

We recently observed that the propensity to induce MDSC accumulation varies greatly between different mammary tumors. In investigating the cause of such diversity, we found a link between mTOR pathway activity in breast cancer cells and MDSC recruitment.1 Genetic or pharmacological inhibition of the mTOR pathway in tumor cells reduced MDSC formation. Furthermore, replenishing MDSCs in these models rescued tumor growth and metastasis. The primary downstream mediator of the mTOR pathway responsible for inducing MDSC formation was found to be G-CSF. Inhibition of upstream receptor tyrosine kinases (e.g., FGFR and EGFR) as well as TORC1 reduced expression of G-CSF. The mTOR pathway controls cap-dependent translation of many oncogenes.2 However, our data suggested that mTOR regulation of G-CSF may be at the transcription level. The activity of certain transcription factors such as STAT3 and SREBP1/2 are known to be regulated in part by mTOR.2 Further studies will be required to examine if these transcription factors directly regulate the expression of G-CSF.
Circumstantial evidence also suggested that the FGFR/mTOR/G-CSF pathway had a higher activity in tumor-initiating cells (TICs). The microenvironment niche is known to play a decisive role in shaping TIC fate,3 intracellular cytokine staining and ELISA for G-CSF revealed an increased G-CSF expression in TICs. Moreover, breast-tumor cells under 3D TIC-enriching conditions promoted MDSC formation in cultured primary bone marrow. Reciprocally, co-culture of tumor-cells with MDSCs increased mammosphere formation, expression of TIC markers, and most importantly, TIC frequencies as measured by limiting transplantation assays. Mechanistically, MDSCs impact TICs at least in part by activating the notch pathway. Overall, these results suggest that MDSCs and TICs have reciprocal effects on each other, thereby establishing a feed-forward loop important for the evolution of these tumors as detailed below.
As illustrated in the Figure 1, GCSF produced by tumors is secreted into the blood stream and drives MDSC accumulation first in the bone marrow. MDSCs are mobile and can infiltrate various distant organs, where they may promote tumor initiation. Indeed, previous studies demonstrated that bone marrow-derived myeloid lineage cells establish a pre-metastatic niche,4 as well as facilitating metastasis by their regulation of systemic immune function. If metastasis initiating tumor cells (MICs) rely on MDSCs, one intriguing question is whether the tissue tropism of MDSCs might impact the known tissue tropism of tumor metastases. Another important question is how closely related cell types with pro- and anti-tumor activities, respectively (polymorphonuclear MDSCs and TANs vs normal neutrophils) are regulated in vivo.5 A better characterization of MDSC heterogeneity will be required in order to answer both of these questions. Future experiments will be required to distinguish MDSC subsets and to determine their activities in both local and systemic immune suppression, TIC and MIC support, and – potentially – metastasis tissue tropism. Identification and characterization of “criminal” MDSC subpopulations may potentially allow their more specific targeting while minimizing bystander damage to other myeloid cells.
Figure 1.
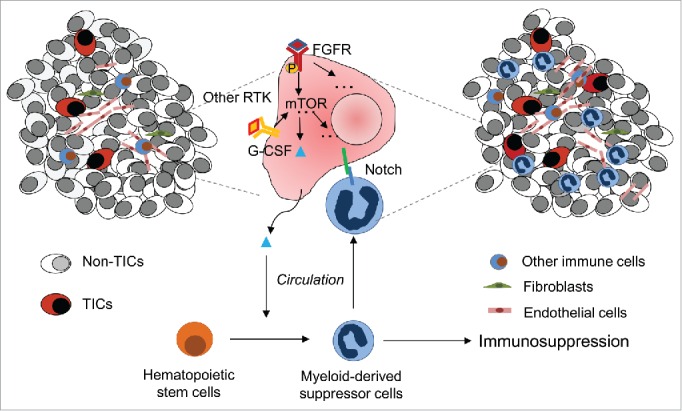
Model of interaction between MDSCs and TICs. Elevated oncogenic receptor tyrosine kinase-mTOR signaling in TICs leads to G-CSF release into the circulation. When reaching bone marrow, G-CSF induces formation of MDSCs. MDSCs suppress T-cell and NK cell responses against tumor and promote angiogenesis. They also directly interact with TICs and enhance stem cell properties via notch signaling.
Our findings also provided important information for the diagnosis and treatment of breast cancer patients. Breast tumors with high mTOR activity in tumors also exhibit a high MDSC infiltration index. FGFR, an upstream activator of mTOR signaling was similarly correlated to MDSCs. These phenotypes were linked to poor prognosis, which in the case of breast cancer suggests a correlation with distant metastasis to vital organs. To diagnose the presence of MDSCs, it may be possible to determine a MDSC signature expression in tumor biopsies. However, this may be difficult to implement as routine diagnostic tool. Instead, the blood levels of G-CSF levels may serve as surrogate marker. Although definitive IHC markers are still lacking for robust detection of MDSC infiltration, S100A8 may help to detect myeloid lineage cells in tumor sections.
Treatments inhibiting MDSC formation potentially may be achieved in a relatively tumor-centric fashion by blocking the hyperactive pathways in tumor cells that lead to G-CSF over-production. Specifically targeting these pathways in tumor cells may allow the continued production of physiologically important low levels of G-CSF by other cell types. In addition, prevention of an MDSC : TIC interaction, e.g. via interruption of notch ligand : receptor pairing, or chemokine-mediated recruitment 1 may decrease tumor initiating cells both at the primary and at the metastatic sites. Finally, the success of low-dosage treatment with several chemotherapy drugs may in part result from the eradication of MDSCs.6
Recently, durable therapeutic responses have been achieved in a subset of patients of various cancer types by targeting immune checkpoint in T-cells (anti-PD1, anti-CTLA4 antibody treatments). Whether the same strategy can be applied to more cancer types and how to increase response rates remain to be addressed. MDSCs are by definition immunosuppressive. The suppression of T cells by MDSCs needs to be overcome in anti-PD1/anti-CTLA4 resistant cancers.7 Accordingly, our data demonstrated that inhibiting MDSC formation through Raptor downregulation in tumor cells reduced the percentage of PD1+ T-cells. Thus, the application of immune checkpoint blockade therapies may benefit from treatments that simultaneously reduce MDSC.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors acknowledge the joint participation by Diana Helis Henry Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with Baylor College of Medicine.
Funding
X. H.-F. Z. is supported by NCI CA183878, Breast Cancer Research Foundation, US Department of Defense DAMD W81XWH-13-1-0195, Susan G. Komen CCR14298445, and McNair Medical Institute. T.W. is supported by Helis Foundation. J. M. R. was supported by NIH grant CA148761 and CA16303.
References
- [1].Welte T, Kim IS, Tian L, Gao X, Wang H, Li J, Holdman XB, Herschkowitz JI, Pond A, Xie G, et al.. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat Cell Biol 2016; 18(6):632-44; advance online publication; PMID:27183469; http://dx.doi.org/ 10.1038/ncb3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:274-93; PMID:22500797; http://dx.doi.org/ 10.1016/j.cell.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015; 16:225-38; PMID:25748930; http://dx.doi.org/ 10.1016/j.stem.2015.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kaplan RN, Psaila B, Lyden D. Bone marrow cells in the ‘pre-metastatic niche’: within bone and beyond. Cancer Metastasis Rev 2006; 25:521-9; PMID:17186383; http://dx.doi.org/ 10.1007/s10555-006-9036-9 [DOI] [PubMed] [Google Scholar]
- [5].Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM. Polarization of tumor-associated neutrophil phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell 2009; 16:183-94; PMID:19732719; http://dx.doi.org/ 10.1016/j.ccr.2009.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res 2005; 11:6713-21; PMID:16166452; http://dx.doi.org/ 10.1158/1078-0432.CCR-05-0883 [DOI] [PubMed] [Google Scholar]
- [7].Sharma P, Allison JP. The future of immune checkpoint therapy. Science 2015; 348:56-61; PMID:25838373; http://dx.doi.org/ 10.1126/science.aaa8172 [DOI] [PubMed] [Google Scholar]