

Senescence is a cellular response, whereby damaged cells cease to proliferate following exposure to endogenous and exogenous stressors, including telomere dysfunction, oncogene activation, genotoxic stress and chromatin remodelling. Despite being a tumor suppressor mechanism by preventing proliferation of potentially harmful cells, senescence is also a driver of age-related tissue dysfunction and pathology. This is in part due to the development of a powerful secretory phenotype involving overproduction of growth factors, proteases and pro-inflammatory cytokines, collectively known as the senescence-associated secretory phenotype (SASP).1 Moreover, these cells also produce and secrete higher levels of reactive oxygen species (ROS). Both the SASP and ROS can stabilize senescence and induce senescence in neighboring cells.1 Thus, there is increasing interest in understanding the pathways that regulate senescence, particularly those that influence its pro-oxidant and pro-inflammatory phenotypes.
Mitochondria have been implicated in cellular senescence, mostly since they are considered the main generators of ROS.2 However, reports have also emphasized the importance of non-mitochondrial ROS sources, redox stress and deficits in antioxidant defenses in senescence induction.1 Therefore, it remained unknown whether mitochondria are truly necessary for senescence and, if so, what mechanisms are involved. Answering these questions is challenging for several reasons: i) conventional assays to assess intracellular ROS are relatively unspecific and with several methodological pitfalls; ii) processes occurring within mitochondria are diverse and complex and iii) intervening in individual aspects of mitochondrial biology results unequivocally in adaptive responses which alter mitochondrial function in different ways. In our study, published in The EMBO Journal, we designed a proof-of-principle experiment that ultimately disclosed the requirement of mitochondria for senescence (Fig. 1).3 We utilised the property of the ubiquitin E3 ligase Parkin to induce widespread mitophagy upon mitochondrial membrane depolarization in order to generate human fibroblasts without mitochondria. Furthermore, we optimised culture conditions to keep mitochondria-depleted cells viable for a sufficient amount of time to investigate their role in senescence.3 The physical loss of mitochondria was confirmed by 3D electron microscopy and undetectable mitochondrial respiration, proteins or mitochondrial DNA. Using this powerful method, we found that following a variety of senescence triggers (e.g. oxidative stress, oncogene activation and replicative exhaustion), features of cellular senescence were significantly reduced in mitochondria-depleted cells, particularly the pro-oxidant and pro-inflammatory phenotypes, while energy levels were preserved with increased compensatory glycolysis. Importantly, global transcriptome analysis revealed that a substantial fraction of senescence-associated changes were mitochondria-dependent. Notably, while expression of cyclin-dependent kinase inhibitors p16ink4a and p21CIP was reduced, mitochondria-depleted senescent cells did not resume proliferation. Based on this data, we suggest that mitochondria may be highly promising targets for anti-senescence therapies. By targeting them, we may be able to suppress the detrimental SASP known to induce aging and cancer, while maintaining the tumor suppressor capabilities of senescent cells.
Figure 1.
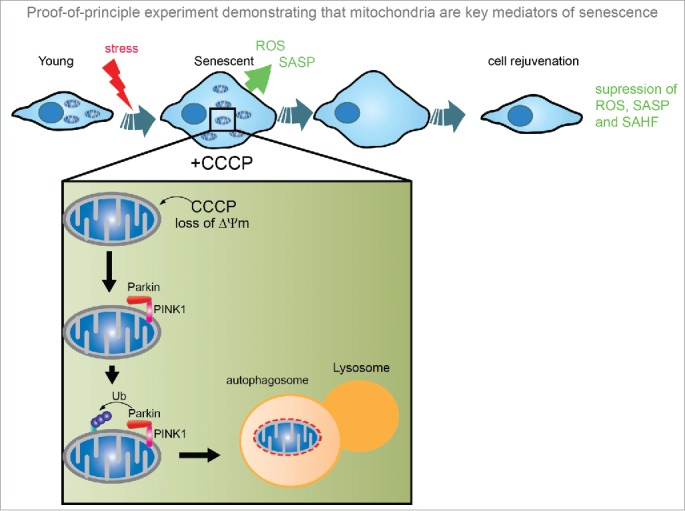
Proof-of-principle experiment demonstrating that mitochondria are key mediators of senescence. Using the parkin-mediated mitophagy system, cells overexpressing the ubiquitin ligase Parkin are treated with CCCP, a mitochondrial membrane uncoupler, which induces mitochondrial depolarisation. Cytosolic Parkin translocates to depolarised mitochondria, and binds to PINK1 leading to ubiquitination of outer mitochondrial membrane proteins, which are recognized by the proteasome and autophagy machineries. Removal of mitochondria from senescent cells through this method suppressed the development of a number of senescence-associated features, including the SASP, ROS and formation of SAHF. ROS, reactive oxygen species; SASP, senescence-associated secretory phenotype; SAHF, senescence-associated heterochromatin foci; CCCP, Carbonyl cyanide m-chlorophenyl hydrazine.
Mitochondrial biogenesis in the pathways driving senescence
The role of mitochondrial homeostasis, mitochondrial metabolites and ROS generation in the process of cellular senescence has been subject of debate.1 We have previously observed that senescent cells present increased mitochondrial mass driven by mitochondrial biogenesis, which results in increased oxygen consumption per cell.2 We have also uncovered a novel senescence regulatory pathway where activation of the ATM, Akt and mTOR phosphorylation cascades downstream of a DNA Damage Response (DDR) triggers PGC-1β-dependent mitochondrial biogenesis. Importantly, modulation of these pathways impacts on mitochondrial biogenesis, senescence and the SASP both in vitro and in vivo.3 Other recent studies have further highlighted mTOR as a SASP suppressor, by alternative mechanisms such as decreasing translation of the proteins IL1A and MAPKAPK2,4,5 further emphasizing mTOR as an anti-senescence target.
Increasing evidence implicates cellular senescence in physiological and pathological settings, such as aging and cancer. In mice, p16Ink4a-positive cells accumulate with age and negatively influence lifespan by promoting age-dependent changes. Notably, inducible elimination of p16Ink4a-positive senescent cells delays the acquisition of age-related pathologies,6 proposing senescent cell elimination as a therapeutic approach to extend healthspan. Another way of alleviating the deleterious effects of senescent cells is to develop drugs that target pathways regulating cellular senescence and its unfavorable phenotypes, namely the SASP and ROS. Our observations place mitochondria centrally in the pathways regulating the pro-aging features of senescence: mitochondria biogenesis follows DDR activation with intermediate ATM-Akt-mTOR phosphorylation to induce pro-oxidant and SASP signals.3 Further studies will be required to delineate the precise mitochondrial signals that trigger the senescence program. Altogether, these findings place mitochondria as potential therapeutic targets for dampening the detrimental side of senescence during aging and identify new regulatory pathways of mitochondrial homeostasis.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Correia-Melo C, Passos JF. Mitochondria: Are they causal players in cellular senescence? Biochimica Et Biophysica Acta (BBA) - Bioenergetics 2015; 1847:1373-9; PMID:26028303; http://dx.doi.org/ 10.1016/j.bbabio.2015.05.017 [DOI] [PubMed] [Google Scholar]
- [2].Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, Miwa S, Olijslagers S, Hallinan J, Wipat A, et al.. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol 2010; 6:347; PMID:20160708; http://dx.doi.org/ 10.1038/msb.2010.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Correia-Melo C, Marques FDM, Anderson R, Hewitt G, Hewitt R, Cole J, Carroll BM, Miwa S, Birch J, Merz A, et al.. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J 2016; 35(7):724-42; PMID:26848154; http://dx.doi.org/ 10.15252/embj.201592862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, Raguz S, Acosta JC, Innes AJ, Banito A, et al.. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol 2015; 17:1205-17; PMID:26280535; http://dx.doi.org/ 10.1038/ncb3225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson-Edell KA, Liu S, et al.. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol 2015; 17:1049-61; PMID:26147250; http://dx.doi.org/ 10.1038/ncb3195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, et al.. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 2016; 530:184-9; PMID:26840489; http://dx.doi.org/ 10.1038/nature16932 [DOI] [PMC free article] [PubMed] [Google Scholar]