

Abstract
Bacterial chromosomes are most often circular DNA molecules. This can produce a topological problem; a genetic crossover from homologous recombination results in dimerization of the chromosome. A chromosome dimer is lethal unless resolved. A site-specific recombination system catalyses this dimer-resolution reaction at the chromosomal site dif. In Escherichia coli, two tyrosine-family recombinases, XerC and XerD, bind to dif and carry out two pairs of sequential strand exchange reactions. However, what makes the reaction unique among site-specific recombination reactions is that the first step, XerD-mediated strand exchange, relies on interaction with the very C-terminus of the FtsK DNA translocase. FtsK is a powerful molecular motor that functions in cell division, co-ordinating division with clearing chromosomal DNA from the site of septation and also acts to position the dif sites for recombination. This is a model system for unlinking, separating and segregating large DNA molecules. Here we describe the molecular detail of the interaction between XerD and FtsK that leads to activation of recombination as deduced from a co-crystal structure, biochemical and in vivo experiments. FtsKγ interacts with the C-terminal domain of XerD, above a cleft where XerC is thought to bind. We present a model for activation of recombination based on structural data.
The majority of bacterial species have circular DNA genomes. Prior to cell division each circular chromosome must be entirely replicated, unlinked and segregated to ensure that each daughter cell inherits a full genome complement. During or following replication DNA repair processes involving homologous recombination can produce a chromosomal crossover, and any odd number of these events between the circular DNA molecules results in a chromosome dimer1. Evidence suggests that this occurs with a probability of 17–40% for each cell cycle2,3. A chromosome dimer is lethal if unresolved, but most bacteria encode for an efficient site-specific recombination system to convert the chromosome dimers back to monomers, the Xer site-specific recombination system4.
Most bacteria encode two tyrosine family recombinases XerC and XerD which bind to the specific chromosomal site dif located in the replication terminus region5,6, although there are notable exceptions where a single Xer protein carries out the reaction7,8,9. The Escherichia coli dif site consists of two 11 bp sites that are an imperfect inverted repeat, separated by a 6 bp spacer, or central region5. XerC and XerD bind to these sites, with the differences between the repeats imparting specificity to one or other recombinase; XerC binds to one repeat and XerD to the other10. Two dif sites bound by XerCD can then be synapsed by protein-protein interactions between the two dif-XerCD complexes forming a pseudo-tetrameric structure11,12 with predicted cyclic interactions between monomers as seen in the synapses of other tyrosine recombinases Cre, λ Int and Flp13,14,15,16. However, the synapse of (XerCD-dif)2 is not by itself catalytically competent; it is not until FtsK is present that cleavage by XerD occurs12,17. After interaction with FtsK, XerD carries out the first pair of strand exchanges to produce a Holliday junction intermediate, that is then resolved by a XerC-mediated pair of strand exchanges to produce a recombinant product17.
The FtsK protein is a powerful DNA translocase that is involved in co-ordinating cell division with DNA unlinking and segregation to ensure that the closing septum does not trap or guillotine chromosomal DNA18,19. FtsK was first identified as a protein involved in cell division20 and has been found to interact with several divisome components, leading to the localisation of FtsK to the site of cell division and the recruitment of downstream divisome components21,22,23. The N-terminal domain of FtsK is membrane bound and hexamerises at mid-cell prior to cell division23,24. The C-terminal domain of FtsK forms a DNA translocase motor, which also hexamerises, and was found to be essential for chromosome dimer resolution through activation of the XerD recombinase25,26,27. Further refinement of the mechanism of activation of XerD showed that it is only the very C-terminal domain of FtsK, called γ, that is absolutely required for recombination17. FtsKγ interacts directly with XerD to stimulate its catalytic activity, leading to XerD cutting and exchanging the first pair of strands within a recombinase synapse between dif sites17.
The FtsKγ domain also plays another role in orienting the FtsK motor to translocate toward dif sites; FtsKγ binds specifically to 8 bp sequences termed KOPS that are highly skewed on each chromosome arm to point towards the dif site28,29,30. Three FtsKγ domains bind specifically to each KOPS site, which leads to loading and hexamerisation of the FtsK motor domains to one side of the KOPS sequence so that subsequent translocation is always directed towards the dif site31.
Here we present the structure of the FtsKγ domain interacting with the XerD recombinase and provide biochemical evidence to support the involvement of amino acids at the interaction surfaces in the activation of recombination. A mutant in XerD that disrupts the interaction with FtsKγ is severely impaired for recombination activity on plasmid substrates and is also incapable of supporting chromosome dimer resolution in cells. Further, we demonstrate that a 9 amino acid stretch from FtsKγ that encompasses the interaction surface is sufficient for activation of recombination. It is of note that the same region of FtsKγ that binds KOPS-DNA (between helices 2 and 3) is also responsible for the protein-protein interaction with XerD to activate recombination. Finally, we also provide structure-based models to explain how the FtsKγ-XerD interaction can influence the XerCD-dif synapse to promote recombination.
Results
Structure reveals the XerD-FtsKγ interaction
The activation of recombination at dif requires the interaction of XerD with the γ domain of FtsK. To understand how this interaction can lead to activation of recombination, a fusion protein between the C-terminal domain of XerD, which contains the catalytic residues required for DNA cleavage and strand exchange, and FtsKγ was produced (XerDC–γ) and crystallized. The full length XerD-FtsKγ fusion protein has previously been shown to be a functional, self-activating, recombinase protein at dif sites17. Attempts to crystallize either XerD with the FtsKγ domain as separate proteins, or to crystallize the full-length XerD-FtsKγ fusion protein both failed to yield high quality crystals. The structure of the XerDC-FtsKγ fusion protein was solved by molecular replacement and refined to a resolution of 2.3 Å (Fig. 1), using the previously reported XerD structure (1A0P) and the NMR structure of the E. coli FtsKγ domain (2VE8) as search models30,32. The asymmetric unit contained a single XerDC–γ complex, but the flexible linker joining the two protein domains was not visible in the density. Details of the linker sequence and all amino acids for which there is insufficient electron density are shown in supplementary data (Figure S1). The linker is 16 amino acids long, which is of sufficient length that the site of interaction between the XerD and FtsKγ portions of the fusion protein should not be limited. Therefore, the linker may have aided co-crystallisation of the two protein domains but its presence should not interfere with interpretation of the interaction. In addition, biochemical investigations were conducted that confirmed the putative interaction (see below).
Figure 1. Crystal structure of XerDC-FtsKγ interaction.

(A) The structure of XerDC (orange) interacting with FtsKγ (green). Helices for FtsKγ are marked. Dotted lines show connections for which the electron density was not observed. The N- and C- termini are marked. (B) Close up of the interacting amino acids at the interface between XerDC and FtsKγ. Hydrogen bonds are shown as dashed lines. (C) Comparison of the amino acids from E. coli FtsKγ that interact with XerDC (this study) and the amino acids from P. aeruginosa FtsKγ that interact with KOPS DNA31. Filled circles above or below the amino acid represent interactions from the amino acid side-chain, and open circles are backbone contacts.
Both the XerDC domain and the FtsKγ domain are structurally very similar to the previously determined individual proteins, with no major conformational changes revealed in either protein domain; overall the XerDC domain only varied by an average RMSD of 0.93 Å on matched Cα atoms, while the RMSD between matched Cα atoms for the FtsKγ domain was 0.77 Å. The density observed for FtsKγ was weaker than that for XerDC and increasingly diminished for atoms that were distal from its interaction with XerD, resulting in the final refined model having chain breaks in this region. However, the density for the interface between the two binding partners was sufficient to facilitate an in depth examination of the key molecular contacts (see Figure S2).
The FtsKγ interaction with XerDC is a 563 Å2 interface that involves two helices on the surface of XerDC (Fig. 1B). These two helices, αE and αH, are on the opposite face from the putative DNA-binding surface of XerD and the active-site residues32. This suggests that the interaction seen in the crystal of XerD with FtsKγ can readily occur whilst XerD is bound to the dif site DNA16, and activation of XerD by FtsKγ is likely to be allosteric. The interaction site on FtsKγ involves the ends of helices α2 and α3 and the loop that joins them30,31. It is noteworthy that this is also a region involved in binding of FtsKγ to KOPS DNA, and the amino acids that contact XerD are a subset of those seen to contact DNA (Fig. 1C); not only is FtsKγ a bi-functional domain with protein-protein and protein-DNA interactions, it uses the same surface to carry out both functions. Mechanistically, this implies that FtsKγ cannot interact with KOPS DNA and XerD simultaneously. Further, it may make interpretation of the results of assays using mutations in the FtsKγ domain difficult; recombination efficiency is often used as a readout but determining whether a mutation affects DNA loading of FtsK or XerD activation or both would be problematic.
Altering residues within the XerD–FtsKγ interface reduces recombination
Although the structure of the fusion protein reveals an interaction surface that is consistent with the proposed role of FtsKγ interacting with and activating XerD whilst it is bound to DNA, there was no obvious structural change in XerD that would account for its activation; the active site residues were in the same, inactive, conformation as seen in the XerD structure without γ present32. Therefore, it was necessary to confirm whether the observed interactions were indeed responsible for activation of recombination by XerD. The FtsKγ and XerD domains are seen to interact via 6 hydrogen bonds and a salt bridge (Table 1) (Fig. 1B), and the residues responsible for these interactions were targeted for site-directed mutagenesis. Each amino acid was changed to alanine, or other amino acids in addition as noted (Fig. 2). Several other residues on the surface of the FtsKγ domain close to the site of interaction with XerD, but not seen to be directly involved in binding to XerD, were also changed by site-directed mutagenesis: R1280, G1318 and R1297. These mutations should not directly affect the XerD-FtsKγ interaction and were predicted to retain close to wild-type activity, acting as negative controls for the recombination assay. Similarly, the amino acid W188 of XerD, located close to the site of interaction with FtsKγ but not directly involved, was altered. Since the residues of FtsKγ seen to interact with XerD were also involved in contacting DNA during loading of the FtsK motor, it would be impossible to separate the DNA binding function of the residues from activation of XerD. Therefore, to avoid any possible ambiguity in the interpretation of the mutant data, each FtsKγ mutant was produced in a fusion protein with XerC so that loading and translocation of the FtsK motor would be unnecessary, and the XerD was wt. Each XerD mutant was produced in a XerD-γ fusion protein. Both fusion proteins are thought to work by increasing the local concentration of FtsKγ so that the interaction with XerD is more efficient; which recombinase protein the FtsKγ domain is attached to does not affect the reaction.
Table 1. Intermolecular contacts between XerDC and FtsKγ.
| XerDC | FtsKγ | Interaction | Distance (Å) |
|---|---|---|---|
| Glu119Oε1 | Asn1296Nδ2 | H-Bond | 2.93 |
| Glu119 | Asn1296 | VDW | |
| Gln123 | Arg1292, Ile1293, Gly1294 | VDW | |
| Ile127 | Arg1292 | VDW | |
| Glu184Oε2 | Gln1288Nε2, Arg1289Nη1, Arg1289Nη2 | H-Bonds | 3.22, 2.53, 2.98 |
| Glu184Oε2 | Arg1289Nη1, Arg1289Nη2 | Salt-Bridges | 2.53, 2.98 |
| Glu184 | Gln1288, Arg1289 | VDW | |
| Tyr187Oη | Arg1292Nη1 | H-Bond | 2.81 |
| Tyr187 | Arg1289, Arg1292, | VDW | |
| Trp188 | Arg1292 | VDW | |
| His257 | Asn1296 | VDW |
Atomic contacts determined using the CCP4i implementation of CONTACT.
Van der Waals interactions defined as non-hydrogen bond contact distances of 4 Å or less.
Hydrogen bond interactions are defined as contact distances of 3.3 Å or less.
Salt-bridge interactions are defined as contact distances of 4.5 Å or less.
Figure 2. Recombination activity of XerD and FtsKγ interaction mutants is reduced.

(A) Recombination in vivo from XerCγ or XerDγ fusion proteins with mutations as denoted in the FtsKγ domain. Mutations in control residues not seen to be involved in contacts in the crystal structure are denoted with an asterisk (*). (B) Recombination in vivo from XerDγ fusion proteins with amino acid substitutions in the XerD portion as noted. Recombination in vitro using purified fusion proteins from mutations in the ftsKγ domain (C) or in the xerD domain (D). (E) Two views of XerD represented as space filling models. On the left the amino acids that interact with FtsKγ are highlighted in green and the individual amino acids are labelled. On the right the relative effect upon recombination efficiency of each amino acid mutation described here is colour coded: red (largest effect) through to yellow (smallest effect).
The resultant XerC-γ or XerD–γ fusion proteins were tested for their recombination activity in vivo; each was independently produced in a strain lacking the C-terminus of FtsK where the fusion protein is the only source of FtsKγ. Also present in the strain was a reporter plasmid bearing 2 dif sites in direct repeat17. In these conditions the “wt” XerC-γ and XerD-γ fusion proteins have previously been shown to be sufficient to efficiently catalyse recombination on the reporter plasmid17. Recombination of the reporter plasmid by each mutant was quantified and compared to the relative level seen with the wild-type fusion protein (Fig. 2A,B). Typical gels are also shown in supplementary data (Figure S3). It is clear that several of the amino acid changes at the XerD-FtsK interaction surface reduce the recombination efficiency in vivo. The amino acids whose alteration showed the greatest effect on recombination are E183/4 from XerD and R1289 from FtsK. The E184-R1289 pair interact with each other via a salt bridge and hydrogen bonding in the crystal structure (Fig. 1B) that, based on recombination efficiency, appears to be a critical factor in the interaction. Furthermore, consistent with this interaction, mutation to change the arginine of this pair to the oppositely charged glutamate (R1289E) had an even greater negative effect upon recombination, as would be expected from the structure where two negatively charged amino acids would now be juxtaposed. Changes in the FtsKγ residues not observed to interact with XerD in the crystal structure resulted in wild type levels of recombination, confirming the specificity of the interaction (Fig. 2). The relative influence of amino acids on the XerD surface is colour coded in Fig. 2E.
In the structure, it does not appear that XerD E183 is close enough to make meaningful interactions with the FtsKγ domain, and yet its mutation has a large effect on recombination, and the double E183/184 mutant is even more affected than either single mutant. Therefore, E183 must also have a role in the interaction that leads to activation of recombination. The terminal oxygen of E183 is around 5 Å from the guanadinium group nitrogen of R1289, but rotation around the glutamate side-chain carbon linkages would allow a much closer approach between these two residues; it is possible that the role of interaction with R1289 of FtsK is shared between these two glutamates. Alternatively, the mutation of E183 may not affect the interaction with FtsKγ but rather it could affect the interaction with XerC that leads to recombination (see Discussion).
Mutants of each XerC/D-FtsKγ fusion protein were overproduced and purified, as previously described17. These proteins were then used for in vitro recombination reactions in the presence of XerC and a model plasmid containing two dif sites. The relative levels of recombination were again compared to wild-type proteins (Fig. 2C,D). Similar results to those seen in vivo were obtained with mutants in the E183/4 pair of XerD and R1289 from FtsK having the greatest effect on recombination.
Since the mutants in XerD for both in vivo and in vitro recombination were produced in a fusion protein this may have artificially raised the level of recombination seen; the covalent linkage would increase the local concentration of XerD and FtsKγ which could potentially reduce the effect of mutations which lower the affinity of the interaction between the two proteins. Further, in vivo, the strain also produced wt XerD from the chromosome and this may have contributed to the background level of recombination, seemingly reducing the effect of each mutation. The fact that a strong effect was still seen for some of the mutants (E184/R1289) shows that these are interactions which contribute greatly to the required association of these two proteins.
Taken together the in vitro and in vivo recombination data supports the interaction surface seen in the crystal structure being vital for the activation of recombination.
A minimal FtsKγ peptide sufficient for recombination
A fusion protein between XerC and FtsKγ has previously been seen to activate recombination at dif as well as, or even more efficiently than, a XerD-FtsKγ fusion (Fig. 1B)17. Therefore, in an attempt to confirm that the proposed interactions revealed by the structural data was responsible for the activation of recombination, a fusion protein was generated carrying only 9 amino acids of the FtsKγ subdomain, FtsK residues 1289–1297 (RQFRIGYNR), that contains the FtsKγ interaction surface observed in the crystal structure, attached to the end of a flexible linker at the C-terminus of XerC. This fusion protein was then used in the in vivo recombination assay described above. Upon induction a clear increase in recombination was observed, confirming that these few amino acids from the FtsK protein are sufficient to stimulate recombination by XerCD at dif (Fig. 3). The 9 amino acid peptide fused to XerC can clearly support recombination. It is perhaps not surprising that it did so far less well than the entire γ domain fused to XerC. Indeed, the XerCγ fusion is sufficiently active that low level production prior to addition of arabinose was enough to recombine the majority of the substrate at time zero, although further recombination was seen over time (Fig. 3A). Expression of XerC alone in this background (i.e. no source of FtsKγ) showed no increase in recombination over the time course (data not shown) as has been seen previously17.
Figure 3. A 9 amino acid stretch of FtsKγ is sufficient to stimulate recombination at dif.

(A) A 2x dif reporter plasmid (represented as a circle with two triangles) is in a strain lacking the C-terminus of FtsK. Recombination of the plasmid to delete one dif site and produce a smaller DNA circle can occur by expression of either an XerC-FtsKγ fusion (as shown previously17) or by expression of XerC-fused to just 9 amino acids from FtsKγ (XerC-γ peptide). Overproduction of the XerC variant was induced at time zero and samples were taken at times indicated. Note that XerC-γ is much more efficient at promoting recombination than the XerC-γ peptide, but expression of both proteins increased recombination over time. (B) Quantification of the level of recombination over time for expression of the XerC-γ peptide fusion protein, showing the average of three independent experiments (error bars are SEM).
The XerD E183A/E184A mutations do not complement a xerD deletion
In order to examine whether the proposed critical amino acids in the FtsKγ-XerD interaction were required for chromosome dimer resolution in growing cells, a co-culture assay was employed33. Cells which are phenotypically Xer− show reduced growth compared to a wild-type because chromosome dimer formation leads to cell death in a proportion of these cells33,34. Two strains, isogenic except that one is ΔxerD, were compared for relative growth. A plasmid expressing either wt XerD or a XerD mutant was present in the xerD strain to gauge whether the protein could complement the deletion phenotype. Specifically, the XerD E183A, E184A or EE183/4AA mutants were used, as well as a plasmid expressing XerC as a negative control. Note that this assay produced the mutant XerD proteins rather than the XerD-FtsKγ fusion mutants used previously, meaning that chromosomal recombination at dif was reliant on the native FtsK protein. Equalised numbers of cells of each strain were inoculated together into the same medium, and were cultured together for ~20 generations. After this period of growth the relative proportion of each strain in the culture was measured by plating onto appropriate selective media. This gives a good readout of how well each XerD protein can complement the ΔxerD mutation, and reduce the growth deficiency of Xer− cells.
The strain expressing wt XerD largely complemented the xerD mutation resulting in a relatively minor loss of fitness in this strain compared to wt (Fig. 4). Expression of XerC in this strain did not compensate for the lack of XerD, as expected, and led to a ~104 fold drop in relative cell numbers over 20 generations (Fig. 4). Expression of the XerD E183/4 mutants also largely failed to complement the xerD mutation and resulted in a large reduction in fitness of the xerD strain relative to the wild-type. As seen previously (Fig. 2) the XerD EE183/4AA double mutant appeared more severely affected than either single mutant and resulted in a reduction in fitness close to that of the XerC negative control. This assay confirms that the identified XerD-FtsK interaction is required for recombination at the chromosomal dif site during dimer resolution in vivo.
Figure 4. XerD E183A and E184A mutants do not function in chromosome dimer resolution.
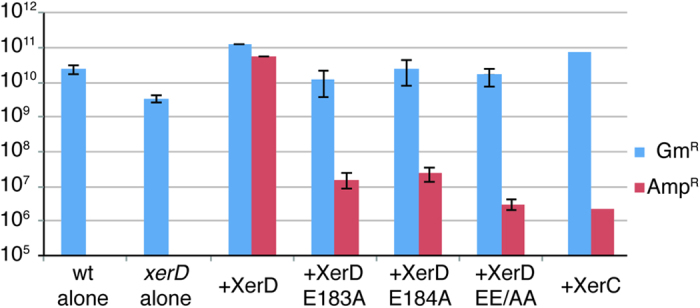
The graph shows cell counts from a co-culture assay after 20 generations of growth. The two strains used were isogenic and gentamicin resistant (GmR), apart from one having a xerD deletion. Growth of the two individual strains separately (with no plasmid) are shown at the left. For co-culture, xerD mutant cells carrying the relevant XerD expression vector as noted were also ampicillin resistant (AmpR). The ratio of ampicillin resistant cells to gentamicin resistant cells shows the degree of complementation achieved by expression of the XerD variant during co-culture. Results are the average of three independent experiments (error bars are SEM).
Discussion
The interaction between the recombinase XerD and the DNA translocase FtsK is vital for chromosome dimer resolution in bacteria4,19. This interaction leads to activation of the catalytic activity of XerD, with concomitant DNA cleavage and strand exchange as the first stage of the site-specific recombination reaction at dif17. Structural data presented here show how the interaction between the two proteins occurs; the FtsKγ domain binds on one surface of XerD whilst dif DNA would be present on the opposite face of the XerD protein (Fig. 1). Mutagenesis resulting in changes to the amino acids involved in the FtsKγ-XerD interaction confirmed that the observed interactions are required for the activation of the XerD catalytic activity. Further, the presence of just 9 amino acids of the FtsKγ domain that encompasses the XerD-interaction surface were sufficient to stimulate recombination.
A number of studies have used inter-species recombination assays using Xer recombinases and FtsKs from different organisms both for in vivo and in vitro recombination assays. For example the P. aeruginosa FtsK has been seen to activate E. coli XerD26. This is consistent with the almost perfect conservation of the amino acids in FtsKγ domains from both organisms that contact XerD (see Fig. 1C). Interactions between the Xer and FtsK proteins from Haemophilus influenzae and Lactococcus lactis have also been examined, and were found to exhibit some degree of species specificity9,35. The FtsKγ domains from these organisms are reasonably well conserved (Supplementary data, Figure S6) with four of the 5 amino acids involved in contacting XerD in the crystal structure from E. coli being the same. However, the interacting residues in XerDs from these organisms are more divergent (Figure S6): of the 5 amino acids found to be important for activation of XerD there are 3 identical and 1 similar in the H. influenzae protein whereas the L. lactis XerS (there is only a single Xer recombinase in this species) has only 1of the 5 amino acids identical to E. coli (Figure S6). Again, this is mirrored in the cross-species activities seen; E. coli FtsK can activate H. influenzae XerD35, but the E. coli FtsKγ alone cannot activate XerS from L. lactis9.
Synapsis between two XerCD-dif complexes has been observed in the absence of the FtsKγ domain11,12. Indeed the data suggest that the synapse adopts a conformation where XerC is inactive and XerD could be active, yet XerD shows no catalytic activity. The addition of FtsK induces a slight conformational change in the synapse that was interpreted as the transition to a XerD-activated (D*) conformation, and leads to formation of the Holliday junction intermediate catalysed by XerD-mediated strand exchanges12,17. We have shown the molecular detail of the interaction between XerD and FtsK, and that disruption of this interaction leads to the failure to activate XerD. However, there is no obvious conformational change in the XerD protein when it interacts with FtsKγ alone. Therefore, the observed transition of the inactive XerCD-dif synapse to the D* state must require interaction with DNA or with XerC, or likely both, in addition to the FtsKγ interaction. Indeed, this makes sense mechanistically; catalysis is only activated within the properly assembled XerCD-dif synapse reducing the occurrence of inappropriate XerD-mediated cleavage of the DNA.
By comparing the XerDC–γ active-site structure to that of activated Cre or λ Int, it is clear that two key amino-acids of the XerD active-site are inappropriately positioned for catalysis13,15: His270 and Tyr279 (Fig. 5A). As previously proposed, a conformational change of the two C-terminal helices of XerD, helix M and N, would produce the active conformation seen in other tyrosine recombinases36 (see Fig. 5B). This conformational change would lead to an active site poised for catalysis but would not interfere with the observed XerD-FtsKγ interactions. It is likely that it is this state, where XerD is poised for catalysis but inactive that is acted upon by FtsKγ to activate the catalytic activity of XerD, as proposed by in vitro studies12.
Figure 5. Models of XerD activation.
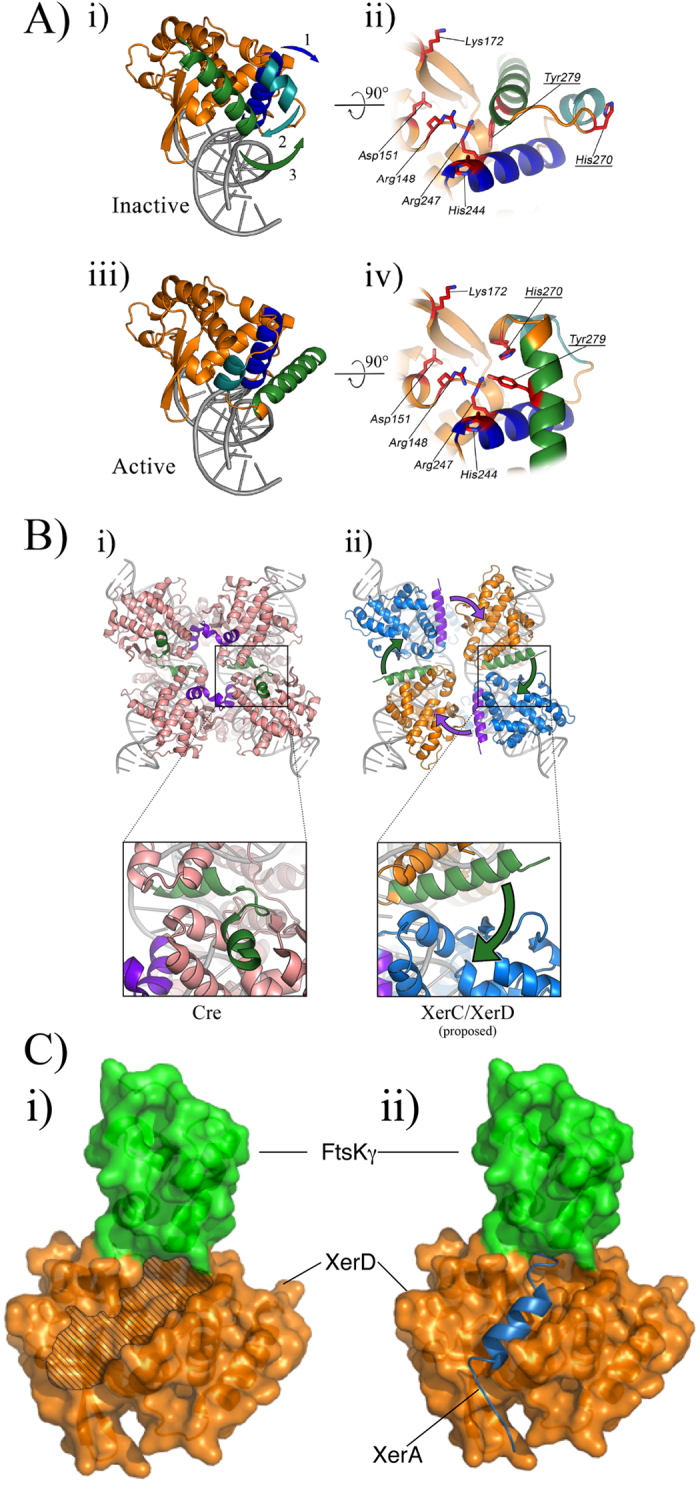
(i) XerD is shown as a ribbon, modelled onto DNA by alignment to the Cre-loxP synaptic structure16, and (ii) shows a close up of the active site residues in their inactive state, as seen in the XerDC-FtsKγ structure. The arrows in (i) indicate movement of helices in order to re-arrange the active site residues to the active configuration as seen in the Cre recombinase and result in the arrangement seen in (iii) and the active site poised for cleavage of DNA (iv). The helices involved in this movement are helix L (blue) helix M (green) and helix N (blue-green). (B) (i) The Cre synaptic structure is shown with the very C-terminal helices coloured to emphasise their interaction with the partner recombinase in a cyclic manner. (ii) 4 monomers of the “active” XerD conformation from (A) are shown superimposed on the position of the Cre monomers from the synapse in (i). The monomers are coloured to represent XerD (orange) and XerC (blue) in a XerD-active synapse. In order to achieve the same cyclic interactions as seen with Cre the C-terminal N-helices of each XerD monomer must break36 and be donated into the adjacent recombinase partner as indicated by the arrows. (C) (i) A model representing the “active” arrangement of XerD from (A), shown in orange, with the FtsKγ domain in green. The groove in which the C-terminus of XerC is thought to bind is shown by the hatched region and extends to the interaction site of XerD with FtsKγ. (ii) The activated XerD conformation and the XerA structure were overlaid and, the position of the C-terminal tail of XerA is shown (blue helix) occupying the cleft in XerD.
Another important feature of the Cre and λ Int synapses is the cyclic interaction between monomers obtained by donation of their C-termini to the adjacent monomer13,15, and these interactions determine which pair of recombinases within a synapse are active and which pair are inactive at any given time. The data from studies on the conformation of XerCD-dif synapses along with data that shows that the very C-termini of both XerC and XerD are important for this reciprocal control of partner activation36,37, suggest that the overall conformation of the synapse closely agrees with those seen for λ Int and Cre11,12.
By modelling the structure of XerD onto the active structures of Cre or λ Int the XerD M and N helices can be re-arranged to produce an active-site close to those seen for other tyrosine recombinases36; this also has the effect of moving the end of helix N to be on the correct side of the molecule to contact the partner recombinase, XerC (Fig. 5B). This movement also removes helix-N from a position where it might sterically hinder the interaction with the C-terminus coming from the partner recombinase. It has also been previously noted that, in order to make the same cyclic contacts as seen with Cre, the N-helix of XerD has to break to reach into the pocket of the partner XerC molecule36. This can readily be modelled using the XerD/XerD-FtsKγ-structure superimposed on the Cre-loxP synaptic structure (Fig. 5B). There is evidence to suggest this model reflects the physical reality; there is a cleft in the top surface of XerD where the C-terminus of the XerC partner recombinase is proposed to interact (Fig. 5B,C), and amino acids within this cleft have been shown to play an important role in controlling the catalytic activity of the interacting partner recombinase36. Further, in the structure of the archaeal Xer homologue, XerA, the very C-terminal helix (helix N) is also seen to occupy this cleft38, although in that structure it is folded back in cis rather than coming from the partner recombinase. Superimposition of the XerA C-terminus onto the FtsKγ-XerD structure shows XerA extending to a position directly underneath the FtsKγ interaction site (Fig. 5C). The Pyrococcus abyssi XerA has a C-terminal tail of similar length to that of XerC from E. coli and could reasonably be expected to be a close model (Figure S4). We can, therefore, be confident that this is an important site of interaction between XerC and XerD, and that the FtsKγ domain is positioned to modulate this interaction.
The C-terminal tail of XerC is positively charged with two lysine residues and an arginine among the last 4 amino acids (see Supplementary Fig. 1). The negatively charged E183 from XerD is at the surface, close to the end of the groove where the XerC tail is proposed to occupy, and would be available for charge-based interactions with the C-terminal tail of XerC (see Figure S5). Indeed, mutation that changes E183 greatly reduces recombination at dif, and this mutation is synergistic with mutation that alters E184, which directly contacts FtsKγ (Fig. 2). We propose that the reduction in recombination activity from alteration of E183 is a consequence of loss of interaction with the XerC partner recombinase, whereas mutation to change E184 results in loss of interaction with FtsK, and that both these interactions are necessary for efficient activation of XerD-mediated cleavage of dif DNA.
The proximity of the binding site of FtsKγ to the proposed position of the XerC tail in the XerD acceptor cleft could thus be the key to activation of the catalytic activity of XerD. The interaction of XerC, XerD and dif is sufficient to produce a synapse with the DNA bent as though XerD would be the active monomer12, yet no catalysis is seen without FtsK. A subtle change in the synapse occurs upon interaction with FtsK leading to XerD-mediated strand exchanged to form a Holliday junction (Fig. 6). We propose that the presence of FtsKγ alters the interaction of the very C-terminus of XerC in the acceptor cleft of XerD such that the synapse can now adopt the activated D* conformation12. Our model now provides a platform for exciting future experiments to determine how FtsKγ influences XerC-D interaction in the cleft and whether interaction of the very C-terminus of XerC with the FtsKγ domain is required for activation of recombinase activity.
Figure 6. Schematic model of recombination.

XerC (blue) and XerD (green) bind to the two halves of the dif site. Initially XerD is in the inactive state (dark green) as seen in the crystal structure (Fig. 1). Upon re-modelling of the three C-terminal helices of XerD as described, the active site is now close to the cleavage competent state (depicted by light green XerD) and the very C-terminal helix (helix N) rotates so that it can now interact with the XerC binding partner. At synapsis, two XerCD-dif sites come together and the potential for a pseudo-fourfold symmetric arrangement of interactions is present, with the N-helices of XerC stretching across synaptic partners to the neighbouring XerD monomers. Upon interaction of FtsKγ there is a modest re-modelling of the complex to increase the bending of the DNA. The FtsKγ domain interacts above the cleft in XerD in which the XerC N-helix sits. Only when all these conditions are achieved does XerD become catalytically active, as denoted by the black asterisk.
Methods
Cloning and mutagenesis
The XerDC-FtsKγ fusion protein (XerD residues 111 to 298 followed by a 14 amino acid linker (GGGSEGGGSEGGSG) +2 amino acids (SR) from the linking XbaI restriction enzyme site followed by FtsK residues 1261 to 1329) was amplified from the full length XerD-FtsKγ17 using Phusion polymerase and cloned into pBad24 between restriction sites for EcoRI and HindIII. The full amino acid sequence of the fusion protein is shown in supplementary data (Figure S1).
xerD-ftsKγ mutants were made by a two-step overlap PCR process, using mutagenic DNA primers and Phusion DNA polymerase (sequences available on request), and cloned into pBAD24. Selected mutant sequences were subcloned into pBAD24-XerD by PCR from the relevant mutant fusion using XerD primers as described previously17, or by site-directed mutagenesis using the Q5 Site-Directed Mutagenesis Kit (New England Biolabs). The xerC–ftsKγ-peptide fusion sequence was cloned by cutting the pBad24 XerC-FtsKγ fusion17 with XbaI and HindIII to remove the FtsKγ domain, and then ligating phosphorylated and annealed oligonucleotides with appropriate overlaps into this vector. All clones produced were verified by sequencing at the Australian Genome Research Facility.
Protein purification
XerDC-FtsKγ fusion overproduction and purification using Ni2+ resin was as previously described17. Eluted protein was then loaded onto a 1 ml HiTrap heparin HP column (GE Healthcare) in buffer A (25 mM Tris (pH 7.5), 100 mM NaCl, 1 mM EDTA, 1 mM DTT) and eluted with a gradient to buffer B (Buffer A + 1 M NaCl). Protein was then further purified on a 1 ml HiTrap Q HP using the same buffer A and B as above. Eluted protein was concentrated and buffer exchanged into 25 mM Tris-HCl (pH 7.5), 150 mM MgCl2, 1 mM DTT, using a VivaSpin 6 centrifugal concentrator (GE Healthcare) to a final concentration of ~9 mg/ml.
All other proteins and fusion proteins for in vitro recombination assays were overproduced and purified as previously17.
Crystallization and data collection for XerDC-FtsKγ
Initial hits were identified using 200 nl protein drops, using commercial screens (Molecular Dimensions) mixed by Mosquito nano-litre robot (TTP Labtech). Following initial screening, trigonal bipyramidal crystals were obtained by 2μl + 2μl hanging drop vapour diffusion in a solution of 100 mM Bicine (pH9), 10% (w/v) polyethylene glycol (8000 g/mol) over 2–5 days. Crystals were washed in paratone-N and flash frozen in liquid N2. Data was collected on MX2 beamline at the Australian Synchrotron, using a wavelength of 0.9184 Å39.
Structure solution and refinement
The structure was solved by molecular replacement using the known structures of E. coli XerD (PDB: 1A0P) and E. coli FtsKγ (PDB: 2J5P) as search models (note that only the C-terminus of the XerD structure was used, amino acids 111–298). The program PHASER40 placed a single molecule of XerDC-FtsKγ in the asymmetric unit. The structure was built using Arp/Warp41 and refined using PHENIX42 and COOT43 (see Table 2 for refinement statistics) to 2.3 Å. 98% of bonds were in the Ramachandran favoured conformation, with 0% outliers.
Table 2. Data collection and refinement statistics (molecular replacement).
| XerDC-FtsKγ | |
|---|---|
| Data collection | |
| Space group | P 65 |
| Cell dimensions | |
| a, b, c (Å) | 83.44, 83.44, 88.66 |
| α, β, γ (°) | 90, 90, 120 |
| Resolution (Å) | 56.02–2.30 (2.38–2.30)* |
| Rmerge | 0.12 (1.66) |
| Rpim | 0.04 (0.54) |
| CC(1/2) | 1.00 (0.53) |
| I/σI | 13.8 (2.1) |
| Completeness (%) | 100 (100) |
| Redundancy | 11.0 (11.2) |
| Refinement | |
| Resolution (Å) | 36.13 - 2.3 (2.38–2.30) |
| No. reflections | 15639 (1566) |
| Rwork/Rfree | 0.191 (0.287)/0.229 (0.303) |
| CCwork | 0.84 (0.65) |
| CCfree | 0.85 (0.78) |
| CC* | 0.94 (0.80) |
| No. atoms | 1976 |
| Protein | 1915 |
| Ligand/ion | — |
| Water | 61 |
| B-factors | Overall 67.5 |
| Protein | 67.9 |
| Ligand/ion | — |
| Water | 52.6 |
| R.M.S. deviations | |
| Bond lengths (Å) | 0.01 |
| Bond angles (°) | 1.16 |
Values in parentheses are for highest-resolution shell.
All structural alignments and structure figures were produced using PyMol (Schrödinger, LLC).
Recombination assays
In vivo recombination assays using XerD-γ fusion proteins were carried out as previously published17,30, but details are given in supplementary material. A similar procedure was used for assessing recombination from XerC-FtsKγ peptide fusions; E. coli strain GR51 (AB1157 xerC ftsK)44, was transformed with pBAD24 derived expression vectors carrying wild type or mutated variants of xerC-γ fusion gene along with plasmid resolution reporter, pRB10, a pSC101 derivative (6 kb, SpR) carrying two directly repeated dif sites, flanking the KmR gene cassette. pRB10 is almost identical to the pFX142 reporter used previously except that pRB10 lacks the duplication of restriction enzyme sites surrounding the two dif sites found in pFX142. Transformants were grown in LB with selection; following 16 h incubations plasmid DNA was recovered from the cultured cells and examined by agarose gel electrophoresis followed by SYBR green staining. Levels of parental and recombinant sized plasmid were quantified using ImageQuant software (GE Healthcare) and percentage recombination calculated.
In vitro recombination, using the 2x dif reporter plasmid pSI56, was as previously described17.
Co-culture growth competition
Assays were carried out essentially as previously published17,34,45. A 1:1 mixture of the two relevant strains (WX31 (AB1157 lac::tetO180 GmR) and WX31 ΔxerD containing the relevant expression vector was prepared and grown in LB at 37 °C to stationary phase (~20 generations)3,17. The relative abundance of each of the two strains was determined by comparing dilutions plated to select for the XerD expression plasmid (gentamycin + ampicillin), with similar dilutions plated without selection for the plasmid, where both strains will grow (gentamycin alone). The relative colony counts on each plate were determined.
Additional Information
Accession codes: Coordinates and structure factors for the XerDC-γ complex have been deposited in the Protein Data Bank under the accession code 5DCF.
How to cite this article: Keller, A. N. et al. Activation of Xer-recombination at dif: structural basis of the FtsKγ–XerD interaction. Sci. Rep. 6, 33357; doi: 10.1038/srep33357 (2016).
Supplementary Material
Acknowledgments
We would like to thanks the beamline scientists on MX2 at the Australian Synchrotron for their assistance. This work was funded by NHMRC Project Grant (APP1005697) and Australian Research Council Future Fellowship (FT120100153) to I.G., and research in the Sherratt laboratory was supported by the Wellcome Trust (SIA 099204/Z/12Z). JL was supported by the Medical Research Council (MC_U105184326) and the Wellcome Trust (095514/Z/11/Z).
Footnotes
Author Contributions Crystallography: Y.X. carried out protein purification and crystallisation, I.G. and A.N.K. collected diffraction data, A.N.K. and J.L. solved the structure (Figure 1). I.G., P.J.L., L.K.A. and D.J.S. designed protein variants and I.G., S.B., R.B. and J.R. carried out mutagenesis, purified proteins and carried out recombination assays (Figures 2 and 3). S.B., I.G., P.J.L., L.K.A. and D.J.S. analysed data. S.B. carried out co-culture assays (Figure 4). I.G. wrote the manuscript and A.N.K. produced structural images for figures. All authors reviewed the manuscript.
References
- McClintock B. A Correlation of Ring-Shaped Chromosomes with Variegation in Zea Mays. Proc Natl Acad Sci USA 18, 677–681 (1932). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel B., Recchia G. D., Penel-Colin M., Ehrlich S. D. & Sherratt D. J. Resolution of holliday junctions by RuvABC prevents dimer formation in rep mutants and UV-irradiated cells. Mol Microbiol 37, 180–191 (2000). [DOI] [PubMed] [Google Scholar]
- Perals K., Cornet F., Merlet Y., Delon I. & Louarn J. M. Functional polarization of the Escherichia coli chromosome terminus: the dif site acts in chromosome dimer resolution only when located between long stretches of opposite polarity. Mol Microbiol 36, 33–43 (2000). [DOI] [PubMed] [Google Scholar]
- Grainge I. Simple topology: FtsK-directed recombination at the dif site. Biochem Soc Trans 41, 595–600, doi: 10.1042/BST20120299 (2013). [DOI] [PubMed] [Google Scholar]
- Blakely G. et al. Two related recombinases are required for site-specific recombination at dif and cer in E. coli K12. Cell 75, 351–361 (1993). [DOI] [PubMed] [Google Scholar]
- Kuempel P. L., Henson J. M., Dircks L., Tecklenburg M. & Lim D. F. dif, a recA-independent recombination site in the terminus region of the chromosome of Escherichia coli. New Biol 3, 799–811 (1991). [PubMed] [Google Scholar]
- Carnoy C. & Roten C. A. The dif/Xer recombination systems in proteobacteria. PLoS One 4, e6531, doi: 10.1371/journal.pone.0006531 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debowski A. W. et al. Xer recombinase and genome integrity in Helicobacter pylori, a pathogen without topoisomerase IV. PLoS One 7, e33310, doi: 10.1371/journal.pone.0033310 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolivos S., Pages C., Rousseau P., Le Bourgeois P. & Cornet F. Are two better than one? Analysis of an FtsK/Xer recombination system that uses a single recombinase. Nucleic Acids Res 38, 6477–6489, doi: 10.1093/nar/gkq507 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakely G. & Sherratt D. Determinants of selectivity in Xer site-specific recombination. Genes Dev 10, 762–773 (1996). [DOI] [PubMed] [Google Scholar]
- Diagne C. T. et al. TPM analyses reveal that FtsK contributes both to the assembly and the activation of the XerCD-dif recombination synapse. Nucleic Acids Res 42, 1721–1732, doi: 10.1093/nar/gkt1024 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawadzki P. et al. Conformational transitions during FtsK translocase activation of individual XerCD-dif recombination complexes. Proc Natl Acad Sci USA 110, 17302–17307, doi: 10.1073/pnas.1311065110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas T. et al. A structural basis for allosteric control of DNA recombination by lambda integrase. Nature 435, 1059–1066, doi: 10.1038/nature03657 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Narendra U., Iype L. E., Cox M. M. & Rice P. A. Crystal structure of a Flp recombinase-Holliday junction complex: assembly of an active oligomer by helix swapping. Mol Cell 6, 885–897 (2000). [PubMed] [Google Scholar]
- Guo F., Gopaul D. N. & van Duyne G. D. Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature 389, 40–46, doi: 10.1038/37925 (1997). [DOI] [PubMed] [Google Scholar]
- Guo F., Gopaul D. N. & Van Duyne G. D. Asymmetric DNA bending in the Cre-loxP site-specific recombination synapse. Proc Natl Acad Sci USA 96, 7143–7148 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainge I., Lesterlin C. & Sherratt D. J. Activation of XerCD-dif recombination by the FtsK DNA translocase. Nucleic Acids Res 39, 5140–5148, doi: 10.1093/nar/gkr078 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozat E. & Grainge I. FtsK DNA translocase: the fast motor that knows where it’s going. Chembiochem 11, 2232–2243, doi: 10.1002/cbic.201000347 (2010). [DOI] [PubMed] [Google Scholar]
- Sherratt D. J., Arciszewska L. K., Crozat E., Graham J. E. & Grainge I. The Escherichia coli DNA translocase FtsK. Biochem Soc Trans 38, 395–398, doi: 10.1042/BST0380395 (2010). [DOI] [PubMed] [Google Scholar]
- Begg K. J., Dewar S. J. & Donachie W. D. A new Escherichia coli cell division gene, ftsK. J Bacteriol 177, 6211–6222 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper G. C., McLennan N., Begg K., Masters M. & Donachie W. D. Only the N-terminal domain of FtsK functions in cell division. J Bacteriol 180, 4621–4627 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubarry N., Possoz C. & Barre F. X. Multiple regions along the Escherichia coli FtsK protein are implicated in cell division. Mol Microbiol 78, 1088–1100, doi: 10.1111/j.1365-2958.2010.07412.x (2010). [DOI] [PubMed] [Google Scholar]
- Yu X. C., Tran A. H., Sun Q. & Margolin W. Localization of cell division protein FtsK to the Escherichia coli septum and identification of a potential N-terminal targeting domain. J Bacteriol 180, 1296–1304 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisicchia P., Steel B., Mariam Debela M. H., Lowe J. & Sherratt D. The N-terminal membrane-spanning domain of the Escherichia coli DNA translocase FtsK hexamerizes at midcell. MBio 4, e00800–00813, doi: 10.1128/mBio.00800-13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aussel L. et al. FtsK Is a DNA motor protein that activates chromosome dimer resolution by switching the catalytic state of the XerC and XerD recombinases. Cell 108, 195–205 (2002). [DOI] [PubMed] [Google Scholar]
- Massey T. H., Mercogliano C. P., Yates J., Sherratt D. J. & Lowe J. Double-stranded DNA translocation: structure and mechanism of hexameric FtsK. Mol Cell 23, 457–469, doi: 10.1016/j.molcel.2006.06.019 (2006). [DOI] [PubMed] [Google Scholar]
- Steiner W., Liu G., Donachie W. D. & Kuempel P. The cytoplasmic domain of FtsK protein is required for resolution of chromosome dimers. Mol Microbiol 31, 579–583 (1999). [DOI] [PubMed] [Google Scholar]
- Bigot S. et al. KOPS: DNA motifs that control E. coli chromosome segregation by orienting the FtsK translocase. EMBO J 24, 3770–3780, doi: 10.1038/sj.emboj.7600835 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy O. et al. Identification of oligonucleotide sequences that direct the movement of the Escherichia coli FtsK translocase. Proc Natl Acad Sci USA 102, 17618–17623, doi: 10.1073/pnas.0508932102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivanathan V. et al. The FtsK gamma domain directs oriented DNA translocation by interacting with KOPS. Nat Struct Mol Biol 13, 965–972, doi: 10.1038/nsmb1158 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe J. et al. Molecular mechanism of sequence-directed DNA loading and translocation by FtsK. Mol Cell 31, 498–509, doi: 10.1016/j.molcel.2008.05.027 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanya H. S. et al. Crystal structure of the site-specific recombinase, XerD. EMBO J 16, 5178–5187, doi: 10.1093/emboj/16.17.5178 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigot S., Corre J., Louarn J. M., Cornet F. & Barre F. X. FtsK activities in Xer recombination, DNA mobilization and cell division involve overlapping and separate domains of the protein. Mol Microbiol 54, 876–886, doi: 10.1111/j.1365-2958.2004.04335.x (2004). [DOI] [PubMed] [Google Scholar]
- Sivanathan V. et al. KOPS-guided DNA translocation by FtsK safeguards Escherichia coli chromosome segregation. Mol Microbiol 71, 1031–1042, doi: 10.1111/j.1365-2958.2008.06586.x (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates J., Aroyo M., Sherratt D. J. & Barre F. X. Species specificity in the activation of Xer recombination at dif by FtsK. Mol Microbiol 49, 241–249 (2003). [DOI] [PubMed] [Google Scholar]
- Hallet B., Arciszewska L. K. & Sherratt D. J. Reciprocal control of catalysis by the tyrosine recombinases XerC and XerD: an enzymatic switch in site-specific recombination. Mol Cell 4, 949–959 (1999). [DOI] [PubMed] [Google Scholar]
- Spiers A. J. & Sherratt D. J. C-terminal interactions between the XerC and XerD site-specific recombinases. Mol Microbiol 32, 1031–1042 (1999). [DOI] [PubMed] [Google Scholar]
- Serre M. C. et al. The carboxy-terminal alphaN helix of the archaeal XerA tyrosine recombinase is a molecular switch to control site-specific recombination. PLoS One 8, e63010, doi: 10.1371/journal.pone.0063010 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhillips T. M. et al. Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. J Synchrotron Radiat 9, 401–406 (2002). [DOI] [PubMed] [Google Scholar]
- McCoy A. J. et al. Phaser crystallographic software. J Appl Crystallogr 40, 658–674, doi: 10.1107/S0021889807021206 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer G., Cohen S. X., Lamzin V. S. & Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc 3, 1171–1179, doi: 10.1038/nprot.2008.91 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221, doi: 10.1107/S0907444909052925 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P. & Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132, doi: 10.1107/S0907444904019158 (2004). [DOI] [PubMed] [Google Scholar]
- Recchia G. D., Aroyo M., Wolf D., Blakely G. & Sherratt D. J. FtsK-dependent and -independent pathways of Xer site-specific recombination. EMBO J 18, 5724–5734, doi: 10.1093/emboj/18.20.5724 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capiaux H., Lesterlin C., Perals K., Louarn J. M. & Cornet F. A dual role for the FtsK protein in Escherichia coli chromosome segregation. EMBO Rep 3, 532–536, doi: 10.1093/embo-reports/kvf116 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.