicly available resources. During the second step, a variety of sequence-based and structural features are extracted using third-party software. A two-step feature-selection procedure is introduced in the third step, where linear SVM-based feature selection36 is first used, followed by incremental feature selection (IFS)37 to characterize the feature subsets that contribute the most information for N- and O-linked glycosylation-site prediction. During the final stage, random forest (RF)-based classifiers are trained using the final selected optimal feature subsets (OFS) for N- and O-linked glycosylation-site prediction. The performance of RF classifiers was extensively evaluated using both cross-validation and independent tests. During this stage, we also compared the performance of our method with that of NGlycPred35, which is the only predictor currently integrating both sequence and structural features for N-linked glycosylation-site prediction.
Figure 1. Overview of the GlycoMinestruct framework.

Four major steps are denoted by different colors: dataset collection and preprocessing (blue), feature extraction (yellow), feature analysis and selection (red), model evaluation (green).
Residue enrichment of sequence motifs for both N- and O-linked glycosylation sites
We first analyzed the amino-acids specificity and enrichment of N- and O-linked glycosylation sites in our curated benchmark datasets. The sequons of N- and O-linked glycosylation sites were presented with a local window size of 14 residues flanking the glycosylation sites (seven residues upstream and downstream of each glycosylation site). pLogo38 was then applied to calculate and draw the sequence logos for N-linked (Fig. 2a) and O-linked (Fig. 2b) glycosylation sites using the human-protein dataset as background for statistical purposes. The sequence logos in Fig. 2 demonstrate the significantly overrepresented and underrepresented amino acids (p = 0.05) for each position of the sequons in the benchmark N- and O-linked glycosylation-site datasets.
Figure 2. Residue specificity and enrichment of sequons.
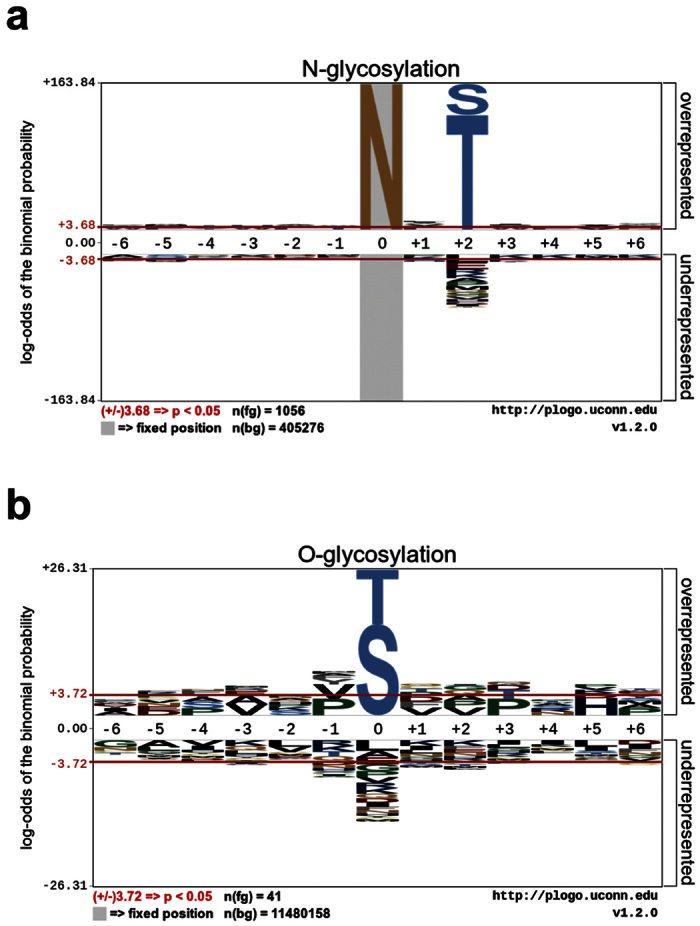
(a) N- and (b) O-linked glycosylation sites with the “human protein dataset” selected as the background set. Sequence logos and statistical test (binomial probabilities and Bonferroni correction) were generated using the pLogo program38.
N-linked and O-linked glycosylation sites show different preferences for neighbouring amino acids (Fig. 2). As expected, for N-linked glycosylation the central position is dominated by asparagine (N) residues; while threonine (T) and serine (S) are preferable residues at the central position of O-linked glycosylation sites. N- and O-linked glycosylation sites further showed different residue preferences at other positions. While threonine (T) and serine (S) were overrepresented in sequence motifs associated with N-glycosylation sites at position +2 (downstream of the N-glycosylation site9), no specific amino acids were found to be overrepresented at other O-linked glycosylation-site positions. The amino acid preferences shown in Fig. 2 represent patterns important for distinguishing N- and O-linked glycosylation sites.
Optimized feature set (OFS)
In addition to reducing the computational complexity of classifiers, effective feature-selection methods can improve the predictive performance of classifiers by eliminating noisy and redundant features. A total of 385 sequence-derived features and 14 structural features were initially extracted using a variety of computational tools for both N- and O-glycosylation. The ‘Methods’ section and Supplementary Table S3 present a detailed description of these features. Applying the proposed two-step feature-selection method led to selection of 14 contributing features for N-linked glycosylation sites, and 11 contributing features for O-linked glycosylation sites. The IFS curves displaying the changes in AUC values during the second step of IFS are shown in Supplementary Fig. S1. The 14 N-linked and 11 O-linked optimal features were selected by five-fold cross-validation using the benchmark datasets. We also performed an independent test using these optimal features and showed that models trained using the two optimal feature sets accurately identified the N- and O-linked glycosylation sites. Tables 1 and 2 provide the lists of selected optimal features for N- and O-glycosylation, respectively. For N-linked glycosylation, the final optimal features included nine sequence-derived features and five structural features, while for O-linked glycosylation, the final optimal features included eight sequence-derived features and three structural features.
Table 1. The selected optimal features for N-linked glycosylation.
| Num. | Feature | Position | Software |
|---|---|---|---|
| V1 | Normalized average hydrophobicity scales | P10 | AAindex81 |
| V2 | Absolute accessibility of non-polar side-chain | P1 | NACCESS84 |
| V3 | PSSM | P250 | PSI-BLAST79 |
| V4 | PSSM | P235 | PSI-BLAST79 |
| V5 | Standard deviation of side-chain depth index | P1 | PSAIA88 |
| V6 | Conformational parameter of beta-turn | P10 | AAindex81 |
| V7 | PSSM | P173 | PSI-BLAST79 |
| V8 | Absolute accessibility of main chain | P1 | NACCESS84 |
| V9 | Mean polarity | P8 | AAindex81 |
| V10 | Log-odds ratio | P1 | DiscoTope87 |
| V11 | Average flexibility indices | P7 | AAindex81 |
| V12 | Mean polarity | P10 | AAindex81 |
| V13 | Absolute accessibility of all-atoms | P1 | NACCESS84 |
| V14 | PSSM | P274 | PSI-BLAST79 |
Features highlighted in italic indicate structural features, while other features not highlighted are sequence-derived features or amino acid properties.
Table 2. The selected optimal features for O-linked glycosylation.
| Num. | Feature | Position | Software |
|---|---|---|---|
| V1 | Conformational parameter of beta-turn | P8 | AAindex81 |
| V2 | PSSM | P38 | PSI-BLAST79 |
| V3 | B factor | P1 | PDB file53 |
| V4 | Normalized average hydrophobicity scales | P8 | AAindex81 |
| V5 | Standard deviation of side-chain depth index | P1 | PSAIA88 |
| V6 | PSSM | P293 | PSI-BLAST79 |
| V7 | PSSM | P248 | PSI-BLAST79 |
| V8 | PSSM | P8 | PSI-BLAST79 |
| V9 | PSSM | P128 | PSI-BLAST79 |
| V10 | Absolute accessibility of main chain | P1 | NACCESS84 |
| V11 | Mean polarity | P8 | AAindex81 |
Features highlighted in italic indicate structural features, while other features not highlighted are sequence-derived features or amino acid properties.
The ‘Num’ column in Tables 1 and 2 indicates the order of the selected features in the OFS, which were ranked by the linear SVM during the first feature selection step to quantify the importance of each individual features. The ‘Position’ column in Tables 1 and 2 indicates the position of a corresponding feature in the local sliding window. Refer to the subsection ‘Feature window’ of ‘Methods’ for the definition of the local sliding window. Among the selected sequence-derived features, PSSM-relevant features for different positions were chosen in the OFS for both N- and O-linked glycosylation sites. PSSM is widely used to characterize the variability of each amino acid in given protein sequences based on multiple-sequence alignment. Previous studies on predicting protein PTM sites, such as phosphorylation39 and ubiquitination40, demonstrated the importance and contribution of PSSM to prediction performance. Similarly, the feature-selection results documented in Tables 1 and 2 revealed that PSSM also plays crucial roles in predicting glycosylation sites, which is consistent with finding from our previous study30. Other sequence-based features were then extracted from the AAindex. A recent study showed that an appropriate degree of hydrophobicity in a glycosylation site is crucial for protein-folding mechanism, indicating a strong relationship between glycosylation and residue hydrophobicity (V1 in Table 1)41.
A conformational parameter involving the β-turn is another feature derived from the AAindex captured as a contributive feature for both N- and O-linked glycosylation prediction. Structural studies showed that turns and bends are regions favorable for harboring glycosylation sites as compared to other secondary structural elements19,42,43. Our feature selection results are consistent with these biological findings.
Tables 1 and 2 show both similarity and diversity of N- and O-linked glycosylation sites in terms of selected structural features. Absolute-accessibility features for different positions calculated by NACCESS (http://www.bioinf.manchester.ac.uk/naccess/) were selected as important features for both N- and O-linked glycosylation sites. Absolute accessibility describes a residue being conformationally accessible as a prerequisite for glycosylation44. Importantly, this study revealed log-odds ratios representing the epitope propensity for B cells as important attributes for N-linked glycosylation prediction. Glycosylated protein antigens play important roles in the immunologic process45 through binding between epitope and B cell. The log-odds ratios presented in Table 1 suggested that epitope propensity was strongly correlated with protein glycosylation. Furthermore, B factors associated with protein structural dynamics were evaluated as being contributory features for O-linked glycosylation prediction (Table 2). Given that glycosylation profoundly affects the protein folding and stability46, the B factor representing thermal motion and used to measure the protein stability, was revealed in our feature-selection results as strongly correlated with protein glycosylation.
Analysis of the composition of selected optimal features indicated that both sequence and structural features contributed to N- and O-linked glycosylation prediction. In the OFS of N-linked glycosylation, a total of 14 optimal features were selected, including nine sequence-derived features and five structural features. Among the nine sequence features, there were five (5/60 = 8.33%) AAindex features and four (4/360 = 1.11%) PSSM features. In the OFS of O-linked glycosylation, eight sequence features and three structural features were finally selected: the eight sequence-derived features include three AAindex (3/60 = 5%) features and five (5/300 = 1.67%) PSSM features. In comparison, sequence-derived features only accounted for 2.3% (9/385) and 2.1% (8/385) of the final selected features for N- and O-linked glycosylation, respectively, while structural features represented ~36% (5/14) and ~21% (3/14), respectively, in the final OFS. Even though the absolute number of the selected sequence-derived features was larger than that of the selected structural features, the relative percentage of the latter was larger than that of the former for both N- (~36%) and O-linked (~21%) glycosylation. This was because the number of initially extracted sequence features was much larger than that of structural features (385 vs. 14). Altogether, these findings suggested that structural features are indispensable and crucial for N- and O-linked glycosylation prediction.
Feature importance and contribution in OFS
Given that the selected features in Tables 1 and 2 may or may not be equally important for glycosylation prediction, we evaluated the importance of individual optimal features in the OFS in terms of their relative contribution to the performance of N- and O-linked glycosylation prediction. Specifically, the importance of each of the features was assessed and ranked based on the average decrease in accuracy of the RF models trained using the independent test after removal of the correspoding feature from the OFS. The results are shown in Fig. 3.
Figure 3. The relative importance and ranking of the selected optimal features.

(a) N-linked glycosylation and (b) O-linked glycosylation based on the average accuracy decrease of models trained after removal of a correspoding feature from the feature set.
The top two features for N-linked glycosylation-site prediction
The two most important structural features for N-linked glycosylation-site prediction (Table 1) were the log-odds ratio (V10, calculated by DiscoTope, which is for discontinuous B cell epitopes prediction) and the absolute accessibility of non-polar side chains (V2, calculated by NACCESS). Removal of each of the two features from the OFS led to the decrease in accuracy of 3.78% and 3.76%, respectively (Fig. 3a). Box plots of these two structural features between N-glycosylation and non-N-glycosylation sites (Supplementary Figs S2j and S2b) showed that N-glycosylation sites had larger average log-odds ratios (−9.596), while non-N-glycosylation sites had an average value of −11.365, suggesting the importance of glycosylation in immunological process. The difference of the average log-odds-ratio values between N-glycosylation sites and non-N-glycosylation sites was statistically significant (p = 0.039). Similarly, in the case of the absolute accessibility of non-polar side chains, N-glycosylation sites also had large average values of 19.504, while non-N-glycosylation sites had lower average values of 17.359, which agree with finding that non-N-glycosylation sites tend to be predicted as solvent-inaccessible. The distribution of the absolute accessibility of non-polar side chains between N-glycosylation sites and non-N-glycosylation sites was also found to be statistically significant (p = 0.003). The boxplots displaying differences between N-linked glycosylation and non-N-glycosylation sites for features listed in Table 1 are shown in Supplementary Fig. S2.
The top two features for O-linked glycosylation-site prediction
Feature importance-ranking analysis indicated that PSSM_P38 (V2, generated by BLAST) and the standard deviation of the side-chain-depth index (V5, calculated by PSAIA) were the two most important features for O-linked glycosylation-site prediction (Fig. 3b). The box plots of these two features for O-glycosylation sites and non-O-glycosylation sites are shown in Supplementary Fig. S3b,e. For the first feature, the distributions of PSSM_P38 values between O-glycosylation and non-O-glycosylation sites were significantly different, (p = 4.98 × 10−8, Supplementary Fig. S3b). The standard deviation of the side-chain-depth index is an important structural feature for O-linked glycosylation-site prediction (Table 2). The box plot in Supplementary Fig. S3e showed that the average depth-index values for O-glycosylation and non-O-glycosylation sites differed significantly (p = 0.003). The larger average depth-index values (0.373) for non-O-glycosylation sites relative to those of O-glycosylation sites (0.297) may be partly explained by tendency of non-O-glycosylation sites to be solvent inaccessible and buried within the protein structure. The boxplots displaying the distributions of other optimal sequence and structural feature values (Table 2) for O-linked glycosylation sites can be found in Supplementary Fig. S3.
Figure 3 was generated based on the ‘Average Accuracy Decrease’ calculated by the Random Forest algorithm after removing a certain feature from the OFS. Supplementary Figs S2 and S3, on the other hand, were drawn based on the t-test to illustrate whether the features from the OFS can significantly distinguish glycosylation sites from non-glycosylation sites (i.e., whether the distribution of the individual feature values among glycosylation sites and non-glycosylation sites was statistically different). It is important to note that these two measures are substantially different and focus on different aspects of the prediction. The t-test focuses exclusively on an individual feature’s capability of discriminating glycosylation sites from non-glycosylation sites; while the Random Forest measures the prediction accuracy decrease after removing the current features and combining the rest as the feature set for retraining the classifiers. Random Forest is a sophisticated algorithm that is capable of calculating information entropy and/or Gini index for accurately classifying the samples. Therefore, the prediction performance of Random Forest does not simply rely on the discriminatory power of an individual feature, but more so on the combination and correlation of all the available features. By way of example, although the hydrophobicity_P10 feature (V1) was capable of distinguishing glycosylation sites from non-glycosylation sites (p-value = 2.35E-43; Supplementary Fig. S2) for N-linked glycosylation, Random Forest could still achieve a better prediction performance (i.e. lower accuracy decrease; Fig. 3a) in the absence of the hydrophobicity_P10 feature (V1) by combining all other available features. Conversely, the lack of the log-ratio feature (V10; p-value = 0.039; Supplementary Fig. S2) for N-linked glycosylation resulted in a worse correlation during the model training using Random Forest and led to the largest average accuracy decrease of 0.0378 (Fig. 3a). In summary, we suggest that these two ranking schemes are both important and that they each focus on and capture different aspects of the prediction.
Performance comparison with other tools
We evaluated and compared site-prediction performance using the OFS, only sequence features, or only structural features based on five-fold cross-validation and independent tests using the benchmark datasets. The AUC values of the models trained with different features for N- and O-glycosylation-site prediction are shown in Fig. 4a. GlycoMinestruct achieved the highest AUC values by combining both structural and sequence features, which suggested that both features played important roles in predicting N- and O-linked glycosylation sites.
Figure 4. ROC curves.

(a) Different GlycoMinestruct models trained with OFSs selected from all features, sequence features only, and structural features only, for N- and O-linked glycosylation sites. (b) N- and O-linked glycosylation-site predictions from GlycoMinestruct (trained with the OFS) and NGlycPred using the independent test dataset.
The Receiver Operating Characteristic (ROC) curves and the corresponding AUC values showed that the models trained using the combination of sequence and structural features improved prediction of both N- and O-linked glycosylation sites as compared with models trained using only structural or sequence features. To further illustrate the predictive performance of GlycoMinestruct, we performed an independent test using the OFS and compared the results with those from NGlycPred35, for N-glycosylation-site prediction. The ROC curves and AUC values of the two methods are shown in Fig. 4b. GlycoMinestruct outperformed NGlycPred for N-linked glycosylation-site prediction. The detailed prediction results in terms of AUC, Matthews correlation coefficient (MCC), ACC, specificity, sensitivity, and precision on both the benchmark and independent datasets are presented in Supplementary Table S1. The performance using the independent test dataset suggested that GlycoMinestruct outperformed NGlycPred in predicting N-linked glycosylation sites with known structural information.
Case study
A case study involving the prediction of N-linked glycosylation sites in two proteins not included in the benchmark dataset illustrated the predictive capability of GlycoMinestruct. The first protein was Toll-like receptor 8 (TLR8; PDB ID: 3WN447; UniProt ID: Q9NR97; Fig. 5a), a key component of innate and adaptive immunity that controls host immune response against pathogens through recognition of molecular patterns specific to microorganisms47. The second protein was α-L-iduronidase (IDUA; PDB ID: 4MJ248; UniProt ID: P35475; Fig. 5b), which contains a complicated structural fold consisting of a triosephosphate isomerase barrel domain harbouring the catalytic site, a β-sandwich domain, and a fibronectin-like domain, and plays an important role in hydrolyzing unsulfated alpha-L-iduronosidic linkages in dermatan sulfate49. Maita et al.50 noted that the crystal structure of α-L-iduronidase indicated that the protein was glycosylated on several sites, but that it contained one consensus asparagine residue in the Asn-X-Ser/Thr motif (Asn-336) that was not glycosylated. The prediction results of GlycoMinestruct and NGlycPred (Fig. 5) demonstrated that GlycoMinestruct correctly identified all experimentally verified glycosylation sites in the two proteins, while NGlycPred failed to predict glycosylation sites Asn-29351 of TLR8 (3WN4, chain A) and Asn-11052 of IDUA (4MJ2, chain A). The N-glycan attached to one predicted IDUA functional site (Asn-372) is crucial to protein function, as it enables the interaction with iduronate analogs in the active site and is required for enzymatic activity48. The consensus Asn-336, which is not glycosylated50, was predicted as such by GlycoMinestruct. A final consensus Asn-X-Ser/Thr residue (Asn-190) that was below the prediction threshold set by GlycoMinestruct was shown to be subject to only partial glycosylation50.
Figure 5. Predicted N-linked glycosylation sites from two case-study proteins using GlycoMinestruct.

(a) Toll-like receptor 8. (b) α-L-iduronidase. Predicted N-glycosylation sites from both GlycoMinestruct and NGlycoPred are colored in yellow, while the sites that were correctly predicted by GlycoMinestruct, but were not predicted by NGlycPred are coloured in red. The illustrations of Pfam domains and N-glycosylation sites of these two proteins shown at the bottom of each panel were rendered using the IBS program98.
Proteome-wide prediction of N- and O-linked glycosylation substrates and sites
In order to test the capability of GlycoMinestruct on systems-level mapping, we performed proteome-wide glycosylation-site prediction. In order to identify novel N- and O-glycosylation substrates and sites, we downloaded and screened the human structural proteome comprising a total of 20,538 human protein structures with resolution better than 3 Å from the PDB database53. To obtain high-confidence prediction results, the N- and O-glycosylation models trained using the corresponding optimal features on the complete training dataset were used, with prediction thresholds adjusted to a 99% specificity level. A summary of the predicted N-linked and O-linked glycosylated substrates and glycosylation sites are shown in Supplementary Table S2. A total of 3386 and 5298 proteins were predicted to be N- and O-glycosylated substrates, respectively, containing 4996 predicted N- and 10529 O-linked glycosylation sites, respectively. As a resource for the community, these proteome-wide results can be downloaded from the GlycoMinestruct website, enabling users to obtain the proteome-wide N- and O-glycosylation-site prediction results for their experimental verification.
Functional enrichment analysis of predicted N- and O-linked glycosylated proteins at the proteome level
To better understand the functional enrichment and systems impact of N- and O-linked glycosylation at the structural proteome level, we used the DAVID software54,55 to perform in-depth bioinformatics analysis of the significantly enriched gene ontology (GO), Kyoto Encyclopedia of Gene and Genomes (KEGG) pathways, and functional annotations in terms of cellular component (GO_CC), biological process (GO_BP), molecular function (GO_MF), and key functional pathways (KEGG_PATHWAY), for N- and O-linked glycosylated proteins, respectively. The overlap between the two lists of N- and O-linked glycosylated proteins indicates that some proteins were predicted to contain both N- and O-linked glycosylation sites. The top 10 significantly enriched GO_CC, GO_BP, GO_MF and KEGG_PATHWAY terms are displayed in Fig. 6a,b. We found that a suitably number of proteins were located within the “extracellular region” and “cytosol” (in terms of GO_CC). During their biogenesis extracellular proteins are first translocated into the endoplasmic reticulum where they may be subject to N-linked glycosylation10, and trafficked via the Golgi compartments where they may be subject to O-linked glycosylation56. The cytosol is another common “cellular component” for glycosylated proteins, being the sub-cellular compartment in which many O-GlcNAc transferases57 and glycosylated enzymes1 reside. We note that for most proteins there exist more than one subcellular location and/or cell component annotations. For example, the cellular component and subcellular location of a protein can be annotated as in the cytoplasm, membrane and nucleus. While this may sometimes reflect experimental difficulties in defining sub-cellular compartments, in at least some cases protein localization changes in response to cellular signals, either regulatory or in disease scenarios, such as is the case for mucin glycoproteins in human cancers, and other factors regulating cell death58. When performing statistical analysis of the GO term enrichment, such multi-location (i.e. “multi-component”) proteins will also be taken into account. It is of particular interest that both N- and O-linked glycosylated proteins were commonly enriched in several KEGG pathways involving complement and coagulation cascades, as well as bladder cancer. There also exist other cancer types that were specifically enriched for N-linked (e.g., pancreatic and prostate) and O-linked (melanoma and renal cell carcinoma) glycosylated proteins. Accordingly, several previous studies also outlined the roles of protein glycosylation and its implications in cancer pathways6, especially prostate cancer59, bladder cancer60 and pancreatic cancer61.
Figure 6. Functional enrichment analysis and classification of N-linked and O-linked glycoproteomes in terms of protein subcellular location, KEGG pathway, molecular function and biological process based on GO annotations.

(a) Subcellular locations and GO terms enriched in N-linked glycosylated proteins. (b) Subcellular locations and GO terms enriched in O-linked glycosylated proteins. (c,d) Distributions of N-linked and O-linked glycosylated proteins categorized based on the numbers of predicted glycosylation sites.
In terms of the biological processes, we found that regulation of cell death (p = 2.20 × 10−16 and p = 1.00 × 10−23 for N- and O-linked glycosylated proteins, respectively) and immune response (p = 3.50 × 10−14 and p = 5.30 × 10−20 for N- and O-linked glycosylated proteins, respectively) were two commonly enriched biological processes shared by N- and O-linked glycosylated proteins. This observation is consistent with a number of immunological studies suggesting that glycosylation plays an essential role in activating and maintaining the immune response62. Additionally, glycosylation was characterized as an important regulator for cell growth and death63, which has been confirmed by our GO-term enrichment analysis. Moreover, we found that phosphorylation (p = 5.50 × 10−18 for N-glycosylated proteins) and related processes were significantly enriched. This highlights the potential for large-scale cross-regulation between glycosylation and phosphorylation in related pathways as previously reported64.
Regarding molecular function, terms related to binding activity were commonly shared by N- and O-linked glycosylated proteins. For example, “purine nucleoside binding” (p = 1.10 × 10−17) and “cofactor binding” (p = 1.60 × 10−17) were the most significant GO_MF terms for N- and O-linked glycosylated proteins, respectively. Binding activities, such as nucleoside binding65, protein binding/inhibiting1,66, and adenosine triphosphate binding67,68 were experimentally validated as being closely associated with glycosylation. Additionally, glycosylation was implicated in the catalytic activities of dipeptidyl-peptidase IV69 and tripeptidyl-peptidase I70, both annotated as “peptidase activity” associated with O-linked glycosylated proteins in Fig. 6b. Analysing the distribution of glycosylated proteins classified based on the number of predicted N- and O-linked glycosylation sites (Fig. 6c,d) revealed that the majority of the glycoproteins contained only one predicted glycosylation site, while a limited number of proteins contained more than six glycosylation sites.
Discussion
Glycosylation is a crucial and ubiquitous type of protein PTM by which carbohydrates are covalently attached to functional groups of a target protein. A better understanding of the most important determinants of protein glycosylation at both the sequence and structure levels required for highly accurate mapping of the human glycoproteome. In this study, we developed a novel bioinformatics tool termed GlycoMinestruct for improved prediction of N- and O-linked glycosylation. It utilizes a variety of complementary sequence-derived and structural features to enable accurate predictions of glycosylation. Using an efficient two-step feature-selection strategy, 14 and 11 optimal features at both the sequence and structural levels were systematically characterized as crucial features for N- and O-linked glycosylation prediction, respectively. The performance of GlycoMinestruct was extensively evaluated using both benchmark and independent test datasets. Five-fold cross-validation and independent testing showed that GlycoMinestruct outperformed NGlycPred, the only available N-linked glycosylation predictor incorporating structural information. Additionally, GO-term analysis revealed commonly and differentially enriched subcellular locations, biological processes, molecular functions, and functional pathways shared between the N- and O-linked glycoproteome. Furthermore, we applied GlycoMinestruct to accurately predict N- and O-linked glycosylation substrates and sites in the human structural proteome. Overall, this study provided a foundation for accurate prediction of the two important types of glycosylation sites in the human proteome. More generally, the techniques and framework of GlycoMinestruct should be also applicable to other types of PTM sites in proteins with available structural information.
A remaining limitation of the current GlycoMinestruct algorithm is that it cannot consider the stoichiometry of modification, i.e. the extent to which any given Asn, Ser or Thr residue will be modified with a glycan. There have been several studies for investigating the stoichiometries of protein phosphorylation71,72,73 and glycosylation24,74,75. However, to the best of our knowledge, there are no systematic datasets for quantitatively modified sites of both N- and O-linked glycosylation in association with the protein stoichiometry, which makes it very challenging to consider such knowledge into the glycosylation predictors at this stage. One note of hope in this regard comes from the case study of α-L-iduronidase where the consensus Asn-190 residue, which is known to be subject to only partial glycosylation50, was predicted as glycosylated by GlycoMinestruct but with a score below the prediction. Perhaps with sufficient data on sites subject to partial occupancy by glycan a dual threshold might be set on predictions to recover “high-stoichiometry” and “partial” extents of glycosylation in the predictor.
Another limitation is that the current algorithm does not consider the biosynthesis pathways as a feature during the model training process, due to the limited availably of annotated entries and the difficulty of extracting such annotations from other third-party databases. We anticipate that with the increasing availability of such biosynthesis pathway data particularly for O-linked glycosylation, further improvement of the performance of our algorithm will become possible.
As an implementation of GlycoMinestruct, an online web server was developed to facilitate high-throughput prediction of N- and O-linked glycosylation sites in human proteins having available structural information. The server is configured using Tomcat 7 (Apache Software Foundation, Forest Hill, MD, USA) and JavaServer Pages (Sun Microsystems, Santa Clara, CA, USA) and is operated under the Linux environment with a 4-TB hard disk and 8 GB memory. The glycosylation site-prediction models used by the server were trained with OFSs on the complete training data used in this study. The server requires users to upload a protein structure file (a. pdb file is preferred), specify the chain name and glycosylation type and provide email addresses. Each submitted job normally takes 4 minutes to complete, and the server will send an email to users once the task is finished (see Supplementary Fig. S4 for the user interface and example prediction output). We hope that this novel approach along with the predicted N- and O-linked glycosylation sites from the human structural proteome address the concerns of the research community29 and provide a solid foundation for development of more accurate glycosylation-site predictors and prioritization of glycosylated candidates for follow-up functional validation.
Methods
Dataset construction
The annotations of C-, N-, and O-linked glycosylation sites were extracted from four major public resources, including UniProt76, PhosphoSitePlus77, SysPTM78, and O-GlycBase (version 6.0). Only experimentally verified glycosylation sites in the human proteins were retained30. To ensure the quality of the curated datasets, any glycosylation sites annotated as “Probable”, “Potential” or “By similarity” were discarded when extracting sequences from UniProt30. All remaining sequences were mapped to the PDB database53 using PSI-BLAST79. PDB entries were selected using the following criteria: (1) X-ray structures only, while nuclear magnetic resonance and electron microscopy structures were excluded; (2) X-ray resolution better than 2.5 Å; (3) structures with missing atoms were removed; and (4) the structure with the highest resolution was selected for protein sequences with more than one mapped PDB structure. The CD-HIT program80 was applied to cluster homologous sequences and reduce sequence redundancy at sequence-identity threshold of 70%35. We obtained 208 N-linked and 29 O-linked glycosylated PDB structures, which corresponded to 570 N-linked and 47 O-linked glycosylation sites, respectively. Initially, we also sought C-linked glycosylation sites within our frame of reference; however, as there was only one PDB structure containing C-linked glycosylation in the datasets, we removed this protein from our analysis and focused on N-linked and O-linked glycosylation prediction.
Regarding the selection of negative data, we extracted information on the relevant amino acid residues (i.e. Asn, Ser and Thr) that were not annotated as glycosylation sites, but that were present in those proteins that contain experimentally verified glycosylation sites. This effort to enhance the reliability of selection of negative sites is based on three criteria: (i) these proteins are biosynthetically relevant to the glycosylation machinery, in that they must be co-located with the relevant machinery i.e. in order to have one or more O-linked glycosylation sites the protein must share sub-cellular location together with an O-glycosidase, (ii) the expression of these glycoproteins must be temporally and developmentally coordinated with the expression of an appropriate glycosidase, and (iii) the experimental data validating glycosylation on at least one site of the given protein is the closest thing available to an experimental validation of non-glycosylation on the other potential sites. However, we also noticed that it was challenging to definitely determine whether the non-glycosylation sites would be glycosylated after being secreted. To the best of our knowledge, there is currently lack of sizable experimental datasets with such annotations.
Another important issue was highly imbalanced datasets, i.e., non-glycosylation sites greatly outnumbered glycosylation sites. If this imbalanced set had been used for model training, the trained models would be highly biased and classify each site in a protein as a non-glycosylation site. To address this imbalance, we used an under-sampling strategy, that enable all experimentally verified N- an O-linked glycosylation sites to be used as positive samples, while the same portion of amino acid residues (i.e., N, S and T) that had not been experimentally verified as glycosylation sites were randomly selected as negative samples from the positive PDB chains (this resulted in positive-to-negative ratio of 1:1). The datasets were further divided into two subsets consisting of benchmark and independent test datasets, which were ~20% of the size of the complete dataset. The benchmark dataset was used for performing five-fold cross-validation and feature selection, while the independent test dataset was used for validation of model performance.
Feature extraction
A variety of sequence and structural features were calculated and extracted in this study. A full list of features can be found in the Supplementary Table S3.
Sequence features
AAindex: hydrophobicity, flexibility, polarity, and β-turn values were extracted from the AAindex database81.
Physicochemical properties: physicochemical properties of proteins were calculated using BioJava82; these properties included pK1 (-COOH), pK2 (-NH3 + ), pKR (R group), pI, hydropathy index, percentage occurrence in proteins, percentage of buried residues, average volume, accessible surface area, van der Waals volume, ranking of amino acid polarities, side-chain polarity, conformational preferences of amino acids (α-helix), and conformational preferences of amino acids (β-strand).
Position-specific scoring matrices (PSSMs): these were calculated by PSI-BLAST79 searches against UniRef90, with three iterations and e-value of 0.00183.
Residue-conservation score: conservation score was derived from the PSSM generated by PSI-BLAST79 and is defined as follows:
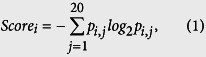 |
where pi,j is the frequency of amino acid j at position i30.
Structural features
Surface accessibility: surface-accessible area of each protein was calculated by NACCESS84 using a probe of radius = 3 Å35. Five classes of surface-accessible area were contained in the output of NACCESS84 and used as structural features in this study, including all-atoms, non-polar side chains, polar side chains, total side chains and main chains.
Secondary structure: secondary structure features were calculated by DSSP85. These included ACC, phi, and psi, and were selected from the output of DSSP as the input features. ACC denotes the solvent accessibility of amino acid residue in terms of the number of water molecules in contact with the corresponding residue, while phi and psi represent two types of International Union of Pure and Applied Chemistry backbone-torsion angles.
Log-odds ratio: log-odds ratio86 is a statistical feature calculated by DiscoTope87.
Depth index: the PSAIA program88 was used to calculate a series of features for depth index, including the average depth index (denoted as ave_dpx), standard deviation of the depth index (sd_dpx), side-chain average depth index (s-ch_ave_dpx), and standard deviation of the side-chain depth index (sd_s-ch_dpx).
B-factor: for each residue, we extracted the B-factor scores of all atoms from protein structure files and calculated their average value89.
Feature window
The location of glycosylation sites may be influenced by surrounding residues at both the sequence and structure level. Therefore, we used sequence and structure windows to encode such features and capture potentially useful information.
Sequence window: to extract the sequence context information surrounding the glycosylation sites, we employed a local sliding window with 2N + 1 = 15 (N = 7, where N denotes the half-window size) residues to represent glycosylation sites. This was used in our previous work and proved to be effective30. In terms of feature nomenclature, each residue was named as PX, where X presents the X-th position of the feature in the local sliding window. The centered glycosylated residue was then denoted as P8. Accordingly, PSSM features, which have a total dimensionality of 15 × 20 = 300, were denoted as P1, P2, …, P300, respectively. Consequently, a total of 385 sequence-based features for each glycosylation and non-glycosylation site were obtained.
Structure window: we adopted a structure window to extract features of spatially neighbouring residues of a potential glycosylation site from protein structures89 using a sphere radius R (R = 10 Å). All spatially proximal residues were included in the structure window if the distance between any atoms of such residues and any atoms of the target residue of interest were less than a threshold, R. After extracting all 14 structural features from each of the spatial residues in the structure window, we then calculated the average values of all structural features for all residues involved within the structure window for each glycosylation/non-glycosylation residue. As a result, a total of 14 structural features for each glycosylation site were obtained.
Feature selection
The proposed feature-encoding schemes led to a high-dimensionality feature vectors, requiring considerable computational time and memory to process. Meanwhile, the initial feature set may contain noisy, redundant and irrelevant features, which will have a potentially negative impact on model performance. In light of this, it was necessary to apply feature-selection methods to reduce the dimensionality of feature vectors by removing redundant and non-contributing features. We used a two-step feature-selection procedure to rank and select the most informative features.
Linear SVM-based feature selection
The first step of feature selection was performed using the linear SVM feature-selection method, which is competitive with traditional feature-selection methods, such as odds ratio and information gain36. For linear-kernel SVMs, the class predictor can be denoted as
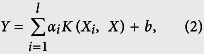 |
For a linear kernel,
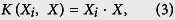 |
and the class predictor can be rewritten as
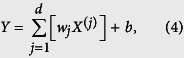 |
where
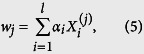 |
where the absolute value |wj| is used as the weight of a feature j. The larger the absolute value of a feature coefficient wj is, the more useful the feature is for the classification36. We used LibSVM90 to calculate |wj|. The top ranked 300 features were then used as the optimal feature candidates (OFCs).
Incremental Feature Selection (IFS)
To determine the final optimal features from the OFCs at the second step of feature selection, an IFS strategy based on a random forest (RF)91 classifier was applied to the benchmark dataset by performing five-fold cross-validation to assess the relative importance and contribution of all OFCs. The IFS procedure can be briefly described as follows. First, it constructs n (n = |OFCs|) feature subsets by adding one feature at a time from OFCs to the candidate feature subset F. Then, the performance of the RF classifier that was trained based on the updated F in each round was evaluated using five-fold cross-validation to avoid over-fitting each time. This process was repeated for 20 rounds, and the average performance was calculated. The i-th feature subset is defined as F = {f | f1, f2, …, fi}30, where fi is the i-th feature from the OFCs. As a result, the feature set with the highest area-under-the-curve (AUC) value amongst the 300 AUC values was selected as the optimal feature set (OFS).
Model training and performance evaluation
We employed the RF algorithm implemented in the R package92 to build glycosylation-site prediction models. RF is an ensemble machine-learning approach based on decision trees and has been successfully applied in many different tasks in protein bioinformatics, such as prediction of RNA-binding sites93, phosphorylation sites94, protease-cleavage sites95 and functional effects of single amino acid variants96. To evaluate the performance of RF classifiers, six performance measures were used, including sensitivity, specificity, precision, accuracy (ACC), the Matthews correlation coefficient (MCC), and AUC. Additionally, the receiver operating characteristic (ROC) curves were also generated, which plotted true-positive rate (TPR) against the false-positive rate (FPR). The ROC curves were drawn and the corresponding AUC values were calculated using the ROCR package97. Refer to the Supplemental Methods for a detailed description of these measures.
Additional Information
How to cite this article: Li, F. et al. GlycoMinestruct: a new bioinformatics tool for highly accurate mapping of the human N-linked and O-linked glycoproteomes by incorporating structural features. Sci. Rep. 6, 34595; doi: 10.1038/srep34595 (2016).
Supplementary Material
Acknowledgments
This work was supported by grants from the National Health and Medical Research Council of Australia (NHMRC) (1092262), National Natural Science Foundation of China (61202167, 61303169) and the Hundred Talents Program of the Chinese Academy of Sciences (CAS). GIW is a recipient of Discovery Outstanding Research Award (DORA) of the Australian Research Council (ARC). JS is a recipient of the Hundred Talents Program of CAS. TL is an ARC Australian Laureate Fellow.
Footnotes
The authors declare no competing financial interests.
Author Contributions S.J., L.T. and Z.Y. conceived, designed and performed the project; L.F. and L.C. performed data collection, feature selection, modelling analyses and implemented the web server; R.J., W.G.I. and L.J. contributed to the discussion of data analysis; All authors wrote and revised the paper.
References
- Spiro R. G. Protein glycosylation: nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology 12, 43R–56R (2002). [DOI] [PubMed] [Google Scholar]
- Moharir A., Peck S. H., Budden T. & Lee S. Y. The role of N-glycosylation in folding, trafficking, and functionality of lysosomal protein CLN5. PLoS One 8, e74299, doi: 10.1371/journal.pone.0074299 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino K., Bones J., Kattla J. J. & Rudd P. M. A systematic approach to protein glycosylation analysis: a path through the maze. Nat Chem Biol 6, 713–723, doi: 10.1038/nchembio.437 (2010). [DOI] [PubMed] [Google Scholar]
- Moremen K. W., Tiemeyer M. & Nairn A. V. Vertebrate protein glycosylation: diversity, synthesis and function. Nature reviews. Molecular cell biology 13, 448–462, doi: 10.1038/nrm3383 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiermaier E. et al. Polysialylation controls dendritic cell trafficking by regulating chemokine recognition. Science 351, 186–190, doi: 10.1126/science.aad0512 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho S. S. & Reis C. A. Glycosylation in cancer: mechanisms and clinical implications. Nature reviews. Cancer 15, 540–555, doi: 10.1038/nrc3982 (2015). [DOI] [PubMed] [Google Scholar]
- Park D. S., Poretz R. D., Stein S., Nora R. & Manowitz P. Association of alcoholism with the N-glycosylation polymorphism of pseudodeficient human arylsulfatase A. Alcoholism, clinical and experimental research 20, 228–233 (1996). [DOI] [PubMed] [Google Scholar]
- Schedin-Weiss S., Winblad B. & Tjernberg L. O. The role of protein glycosylation in Alzheimer disease. The FEBS journal 281, 46–62, doi: 10.1111/febs.12590 (2014). [DOI] [PubMed] [Google Scholar]
- Gavel Y. & von Heijne G. Sequence differences between glycosylated and non-glycosylated Asn-X-Thr/Ser acceptor sites: implications for protein engineering. Protein engineering 3, 433–442 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aebi M. N-linked protein glycosylation in the ER. Biochimica et biophysica acta 1833, 2430–2437, doi: 10.1016/j.bbamcr.2013.04.001 (2013). [DOI] [PubMed] [Google Scholar]
- Van den Steen P., Rudd P. M., Dwek R. A. & Opdenakker G. Concepts and principles of O-linked glycosylation. Critical reviews in biochemistry and molecular biology 33, 151–208, doi: 10.1080/10409239891204198 (1998). [DOI] [PubMed] [Google Scholar]
- Li B. & Kohler J. J. Glycosylation of the nuclear pore. Traffic 15, 347–361, doi: 10.1111/tra.12150 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halim A. et al. Discovery of a nucleocytoplasmic O-mannose glycoproteome in yeast. Proceedings of the National Academy of Sciences of the United States of America 112, 15648–15653, doi: 10.1073/pnas.1511743112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado-Guerrero R. Recent structural and mechanistic insights into protein O-GalNAc glycosylation. Biochemical Society transactions 44, 61–67, doi: 10.1042/BST20150178 (2016). [DOI] [PubMed] [Google Scholar]
- Bard F. & Chia J. Cracking the Glycome Encoder: Signaling, Trafficking, and Glycosylation. Trends in cell biology 26, 379–388, doi: 10.1016/j.tcb.2015.12.004 (2016). [DOI] [PubMed] [Google Scholar]
- Thanka Christlet T. H. & Veluraja K. Database analysis of O-glycosylation sites in proteins. Biophysical journal 80, 952–960 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweiler R., Hermjakob H. & Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochimica et biophysica acta 1473, 4–8 (1999). [DOI] [PubMed] [Google Scholar]
- Nilsson I. M. & von Heijne G. Determination of the distance between the oligosaccharyltransferase active site and the endoplasmic reticulum membrane. The Journal of biological chemistry 268, 5798–5801 (1993). [PubMed] [Google Scholar]
- Petrescu A. J., Milac A. L., Petrescu S. M., Dwek R. A. & Wormald M. R. Statistical analysis of the protein environment of N-glycosylation sites: implications for occupancy, structure, and folding. Glycobiology 14, 103–114, doi: 10.1093/glycob/cwh008 (2004). [DOI] [PubMed] [Google Scholar]
- Morelle W. & Michalski J. C. Analysis of protein glycosylation by mass spectrometry. Nature protocols 2, 1585–1602, doi: 10.1038/nprot.2007.227 (2007). [DOI] [PubMed] [Google Scholar]
- Zhang S. & Williamson B. L. Characterization of protein glycosylation using chip-based nanoelectrospray with precursor ion scanning quadrupole linear ion trap mass spectrometry. Journal of biomolecular techniques: JBT 16, 209–219 (2005). [PMC free article] [PubMed] [Google Scholar]
- Wollscheid B. et al. Mass-spectrometric identification and relative quantification of N-linked cell surface glycoproteins. Nature biotechnology 27, 378–386, doi: 10.1038/nbt.1532 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shubhakar A. et al. High-Throughput Analysis and Automation for Glycomics Studies. Chromatographia 78, 321–333, doi: 10.1007/s10337-014-2803-9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S. & Zhang H. Large-Scale Measurement of Absolute Protein Glycosylation Stoichiometry. Analytical chemistry 87, 6479–6482, doi: 10.1021/acs.analchem.5b01679 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayakumar D., Marathe D. D. & Neelamegham S. Detection of site-specific glycosylation in proteins using flow cytometry. Cytometry. Part A: the journal of the International Society for Analytical Cytology 75, 866–873, doi: 10.1002/cyto.a.20773 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y., Zhou Y., Elliott S., Aebersold R. & Zhang H. Solid-phase extraction of N-linked glycopeptides. Nature protocols 2, 334–339, doi: 10.1038/nprot.2007.42 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. et al. Detection and verification of glycosylation patterns of glycoproteins from clinical specimens using lectin microarrays and lectin-based immunosorbent assays. Analytical chemistry 83, 8509–8516, doi: 10.1021/ac201452f (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuno A. et al. Evanescent-field fluorescence-assisted lectin microarray: a new strategy for glycan profiling. Nature methods 2, 851–856, doi: 10.1038/nmeth803 (2005). [DOI] [PubMed] [Google Scholar]
- Walt D. et al. The National Academies Collection: Reports funded by National Institutes of Health (National Academies Press, 2012). [Google Scholar]
- Li F. et al. GlycoMine: a machine learning-based approach for predicting N-, C- and O-linked glycosylation in the human proteome. Bioinformatics 31, 1411–1419, doi: 10.1093/bioinformatics/btu852 (2015). [DOI] [PubMed] [Google Scholar]
- Gupta R. & Brunak S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pacific Symposium on Biocomputing. Pacific Symposium on Biocomputing 310–322 (2002). [PubMed] [Google Scholar]
- Steentoft C. et al. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. The EMBO journal 32, 1478–1488, doi: 10.1038/emboj.2013.79 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caragea C., Sinapov J., Silvescu A., Dobbs D. & Honavar V. Glycosylation site prediction using ensembles of Support Vector Machine classifiers. Bmc Bioinformatics 8, 438, doi: 10.1186/1471-2105-8-438 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamby S. E. & Hirst J. D. Prediction of glycosylation sites using random forests. Bmc Bioinformatics 9, 500, doi: 10.1186/1471-2105-9-500 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang G. Y. et al. Computational prediction of N-linked glycosylation incorporating structural properties and patterns. Bioinformatics 28, 2249–2255, doi: 10.1093/bioinformatics/bts426 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brank J. & Grobelnik M. Feature selection using linear support vector machines (2002).
- Liu H. A. & Setiono R. Incremental feature selection. Appl Intell 9, 217–230, doi: 10.1023/A:1008363719778 (1998). [DOI] [Google Scholar]
- O’Shea J. P. et al. pLogo: a probabilistic approach to visualizing sequence motifs. Nature methods 10, 1211–1212, doi: 10.1038/nmeth.2646 (2013). [DOI] [PubMed] [Google Scholar]
- Biswas A. K., Noman N. & Sikder A. R. Machine learning approach to predict protein phosphorylation sites by incorporating evolutionary information. BMC bioinformatics 11, 273, doi: 10.1186/1471-2105-11-273 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Zhou Y., Zhang Z. & Song J. Towards more accurate prediction of ubiquitination sites: a comprehensive review of current methods, tools and features. Briefings in bioinformatics 16, 640–657, doi: 10.1093/bib/bbu031 (2015). [DOI] [PubMed] [Google Scholar]
- Lu D., Yang C. & Liu Z. How hydrophobicity and the glycosylation site of glycans affect protein folding and stability: a molecular dynamics simulation. The journal of physical chemistry. B 116, 390–400, doi: 10.1021/jp203926r (2012). [DOI] [PubMed] [Google Scholar]
- Mazumder R., Morampudi K. S., Motwani M., Vasudevan S. & Goldman R. Proteome-wide analysis of single-nucleotide variations in the N-glycosylation sequon of human genes. PloS One 7, e36212, doi: 10.1371/journal.pone.0036212 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avanov A. [Conformational aspects of glycosylation]. Molekuliarnaia biologiia 25, 293–308 (1991). [PubMed] [Google Scholar]
- Lam P. V. et al. Structure-based comparative analysis and prediction of N-linked glycosylation sites in evolutionarily distant eukaryotes. Genomics, proteomics & bioinformatics 11, 96–104, doi: 10.1016/j.gpb.2012.11.003 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfert M. A. & Boons G. J. Adaptive immune activation: glycosylation does matter. Nat Chem Biol 9, 776–784, doi: 10.1038/nchembio.1403 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman A. et al. Glycosylation at Asn91 of H1N1 haemagglutinin affects binding to glycan receptors. The Biochemical journal 444, 429–435, doi: 10.1042/BJ20112101 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokatla H. P. et al. Structure-based design of novel human Toll-like receptor 8 agonists. ChemMedChem 9, 719–723, doi: 10.1002/cmdc.201300573 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bie H. Y. et al. Insights into mucopolysaccharidosis I from the structure and action of alpha-L-iduronidase. Nat Chem Biol 9, 739-+, doi: 10.1038/Nchembio.1357 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bie H. et al. Insights into mucopolysaccharidosis I from the structure and action of α-L-iduronidase. Nature chemical biology 9, 739–745 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maita N. et al. Human alpha-L-iduronidase uses its own N-glycan as a substrate-binding and catalytic module. Proceedings of the National Academy of Sciences of the United States of America 110, 14628–14633, doi: 10.1073/pnas.1306939110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanji H., Ohto U., Shibata T., Miyake K. & Shimizu T. Structural reorganization of the Toll-like receptor 8 dimer induced by agonistic ligands. Science 339, 1426–1429, doi: 10.1126/science.1229159 (2013). [DOI] [PubMed] [Google Scholar]
- Chen R. et al. Glycoproteomics analysis of human liver tissue by combination of multiple enzyme digestion and hydrazide chemistry. Journal of proteome research 8, 651–661, doi: 10.1021/pr8008012 (2009). [DOI] [PubMed] [Google Scholar]
- Berman H. M. et al. The Protein Data Bank. Nucleic acids research 28, 235–242 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W., Sherman B. T. & Lempicki R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols 4, 44–57, doi: 10.1038/nprot.2008.211 (2009). [DOI] [PubMed] [Google Scholar]
- Huang da W., Sherman B. T. & Lempicki R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research 37, 1–13, doi: 10.1093/nar/gkn923 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill D. J., Chia J., Senewiratne J. & Bard F. Regulation of O-glycosylation through Golgi-to-ER relocation of initiation enzymes. The Journal of cell biology 189, 843–858, doi: 10.1083/jcb.201003055 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comer F. I. & Hart G. W. O-Glycosylation of nuclear and cytosolic proteins. Dynamic interplay between O-GlcNAc and O-phosphate. The Journal of biological chemistry 275, 29179–29182, doi: 10.1074/jbc.R000010200 (2000). [DOI] [PubMed] [Google Scholar]
- Traven A., Huang D. C. & Lithgow T. Protein hijacking: key proteins held captive against their will. Cancer cell 5, 107–108 (2004). [DOI] [PubMed] [Google Scholar]
- Drake R. R., Jones E. E., Powers T. W. & Nyalwidhe J. O. Altered glycosylation in prostate cancer. Advances in cancer research 126, 345–382, doi: 10.1016/bs.acr.2014.12.001 (2015). [DOI] [PubMed] [Google Scholar]
- Costa C. et al. Abnormal Protein Glycosylation and Activated PI3K/Akt/mTOR Pathway: Role in Bladder Cancer Prognosis and Targeted Therapeutics. PloS One 10, e0141253, doi: 10.1371/journal.pone.0141253 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassaganas S. et al. Pancreatic cancer cell glycosylation regulates cell adhesion and invasion through the modulation of alpha2beta1 integrin and E-cadherin function. PLoS One 9, e98595, doi: 10.1371/journal.pone.0098595 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto-Hino M. et al. Dynamic regulation of innate immune responses in Drosophila by Senju-mediated glycosylation. Proceedings of the National Academy of Sciences of the United States of America 112, 5809–5814, doi: 10.1073/pnas.1424514112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenstein R. G. & Rabinovich G. A. Glycobiology of cell death: when glycans and lectins govern cell fate. Cell death and differentiation 20, 976–986, doi: 10.1038/cdd.2013.50 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart G. W., Slawson C., Ramirez-Correa G. & Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annual review of biochemistry 80, 825–858, doi: 10.1146/annurev-biochem-060608-102511 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogue D. L., Hodgson K. C. & Cass C. E. Effects of inhibition of N-linked glycosylation by tunicamycin on nucleoside transport polypeptides of L1210 leukemia cells. Biochemistry and cell biology = Biochimie et biologie cellulaire 68, 199–209 (1990). [DOI] [PubMed] [Google Scholar]
- Margraf-Schonfeld S., Bohm C. & Watzl C. Glycosylation affects ligand binding and function of the activating natural killer cell receptor 2B4 (CD244) protein. The Journal of biological chemistry 286, 24142–24149, doi: 10.1074/jbc.M111.225334 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perego P., Gatti L. & Beretta G. L. The ABC of glycosylation. Nature reviews. Cancer 10, 523, doi: 10.1038/nrc2789-c1 (2010). [DOI] [PubMed] [Google Scholar]
- Beers M. F. et al. Disruption of N-linked glycosylation promotes proteasomal degradation of the human ATP-binding cassette transporter ABCA3. American journal of physiology. Lung cellular and molecular physiology 305, L970–L980, doi: 10.1152/ajplung.00184.2013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aertgeerts K. et al. N-linked glycosylation of dipeptidyl peptidase IV (CD26): effects on enzyme activity, homodimer formation, and adenosine deaminase binding. Protein science: a publication of the Protein Society 13, 145–154, doi: 10.1110/ps.03352504 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golabek A. A. et al. Biosynthesis, glycosylation, and enzymatic processing in vivo of human tripeptidyl-peptidase I. The Journal of biological chemistry 278, 7135–7145, doi: 10.1074/jbc.M211872200 (2003). [DOI] [PubMed] [Google Scholar]
- Wu R. et al. A large-scale method to measure absolute protein phosphorylation stoichiometries. Nature methods 8, 677–683, doi: 10.1038/nmeth.1636 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson H., Eyers C. E., Eyers P. A., Beynon R. J. & Gaskell S. J. Rigorous determination of the stoichiometry of protein phosphorylation using mass spectrometry. Journal of the American Society for Mass Spectrometry 20, 2211–2220, doi: 10.1016/j.jasms.2009.08.009 (2009). [DOI] [PubMed] [Google Scholar]
- Witze E. S., Old W. M., Resing K. A. & Ahn N. G. Mapping protein post-translational modifications with mass spectrometry. Nature methods 4, 798–806, doi: 10.1038/nmeth1100 (2007). [DOI] [PubMed] [Google Scholar]
- Rexach J. E. et al. Quantification of O-glycosylation stoichiometry and dynamics using resolvable mass tags. Nature chemical biology 6, 645–651, doi: 10.1038/nchembio.412 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark P. M., Rexach J. E. & Hsieh-Wilson L. C. Visualization of O-GlcNAc glycosylation stoichiometry and dynamics using resolvable poly(ethylene glycol) mass tags. Current protocols in chemical biology 5, 281–302, doi: 10.1002/9780470559277.ch130153 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz U. & UniProt C. From protein sequences to 3D-structures and beyond: the example of the UniProt knowledgebase. Cellular and molecular life sciences: CMLS 67, 1049–1064, doi: 10.1007/s00018-009-0229-6 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck P. V. et al. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic acids research 40, D261–D270, doi: 10.1093/nar/gkr1122 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. SysPTM: a systematic resource for proteomic research on post-translational modifications. Molecular & cellular proteomics: MCP 8, 1839–1849, doi: 10.1074/mcp.M900030-MCP200 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic acids research 25, 3389–3402 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Niu B., Gao Y., Fu L. & Li W. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26, 680–682, doi: 10.1093/bioinformatics/btq003 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima S. et al. AAindex: amino acid index database, progress report 2008. Nucleic acids research 36, D202–D205, doi: 10.1093/nar/gkm998 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland R. C. et al. BioJava: an open-source framework for bioinformatics. Bioinformatics 24, 2096–2097, doi: 10.1093/bioinformatics/btn397 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzek B. E. et al. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932, doi: 10.1093/bioinformatics/btu739 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard S. J. & Thornton J. M. Naccess. Computer Program, Department of Biochemistry and Molecular Biology, University College London 2 (1993). [Google Scholar]
- Joosten R. P. et al. A series of PDB related databases for everyday needs. Nucleic acids research 39, D411–D419, doi: 10.1093/nar/gkq1105 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senn S. Review of Fleiss, statistical methods for rates and proportions. Research synthesis methods 2, 221–222, doi: 10.1002/jrsm.50 (2011). [DOI] [PubMed] [Google Scholar]
- Andersen P., Nielsen M. & Lund O. Prediction of residues in discontinuous B-cell epitopes using protein 3D structures. Protein science: a publication of the Protein Society 15, 2558–2567, doi: 10.1110/ps.062405906 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihel J., Sikic M., Tomic S., Jeren B. & Vlahovicek K. PSAIA - protein structure and interaction analyzer. BMC structural biology 8, 21, doi: 10.1186/1472-6807-8-21 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J., Liu Q., Ellis J. & Li J. Tertiary structure-based prediction of conformational B-cell epitopes through B factors. Bioinformatics 30, i264–i273, doi: 10.1093/bioinformatics/btu281 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C.-C. & Lin C.-J. LIBSVM: a library for support vector machines. ACM Transactions on Intelligent Systems and Technology (TIST) 2, 27 (2011). [Google Scholar]
- Breiman L. Random forests. Mach Learn 45, 5–32, doi: 10.1023/A:1010933404324 (2001). [DOI] [Google Scholar]
- Liaw A. & Wiener M. Classification and regression by randomForest. R news 2, 18–22 (2002). [Google Scholar]
- Liu Z. P., Wu L. Y., Wang Y., Zhang X. S. & Chen L. Prediction of protein-RNA binding sites by a random forest method with combined features. Bioinformatics 26, 1616–1622, doi: 10.1093/bioinformatics/btq253 (2010). [DOI] [PubMed] [Google Scholar]
- Fan W. et al. Prediction of protein kinase-specific phosphorylation sites in hierarchical structure using functional information and random forest. Amino acids 46, 1069–1078, doi: 10.1007/s00726-014-1669-3 (2014). [DOI] [PubMed] [Google Scholar]
- Li B. Q., Cai Y. D., Feng K. Y. & Zhao G. J. Prediction of protein cleavage site with feature selection by random forest. PloS One 7, e45854, doi: 10.1371/journal.pone.0045854 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M. et al. FunSAV: predicting the functional effect of single amino acid variants using a two-stage random forest model. PloS One 7, e43847, doi: 10.1371/journal.pone.0043847 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sing T., Sander O., Beerenwinkel N. & Lengauer T. ROCR: visualizing classifier performance in R. Bioinformatics 21, 3940–3941, doi: 10.1093/bioinformatics/bti623 (2005). [DOI] [PubMed] [Google Scholar]
- Liu W. et al. IBS: an illustrator for the presentation and visualization of biological sequences. Bioinformatics 31, 3359–3361, doi: 10.1093/bioinformatics/btv362 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.