

Abstract
The functional role of cholinergic input in the modulation of sensory responses was studied using a combination of in vivo and in vitro electrophysiology supplemented by mathematical modeling. The electrosensory system of weakly electric fish recognizes different environmental stimuli by their unique alteration of a self-generated electric field. Variations in the patterns of stimuli are primarily distinguished based on their frequency. Pyramidal neurons in the electrosensory lateral line lobe (ELL) are often tuned to respond to specific input frequencies. Alterations in the tuning of the pyramidal neurons may allow weakly electric fish to preferentially select for certain stimuli. Here we show that muscarinic receptor activation in vivo enhances the excitability, burst firing, and subsequently the response of pyramidal cells to naturalistic sensory input. Through a combination of in vitro electrophysiology and mathematical modeling, we reveal that this enhanced excitability and bursting likely results from the down-regulation of an A-type potassium current. Further, we provide an explanation of the mechanism by which these currents can mediate frequency tuning.
INTRODUCTION
It is well known that sensory neurons can give vastly different responses to the same sensory stimulus based on the behavioral context, and there is great interest in understanding the mechanisms by which this occurs (Abbott 2005).
Cholinergic pathways can regulate information processing in several brain areas (reviewed in Everitt and Robbins 1997; Sarter et al. 2005), and studies have shown that cholinergic input can enhance a neuron’s response to specific types of sensory input (Bakin and Weinberger 1996; Gu 2003; Kilgard and Merzenich 1998; Sarter et al. 2005; Tang et al. 1997; Weinberger 2003). Cholinergic input can control a cell’s firing properties in a number of different ways, possibly through the regulation of several ion channels or through the modulation of synaptic properties (review; Lucas-Meunier et al. 2003). However, direct links between the cholinergic modulation of an individual neuron and the effects that such changes would have on responses to external sensory stimuli at the systems level have been more difficult to make.
Weakly electric fish sense distortions in their self-generated electric field caused by nearby objects (Fig. 1, A and B). Electroreceptive neurons on their skin encode these perturbations through changes in firing rate (Bastian 1981), and this information is then relayed to the pyramidal cells within the electrosensory lateral line lobe (ELL) (for a review of the electrosensory system, see Turner and Maler 1999) (Fig. 1C). The electrosensory system displays many similarities with mammalian sensory systems (Berman and Maler 1999; Krahe and Gabbiani 2004; Sadeghi et al. 2007) and benefits from detailed neuroanatomy (Maler et al. 1991), well-characterized physiology (both at the systems and single neuron level), and distinct response patterns to natural sensory input (Bastian et al. 2002; Berman and Maler 1998a–c; Chacron 2006; Chacron et al. 2005). Previous studies have shown that descending glutamatergic pathways can control pyramidal cell responses to sensory input (Chacron et al. 2005; Mehaffey et al. 2005). Here we focus on descending cholinergic input, which likely emanates from a unique pathway (Fig. 1C) (Maler et al. 1981). We show that activation of muscarinic acetylcholine receptors can increase a neuron’s response to low-frequency input through an increase in firing rate and burst fraction that is the result of the downregulation of an A-type potassium current.
FIG. 1.

The electrosensory system. A: weakly electric fish generate an electric field around their body to electrolocate objects. For Apteronotus leptorhynchus, the electric organ discharge (EOD) waveform recorded at 1 point in space is quasi-sinusoidal with a frequency of 600–1,000 Hz (green trace). B: illustration of the Global stimulation geometry used. Amplitude modulations of the animals’ own EOD are delivered via 2 silver-silver-chloride electrodes (G1 and G2) located 19 cm away on each side of the animal. The perturbations of the EOD created are roughly spatially homogeneous on the animal’s skin surface. C: illustration of the relevant circuitry. Electroreceptor afferents on the animal’s skin detect amplitude modulations of the EOD and synapse unto pyramidal cells within the electrosensory lateral line lobe. Pyramidal cells project to the nucleus praeeminentialis (NP). Bipolar cells (BP) and stellate cells feed back, from the NP unto pyramidal cells by way of the tractus stratum fibrosum (StF). Multipolar cells (MP) project to granule cells (GC) within the eminentia granularis posterior (EGP) that feed back unto pyramidal cells via parallel fibers. Finally, pyramidal cells are thought to receive cholinergic muscarinic input from eurydendroid cells within the EGP via vertical fibers. DML, dorsal molecular layer; VML, ventral molecular layer; PCL, pyramidal cell layer; PL, plexiform layer; DNL, deep neuropil layer.
METHODS
In vivo electrophysiology
EXPERIMENTAL SETUP
The weakly electric fish Apteronotus leptorhynchus was used exclusively in this study. Fish were housed in groups of 3–10 in 150-l tanks, water temperature was maintained between 26 and 28°C, and water resistivity varied between 2,000 and 5,000 Ω·cm. Experiments were performed in a 39 × 44 × 12-cm-deep Plexiglass aquarium with water recirculated from the animal’s home tank. Animals were artificially respirated with a continuous water flow of 10 ml/min. Surgical techniques were the same as those described previously (Bastian 1996a,b), and all procedures were in accordance with animal care and use guidelines of McGill University.
Recording
Extracellular recordings from pyramidal neurons were made with metal-filled micropipettes (Frank and Becker 1964). Recording sites as determined from surface landmarks and recording depth were limited to the centrolateral and lateral segments of the ELL only. Extracellular signals were recorded at 10 kHz using a CED 1401 amplifier with spike2 software (Cambridge Electronic Design, Cambridge, UK). Spikes were detected with custom-written software in Matlab (Mathworks, Natick, MA).
STIMULATION
The stimulation protocol was described previously in detail (Bastian et al. 2002). Stimuli consisted of random amplitude modulations (RAMs) of the animal’s own electric organ discharge (EOD) and were generated by multiplying a Gaussian band limited (0–120 Hz, 8th-order Butterworth) white noise with an EOD mimic that consisted of a train of single-cycle sinusoids with frequency slightly higher than that of the EOD and phase locked to the zero-crossings of the animal’s own EOD. The resulting signal was then presented via two silver-silver-chloride electrodes located 19 cm on each side of the animal, giving rise to stimuli that were spatially diffuse (Fig. 1B). Stimulus intensities were similar to those previously used (Bastian et al. 2002; Chacron 2006; Chacron et al. 2005), and typical contrasts used ranged between 5 and 20%. Each stimulus presentation lasted 100 s to obtain sufficient amounts of data. It should be noted that male fish routinely gave behavioral responses (chirps) when the noise stimulus was presented as described previously (Doiron et al. 2003a).
PHARMACOLOGY
Previously established micropressure ejection techniques were used to focally apply drugs within the ELL dorsal molecular layer containing the apical dendritic trees of pyramidal cells (Bastian 1993; Bastian and Nguyenkim 2001; Bastian et al. 2004; Chacron 2006; Chacron et al. 2005). Multibarrel pipettes were pulled to a fine tip and subsequently broken to a total tip diameter of 10 μm. One barrel was filled with a 1 mM solution of carbachol, the other filled with a 1 mM solution of atropine, whereas a third was filled with a 1 mM glutamate solution. After a recording from a well-isolated pyramidal cell was established, the pressure pipette was slowly advanced into an appropriate region of the ELL molecular layer while periodically ejecting “puffs” of glutamate (duration = 100 ms, pressure = 40 psi). As described previously, proximity to a recorded cell will result in short-latency excitation of that cell. After correct placement, carbachol or atropine were then delivered as a series of 10 pulses (duration = 100 ms, pressure = 40 psi). This treatment typically resulted in altered pyramidal cell activity lasting 2–3 min.
DATA ANALYSIS
Responses to random AMs were accumulated as sequences of spike times for each cell. ISI sequences were computed as the time between consecutive spikes and ISI histograms were built with a bin width of 1 ms. We divided the counts by the total count number across all bins times the bin width to obtain the ISI probability density P(I). We obtained a binary sequence X(t) from the spike train sampled at 2 kHz. X(t) and the stimulus S(t) (also sampled at 2 kHz) were then used to estimate the mutual information rate density as: I(f) = −log2[1 − C(f)] (in bit per spike per Hz) (Borst and Theunissen 1999). Here C(f) = |Prs(f)|2/[Pss(f) Prr(f)] is the stimulus-response coherence (Roddey et al. 2000), where Prs denotes the stimulus-response cross spectrum, Pss is the power spectrum of the stimulus, and Prr is the power spectrum of the spike train. All spectral quantities were estimated using multitaper estimation techniques (Jarvis and Mitra 2001). To account for the fact that the mutual information rate density increases with firing rate (Borst and Haag 2001), we normalized it by the mean firing rate fr (measured in spike/s) during stimulation: MI(f) = I(f)/fr. MI(f) is thus measured in bit per spike per Hz. All values are reported as means ± SD.
In vitro electrophysiology
PREPARATION OF SLICES
Fish (A. leptorhynchus) were obtained from local importers and maintained at 26–28°C in fresh water aquaria, in accordance with protocols approved by the McGill University Animal Care Committee. All chemicals were obtained from Sigma (St. Louis, MO) unless otherwise noted. Animals were anesthetized in 0.05% phenoxyethanol, and ELL tissue slices of 300- to 400-μm thickness were prepared as previously described (Turner et al. 1994). Slices were maintained by constant perfusion of artificial cerebrospinal fluid (ACSF, 1–2 ml/min), and superfusion of humidified 95% O2-5% CO2 gas. ACSF contained (in mM) 124 NaCl, 3 KCl, 25 NaHCO3, 1.0 CaCl2, 1.5 MgSO4, and 25 D-glucose, pH 7.4. HEPES-buffered ACSF for pressure ejection of pharmacological agents contained the same elements, with the following differences: 148 mM NaCl, 10 mM HEPES.
RECORDING PROCEDURES
Glass microelectrodes were filled with 2 M KAc (pH 7.4; 90–120 MΩ resistance). Recordings were made from the somata of pyramidal neurons in the centrolateral segment and digitized using a NI PCI-6030E DAQ board (National Instruments, Austin, TX). Intracellular stimuli were delivered, and data were recorded with Clampex 9.0 software (Axon Instruments). Cells were held at a voltage level just below firing threshold. Negative current injections (0.3 nA) were given every 5 s to measure resistance changes during drug application. All drugs [carbachol (1 mM), atropine (1 mM), 4-aminopyridine (4-AP, 1 mM), and TEA (1 mM)] were focally ejected into the dorsal molecular layer (DML) using electrodes of 1-to 2-μm tip diameter and 7–15 psi pressure ejection. Previous studies have estimated a ~10 times dilution factor for ejected toxins (Turner et al. 1994). Pharmacological agents were dissolved in HEPES-buffered ACSF.
DATA ANALYSIS
Data were analyzed using Clampfit 9.0 software (Axon Instruments). Baseline depolarizations were measured as the difference between 1 s membrane potential averages before drug application and after a depolarization plateau had been reached. Burst fractions were the proportion of ISIs that were <10 ms, representing a previously defined burst threshold (Oswald et al. 2004). The ISI histogram (ISIH) was created from the sum of ISI events (bin width = 0.5 ms) from a 1-s depolarization (0.3 nA) for three cells in which carbachol was washed out. The ISIH values were normalized as a fraction of the total event number to account for the increased firing rate after carbachol. Resistance was measured as the average hyperpolarization value 40–50 ms after a negative current injection (0.3 nA). Significance was evaluated using the Student’s t-test (P = 0.05). ISI sequences were computed as the time between consecutive spikes, and ISI histograms were built with a bin width of 1 ms. We divided the counts by the total count number across all bins times the bin width to obtain the ISI probability density P(I). All values are reported as means ± SD.
Model description
We used a previously described two-compartment model of an ELL pyramidal cell (Doiron et al. 2002; Oswald et al. 2004) that contains all the essential elements to reproduce bursting seen experimentally (Doiron et al. 2001; Lemon and Turner 2000). The model neuron is comprised of an isopotential soma (s) and a single dendritic compartment that are joined through an axial resistance of 1/gc (gc: coupling conductance) allowing for the electrotonic diffusion of currents from the soma to dendrite (d) and vice versa. Both compartments contain the essential spiking currents; fast inward Na+ (INa,s, INa,d) and outward delayed rectifying (Dr) K+ (IDr,s, IDr,d), and passive leak currents (Ileak). The presence of spiking currents in the dendrite enables the active backpropagation of somatic action potentials required for bursting. The membrane potentials at the soma, Vs, and the dendrite, Vd, are determined using a Hodgkin–Huxley like formalism (Koch 1999). The original model (Doiron et al. 2002) comprised six nonlinear differential equations. This study expands on this model by incorporating an A-type K+ current, Ia,d, into the dendritic compartment. We note that an A-type potassium current was previously incorporated into a multicompartmental model of pyramidal neuron activity on which the reduced model used here is based (Doiron et al. 2001). The kinetics for the A-type potassium current are qualitatively similar to those used previously. Vs and Vd are described by the following equations
| (1) |
| (2) |
η1(t) and η2(t) are Ornstein-Uhlenbeck processes (Gardiner 1985) to account for sources of intrinsic noise described by
where ξ1,2(t) are independent Gaussian random variables with zero mean and unit SD. Here S(t) is a time varying stimulus which is either low-pass filtered (8th-order Butterworth, 120-Hz cutoff frequency) Gaussian white noise or a sinusoid with a given frequency. The parameter g is a maximal conductance (gmax, mS/cm2), whereas m and s are activation parameters and h, n, and p are inactivation parameters. Each is described by the following equation
| (3) |
where x∞(V) is the infinite conductance curve and τ is the time constant. The infinite conductance curve is modeled as a sigmoid
| (4) |
and the values for τ, V1/2, and k for each current are given in Table 1. Other parameter values are the ratio of somatic to total area: κ = 0.8; the reversal potentials: VNa = 40 mV, VK = −88.5 mV, Vleak = −70 mV; membrane capacitance: Cm = 1 μF/cm2; gleak = 0.18 mS/cm2, and gc = 0.21 mS/cm2; time constant of the noise: τη = 20 ms; intensity of the noise: D = 0.01 (ms)−2. The model equations were integrated using an Euler-Maruyama algorithm with a time step of 0.5 μs.
TABLE 1.
Model parameters
| Current | gmax, mS/cm2 | V1/2, mV | k | τ, m |
|---|---|---|---|---|
| INas[m∞(Vg)] | 35 | −50 | 3 | N/A |
| IDr.s[ng(Vs)] | 15 | −50 | 3 | 0.19 |
| INa,d[md(Vd)/hd(Vd)] | 4 | −40/−52 | 5/−5 | 0.3/1.0 |
| Ia,d[ma(Vd)/ha(Vd)] | 2 | −69/−69 | 4/1 | 1/10 |
| IDr,d[nd(Vd)/Pd(Vd)] | 15 | −40/−65 | 5/−6 | 0.9/3 |
RESULTS
Carbachol increases burst firing in vivo
We obtained extracellular recordings from ELL pyramidal neurons in vivo. The stimulation consisted of random amplitude modulations of the animal’s own electric field that were spatially diffuse, mimicking distortions caused by conspecifics (see Fig. 1B) (Bastian et al. 2002; Chacron 2006; Chacron et al. 2005). To look at the effects of cholinergic input onto pyramidal neurons, we used previously established pharmacological techniques (Bastian 1993; Bastian et al. 2004; Chacron 2006; Chacron et al. 2005) to apply carbachol, a cholinergic receptor agonist, within the ELL dorsal molecular layer (DML) where cholinergic binding sites are located (Phan and Maler 1983). In all cases, this led to an increased firing rate that recovered to values similar to those seen under control conditions (Fig. 2; control: 15.57 ± 6.42 Hz; carbachol: 20.12 ± 9.34 Hz; recovery: 13.56 ± 6.62 Hz). The firing rate under carbachol was significantly higher than the firing rate under control conditions (P = 0.005, paired t-test, n = 14). For the cells that were followed to recovery, the firing rate after recovery was not significantly different from the firing rate under control conditions (P = 0.35, paired t-test, n = 6).
FIG. 2.

Carbachol leads to increased cellular excitability in vivo. A: extracellular recording of spontaneous activity of an electrosensory lateral line lobe (ELL) pyramidal neuron. B: increased firing in response to application of carbachol. C: carbachol leads to a significant increase in the average firing rate (control: 15.57 ± 6.42 Hz; carbachol 20.12 ± 9.34 Hz; P = 0.005, paired t-test, n = 14), which returns to control levels after recovery period (13.56 ± 6.62 Hz, P = 0.35, paired t-test, n = 6).
The increased firing rate was accompanied by an increased fraction of high-frequency ISIs (<10 ms; Fig. 3A). It has been shown previously that ISIs with a value <10 ms were primarily caused by an intrinsic burst mechanism (Doiron et al. 2003b; Oswald et al. 2004). We thus computed the burst fraction (i.e., the fraction of ISIs <10 ms) as done previously (Oswald et al. 2004; see METHODS). On average, carbachol application more than doubled the burst fraction with respect to values under control conditions (Fig. 3C; control: 0.11 ± 0.10; carbachol: 0.24 ± 0.15, P = 0.001, paired t-test, n = 14). For the cells that were followed to recovery, the burst fraction values after recovery were not significantly different from those obtained under control conditions (Fig. 3C; recovery: 0.07 ± 0.08, P = 0.82, paired t-test, n = 6).
FIG. 3.

Muscarinic receptor activation leads to an increase in burst firing in vivo. A: interspike interval (ISI) histograms before (blue), during (red), and after (green) application of carbachol. Carbachol leads to an increase in ISIs <10 ms. B: ISI histograms before (blue), during (red), and after (green) application of carbachol in the presence of atropine. Atropine prevents the effect of carbachol revealing muscarinic receptor activation by carbachol. C: bar graph representing the average increase of the burst fraction (fraction of ISIs <10 ms; control: 0.11 ± 0.10; carbachol: 0.24 ± 0.15, P = 0.001, paired t-test, n = 14) after carbachol and return to control levels after recovery period. (D) Atropine prevents an increase in burst fraction for the population of cells tested.
Because carbachol can activate both nicotinic and muscarinic AChRs, we applied atropine, a muscarinic acetylcholine receptor (mAChR) antagonist, to determine which receptor subtype was primarily activated. Atropine alone did not affect the firing rate (P = 0.56, paired t-test, n = 5) or the burst fraction (compare C and D of Fig. 3, P = 0.65, paired t-test, n = 5) of the pyramidal neurons. Atropine did, however, prevent the carbachol from having an effect on the firing rate (control: 12.96 ± 3.35 Hz; carbachol following atropine: 11.58 ± 3.58 Hz; P = 0.21, paired t-test, n = 7), the ISI distribution (Fig. 3B), or the burst fraction (Fig. 3D; control: 0.14 ± 0.13; carbachol following atropine: 0.13 ± 0.13; P = 0.72, paired t-test, n = 5).
Muscarinic receptor activation alters pyramidal cell tuning in vivo
To quantify the effects of mAChR activation on pyramidal neuron responses to sensory input, we used information theory (Cover and Thomas 1991; Shannon 1948) to compute the mutual information rate density between the sensory stimulus and the pyramidal cell’s spike train in response to that stimulus (Borst and Theunissen 1999). The mutual information rate density is normalized by the neuron’s firing rate and is measured in bit·s−1·Hz−1: thereby quantifying the information transmitted by each action potential as a function of frequency per unit time. Application of carbachol primarily increased the information rate density at low frequencies (Fig. 4A). We computed Mlow, the average mutual information rate density for the frequency range 0–20 Hz (Chacron et al. 2003, 2005) to assess the cell’s low-frequency response and found that carbachol led to a significant increase in Mlow (control: 0.004 ± 0.001 bit per spike per Hz; carbachol: 0.006 ± 0.001; P = 0.009, paired t-test, n = 14). There was no significant change overall in the response to high-frequency stimuli (Fig. 4B) as quantified by Mhigh, the average mutual information rate density for the frequency range 40–60 Hz (P = 0.52, paired t-test, n = 14). Activation of cholinergic input unto pyramidal cells can thus increase their response to low-frequency sensory stimuli.
FIG. 4.
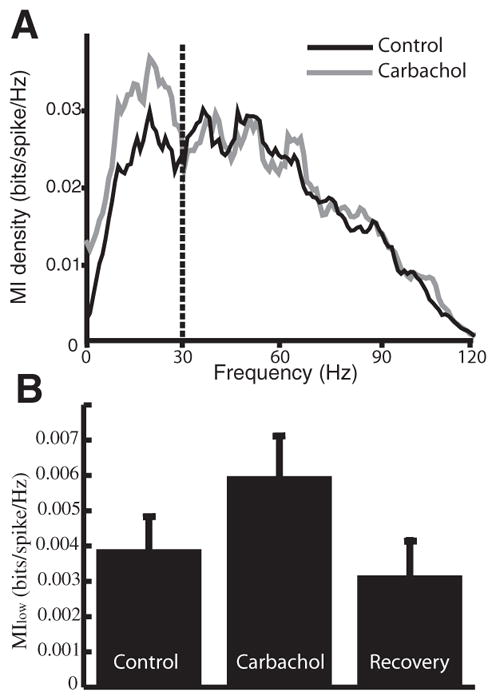
Muscarinic receptor activation increases the response to low-frequency sensory stimuli in vivo. A: mutual information rate density showing an increased response to frequencies <30 Hz. B: bar graph showing the average mutual information rate density for low frequencies (Mlow). Mlow increased after carbachol application and then returned to control levels (control: 0.004 ± 0.001 bits·spike−1·Hz−1; carbachol: 0.006 ± 0.001; P = 0.009, paired t-test, n = 14).
Application of carbachol in vitro increases bursting
mAChR activation has been found to alter the firing properties of individual neurons through the modulation of a number of individual ionic conductances (Akins et al. 1990; Chen and Johnston 2004; Delgado-Lezama et al. 1997; Mittmann and Alzheimer 1998; Nakajima et al. 1986; Stocker et al. 1999; Svirskis and Hounsgaard 1998; Tai et al. 2006). To determine if the effects of mAChR activation resulted from the regulation of one or several ion channels, we used an in vitro ELL slice preparation (Turner et al. 1994, 1996).
Intracellular sharp electrode recordings were obtained from ELL pyramidal neurons. Most of the pyramidal neurons recorded from spontaneously fired action potentials at their resting membrane potential. To silence cell firing, negative current injections were used to hold the membrane potential below the firing threshold (average holding potential: −73 ± 6 mV; n = 13). Carbachol (1 mM) was applied to the DML and this resulted on average in a 3.8 ± 1.8-mV depolarization of the membrane potential (P < 0.001; paired t-test; n = 20; Fig. 5A), which often caused the action potential threshold to be reached. Application of atropine (1 mM) prior to carbachol prevented this depolarization (Fig. 5B; −76.5 ± 4.4 to −77.2 ± 4.2 mV; P = 0.11; paired t-test; n = 4), suggesting the effect is due in large part to mAChR activation. When applied on its own atropine had no effect on the membrane properties (data not shown). Step current injections revealed an increase in firing rate following carbachol (Fig. 5C, control: 39 ± 14; carbachol: 67 ± 23 Hz; P < 0.001; paired t-test; n = 13) as well as an increase in burst fraction (Fig. 5D; 0.26 ± 0.29 to 0.39 ± 0.26; P < 0.01; paired t-test; n = 13). The effects of carbachol on the in vitro slice preparations were thus found to be qualitatively similar to those obtained in vivo as, in both cases, carbachol lead to increased spiking and bursting activity and its effects were prevented by atropine.
FIG. 5.

Carbachol activates muscarinic receptors in vitro. A: carbachol leads to a −4-mV depolarization of the membrane potential from a subthreshold level leading to spiking (spikes truncated at −35 mV). B: atropine eliminates the effects of carbachol, suggesting mAChR activation. C: ISI histogram showing an increase in ISI’s <10 ms after carbachol application. D: significant increase in the burst fraction (fraction of ISIs <10 ms) after carbachol application. E: carbachol also leads to an increased membrane resistance. F: carbachol in the presence of atropine does not increase the membrane resistance.
The membrane depolarization caused by carbachol application in vitro suggests an effect on subthreshold membrane properties, which could be mediated by regulation of one or multiple ion channels. The effects of carbachol were most likely not due to voltage-gated Ca2+, high-threshold K+, or Ca2+-activated K+ channels because these channels are not typically active at these potentials. To further characterize the changes caused by carbachol, negative current pulses (0.3 nA) were applied at regular intervals to measure subthreshold membrane resistance changes in response to drug application. Carbachol increased the membrane resistance on average by 37% (Fig. 5E; control: 15.7 ± 7.1 MΩ; carbachol: 20.1 ± 7.7 MΩ; P < 0.001; paired t-test; n = 12), suggesting the down-regulation of an outward current. The carbachol effect is then unlikely to be mediated by subthreshold inward currents such as persistent sodium.
It was then hypothesized that subthreshold K+ currents were responsible for the effects of carbachol. The negative current injections showed no evidence of a depolarizing rectification characteristic of H-type currents (Ih) (Maccaferri et al. 1993), and previous studies have been unable to demonstrate the presence of H-type potassium currents in pyramidal neurons. However, evidence does exist for an A-type K+ current (IA) in pyramidal neurons (Berman and Maler 1999; Mathieson and Maler 1988), which lead us to speculate that a downregulation of IA was responsible for the effects observed.
Downregulation of a 4-AP-sensitive K+ current by muscarinic input
To determine if the effects of mAChR activation were due to the downregulation of an A-type current, we made use of the potassium channel antagonist 4-AP. Previous results have shown that 4-AP can block the Kv1 and Kv4 channels linked to A-type currents (Coetzee et al. 1999), and 4-AP has already been shown to alter the firing properties of ELL pyramidal neurons (Mathieson and Maler 1988). If mAChR activation downregulates IA, then 4-AP should result in similar changes to the subthreshold membrane kinetics as those seen following carbachol application. When 4-AP (1 mM) was applied to the DML, the membrane potential was depolarized (Fig. 6A; 2.8 ± 0.8 mV; P < 0.001; paired t-test; n = 5) from a holding potential just below threshold (−73 ± 2 mV). Application of 4-AP also increased the resistance, on average, by 48% (Fig. 6B; 16.9 ± 8.9 to 23.5 ± 10.7 MΩ; P = 0.015; paired t-test; n = 7). Neither of the changes after 4-AP were statistically different from the changes seen after carbachol (depolarization P = 0.11; resistance P = 0.30; t-test). Because 4-AP can block a number of K+ channels not typically associated with IA, we also used TEA to evaluate the effects of blocking these channels (Coetzee et al. 1999). TEA (1 mM) had no significant effect on the holding potential (Fig. 6C; control: −74.0 ± 1.2 mV; TEA: −74.1 ± 1.5 mV; P = 0.36; paired t-test; n = 8) or the membrane resistance (Fig. 6D; control: 19.8 ± 8.9 MΩ; TEA: 19.3 ± 9.7 MΩ; P = 0.38; paired t-test; n = 7). However, when carbachol was applied after TEA, a depolarization leading to firing still occurred (2.8 ± 2.0 mV, P = 0.001; paired t-test; n = 7) along with an average 40% increase in membrane resistance (control: 17.5 ± 7.0 MΩ; TEA+carbachol: 23.8 ± 8.1 MΩ; P = 0.002; paired t-test, n = 5). These effects were not statistically different from those obtained with carbachol application alone (membrane depolarization P = 0.22; resistance P = 0.12; t-test). Importantly when 4-AP was applied after carbachol, it did not lead to a further depolarization or change in membrane resistance when the membrane potential was reset to precarbachol values (−73.1 ± 2.5 to −72.8 ± 2.6; P = 0.18; 25.8 ± 3.8 to 25.6 ± 3.9 MΩ; P = 0.35; n = 7). The similarities between the effects of carbachol and 4-AP suggest that the effects of mAChR activation by carbachol are mediated by the downregulation of K+ channels that are blocked by 4-AP and not TEA, such as those linked to IA.
FIG. 6.

A potassium channel blocker can mirror the effects of carbachol. A and B: potassium channel antagonist 4-aminopyridine (4-AP) leads to a depolarization of the membrane potential (A) and an increase in resistance (B). C and D: potassium channel blocker TEA does not affect either the membrane potential (C) or resistance (D).
In ELL pyramidal neurons, the 4-AP-sensitive A-type K+ current was previously shown to control the first spike latency following a step depolarization (Mathieson and Maler 1988). The latent period is controlled by the membrane potential value preceding the depolarization as more hyperpolarized levels will remove the inactivation of IA and thereby increase first spike latency (Connor and Stevens 1971; McCormick 1991; Schoppa and Westbrook 1999). Similar control of first spike latency was shown for ELL pyramidal neurons (Mathieson and Maler 1988).
As such, we investigated whether 4-AP, TEA, or carbachol had effects on first spike latency. The membrane potential was set at the same value (typically −73 mV) both before and after drug application. Carbachol and 4-AP each led to a similar reduction in first spike latency after a step depolarization (Fig. 7A: control: 44.4 ± 22.6 ms; carbachol: 10.4 ± 4.4 ms; P < 0.001; paired t-test; n = 11; B: control: 49.1 ± 19.4 ms; 4-AP: 19.4 ± 10.5 ms; P < 0.001; paired t-test; n = 7). Conversely, first spike latency was not affected by TEA (Fig. 7C; control: 37.4 ± 21.9 ms; TEA: 43.2 ± 34.9 ms; P = 0.13; paired t-test; n = 8) but could be reduced by a subsequent application of carbachol (Fig. 7D; control: 29.4 ± 19.4 ms; TEA+carbachol: 10.8 ± 8.7 ms; P = 0.01; paired t-test; n = 5). These results support the hypothesis that mAChR activation leads to an inactivation of IA.
FIG. 7.

Carbachol and 4-AP lead to a decreased 1st spike latency, suggesting the involvement of an A-type current. A: carbachol leads to a reduced spike latency after a step current injection (0.2 nA). B: 4-AP mirrors the effect of carbachol reducing 1st spike latency in a representative cell. C: TEA does not alter the spike latency. D: carbachol decreases latency even when applied after TEA. E: bar graphs showing a significant decrease in latency after carbachol and 4-AP (control: 44.4 ± 22.6 ms; carbachol: 10.4 ± 4.4 ms; P < 0.001; paired t-test; n = 11: control: 49.1 ± 19.4 ms; 4-AP: 19.4 ± 10.5 ms; P < 0.001; paired t-test; n = 7), whereas TEA did not show a significant decrease in latency (control: 37.4 ± 21.9 ms; TEA: 43.2 ± 34.9 ms; P = 0.13; paired t-test; n = 8).
Modeling the in vitro effects of IA
Because there is not a specific regulator of IA and, as mentioned, 4-AP is a nonspecific potassium channel antagonist, it was important to determine if a downregulation of IA is sufficient to cause the effects after carbachol application. We subsequently incorporated an A-type current into a two-compartment numerical model that was previously developed for ELL pyramidal neurons (Doiron et al. 2002; Oswald et al. 2004). We mimicked the effects of carbachol and 4-AP in the model by setting the maximum conductance of the A-type current to zero from its control value (2 mS/cm2). This resulted in a ~5-mV depolarization of the membrane potential (holding potential = −73 mV) that induced spiking (Fig. 8A). The membrane resistance was larger by 33% without IA, similar to the effects obtained in vitro with carbachol or 4-AP application (compare Figs. 8B with 5E and 6B). We also obtained a greater first spike latency following a step depolarization with IA present (Fig. 8, C and D; with IA: 79.26 ± 15.64 ms; without IA: 11.34 ± 5.56 ms). Finally, the model neuron had a much greater tendency to burst without IA (Fig. 8E). This was quantified by the burst fraction (Fig. 8F), which was much greater without IA (with IA: 0.02; without IA: 0.35). Therefore our results show that the removal of an IA current in a model pyramidal cell can account for all the effects observed with carbachol and 4-AP application in vitro.
FIG. 8.

Modeling the effects of blocking A-type channels. A: removal of IA leads to a ~5-mV depolarization and spiking. At t = 30 ms, we set gA from 2 mS/cm2 to 0. We had I0 = 1.3. B: step hyperpolarization with gA = 2 mS/cm2 (black) and gA = 0 (gray) showing a 33% increase in resistance with D = 0. This was obtained by setting I0 from 1.375 to 0.875. C: step depolarizations with (black, gA = 2 mS/cm2) and without (gray, gA = 0)IA. At t = 130 ms, we increased I0 from 0.5 to 1.7 (gray) and from 1 to 2.3 (black). D: mean latencies obtained under repeated step depolarizations as described in C. E: ISI probability densities obtained under repeated step depolarizations as described in C. F: burst fraction obtained from the ISI probability. Note that S(t) = 0 for all panels.
To understand how IA affects signal transmission, we used noise current injections in the model to mimic sensory stimulation in vivo as done previously (Oswald et al. 2004). It was found that IA significantly reduced the model neuron’s response to low-frequency input, consistent with the in vivo results (Fig. 9A). To understand this effect, we used sinusoidal current injections of different frequencies. For 1 Hz, it was found that removal of IA had little effect on the firing rate (Fig. 9, B and C). This can be understood as follows: during the negative portion of the sinusoid, the membrane is hyperpolarized, and this will tend to de-inactivate IA; however, because of the large period (1 s), IA will subsequently inactivate before the onset of spiking, resulting in no net effect. In contrast, removal of IA significantly increased the firing rate for a 5-Hz sinusoid (Fig. 9D). In this case, IA does not fully inactivate during the shorter depolarizing portion of the sinusoid and thus can have a significant effect on firing rate. Finally, removal of IA had little effect on the firing rate for a 40-Hz sinusoid (Fig. 9E). In this case, the short hyperpolarization is unlikely to de-inactivate the A-type channels. A plot of the model neuron’s firing rate with and without IA as a function of frequency revealed an effect for frequencies contained between 1 and 40 Hz (Fig. 9F).
FIG. 9.

Modeling the effects of A-type channels on signal processing. A: mutual information density with (black) and without (gray) IA. Removal of IA leads to increased information at low (<20 Hz) frequencies. In both cases, we injected 0 mean low-pass filtered (8th-order butterworth, 120-Hz cutoff) Gaussian white noise with SD 0.5. Other parameters were the same as in Fig. 7 except I0 = 0.7, gA = 0 (gray), and I0 = 2, gA = 2 mS/cm2 (black). B: membrane potential with IA and a 1-Hz sinusoidal current injection. C: membrane potential without IA and a 1-Hz sinusoidal current injection. D: membrane potential with (black) and without (gray) IA with a 5-Hz sinusoidal current injection. E: membrane potential with (black) and without (gray) IA with a 40-Hz sinusoidal current injection. F: firing rate as a function of the sinusoidal current’s frequency with (black) and without (gray) IA. For all simulations with sinusoidal current, we had I0 = 0.4 (black) and I0 = 1.4 (gray) as well as D = 0 and the sinusoid’s amplitude was set to 2 for all frequencies.
Our modeling results have thus shown that A-type currents can have significant effects on the tuning of neurons in the low-frequency range. As such, our modeling results provide an explanation for why mAChR activation in vivo increased the cell’s response to low-frequency input only.
DISCUSSION
We have shown that activation of muscarinic receptors can significantly alter the response of ELL pyramidal neurons to sensory input. In vivo application of the cholinergic agonist carbachol increased pyramidal neuron excitability, leading to an increase in spiking and bursting activity. Moreover, it was found that carbachol application onto pyramidal neurons increased the transmission of the low-frequency components of an external electrosensory signal. The effects of carbachol could be prevented by the muscarinic antagonist atropine, suggesting carbachol was primarily activating muscarinic acetylcholine receptors (mAChR).
To understand the cellular mechanisms that mediate these altered responses, we investigated the consequences of cholinergic receptor activation in vitro. We found that carbachol application also increased pyramidal cell excitability and burst firing and that these effects were also prevented by atropine. Activation of mAChRs in vitro led to a ~4-mV membrane depolarization from a level below threshold accompanied by an increase in the subthreshold membrane resistance. These results suggested that cholinergic receptor activation downregulates an outward current that is active in the subthreshold regime. Because the presence of an A-type current was previously shown in ELL pyramidal neurons (Mathieson and Maler 1988), we set out to determine if this current was being down regulated by mAChR activation. It was found that the K+ channel antagonist 4-AP had effects on the subthreshold membrane kinetics similar to those of carbachol. Furthermore, carbachol could prevent the subthreshold effects of 4-AP, suggesting that mAChR activation downregulates 4-AP-sensitive channels. Because 4-AP is a nonspecific potassium channel blocker, we also used the K+ channel blocker TEA to confirm that the effects of carbachol were confined to the K+ channels that are known to mediate A-type currents. TEA had no significant effect on either the holding potential or the membrane resistance, whereas application of carbachol in the presence of TEA led to changes in holding potential and membrane resistance that were comparable to those seen with carbachol application alone. Because previous studies had shown that blocking A-type currents with 4-AP could have a significant effect on first spike latency (Mathieson and Maler 1988), we measured the effects of carbachol and 4-AP on first spike latency. We confirmed the effect of 4-AP previously shown and additionally demonstrated that carbachol could lead to similar decreases in first spike latency, whereas TEA had no significant effect.
Control of a neuron’s frequency tuning by A-type currents
We used a two-compartmental mathematical model to assess if the removal of an A-type current could replicate the effects of carbachol. The model showed that the removal of an A-type current can result in similar changes in burst firing, holding potential, membrane resistance, and first spike latency as those seen under experimental activation of mAChRs, supporting the in vitro findings (Fig. 8). Additionally it was shown that when the model was presented with a noise current injection to mimic sensory stimulation in vivo, removal of IA resulted in an increased response to low-frequency input, which was similar to that seen after carbachol application in vivo. To understand this effect, we used sinusoidal current injections. It was shown that for very low frequencies (<1 Hz) removal of IA had no effect on neuronal response properties. We propose that this is due to the length of the depolarizing phase of the sine wave, which should result in a complete inactivation of IA by the time spike threshold is reached, thus eliminating its effect. Further, it was shown that at frequencies >40 Hz, the removal of IA again had no effect because the period of hyperpolarization is insufficient to de-inactivate the A-type channels. As such, our modeling results suggest that the downregulation of A-type potassium channels is sufficient to explain the increase in the low-frequency tuning, in the range between 1 and 40 Hz, of pyramidal neurons found in vivo after pharmacological activation of mAChRs.
Pyramidal neuron’s response to low frequencies is behaviorally relevant
We have shown that mAChR activation leads to an increased pyramidal neuron response to the low temporal frequency components of sensory input. A number of natural sensory stimuli contain low temporal frequencies. The jamming avoidance response (JAR) (reviewed in Heiligenberg 1991) is a well-described behavior that is triggered by low temporal frequency, spatially diffuse electrosensory stimuli. Specifically, the JAR occurs when a fish encounters a conspecific with an EOD frequency close to its own. These signals can interfere with electrolocation if the frequency difference is <8 Hz (Heiligenberg 1991). The fish with the highest EOD frequency increases its EOD frequency until the signals will interfere less with electrolocation. However, previous in vivo studies have shown that pyramidal neurons generally display poor responses to low-frequency global stimuli (Bastian et al. 2002, 2004; Chacron 2006; Chacron et al. 2003, 2005). Our results show that cholinergic input has the capacity to make pyramidal neurons more responsive these stimuli.
Alternatively, prey can also cause low-frequency stimuli. However, these signals are more spatially localized (Nelson and Maciver 1999). It is therefore also possible that the cholinergic pathway can increase the response of pyramidal neurons to prey stimuli, thereby improving the animal’s prey detection abilities. Although further studies are needed to understand how and when cholinergic input becomes active, it is likely descending input from a higher brain area (see following text) which can be activated in response to behaviorally relevant stimuli.
Muscarinic control of the A-type current
A number of higher order functions can be controlled by cholinergic input including, but not limited to, attention (Bucci et al. 1998; Voytko et al. 1994), learning (Fine et al. 1997; Miranda and Bermudez-Rattoni 1999), and memory (Hasselmo et al. 1992; Sarter et al. 2005). These types of modulations often elevate sensory responsiveness and occur when a cholinergic pathway is activated in conjunction with an external sensory stimulus. An example of such control occurs in the somatosensory cortex where a stimulus response can be enhanced by stimulating descending cholinergic pathways from the cortex during the presentation of an external cue (Donoghue and Carroll 1987; Rasmusson and Dykes 1988). In the visual system, cholinergic input can lead to potentiation through an enhancement of the signal to noise ratio (Gu 2003).
In the hippocampus, LTP may be facilitated when a back-propagating spike is paired with an EPSP (Magee and Johnston 1997). It has been shown that the downregulation of an A-type current can increase the backpropagation of dendritic spikes (Hoffman et al. 1997). It is likely that the downregulation of IA can lead to an increase in the strength of the backpropagating spike and thus facilitate LTP, when paired with an EPSP as described in the preceding text. This may be one mechanism through which IA can regulate higher order processes, such as learning. The dendritic modification of IA by mAChRs that we have shown here results in an increased response to low-frequency input through an increase in bursting. The reduction of IA may result in an increase in backpropagation of dendritic spikes in a similar way to that described for the facilitation of LTP. The backpropagation of dendritic spikes has been shown to control bursting in ELL pyramidal neurons (Lemon and Turner 2000; Turner et al. 1994), and because bursts are strong encoders of low frequencies in this system (Oswald et al. 2004), control of backpropagation by IA may be the mechanism whereby the frequency response is modulated. In fact mathematical modeling has suggested that the removal of a low-threshold potassium current (IKLT) from pyramidal neurons has the same effect as decreasing the size of a sodium spike (Fernandez et al. 2005a), which has been shown to modulate burst frequency (Fernandez et al. 2005b).
Downregulation of A-type currents by cholinergic receptor activation has already been demonstrated in other systems such as the hippocampus (Nakajima et al. 1986) and neostriatum (Akins et al. 1990; Nakamura et al. 1997). Activation of M1 receptors leads to an increase in protein kinase C (PKC) that can lead to a decrease in Kv4-generated currents. The kinetics of Kv4 type channels suggest they can generate an A-type current (Coetzee et al. 1999; Song 2002). Interestingly it has also been shown that PKC may modulate IA through a positive shift in the activation curve (Hoffman and Johnston 1998) instead of the direct reduction in IA that was previously described. In weakly electric fish, it has been suggested that a positive shift in the IA activation curve can prevent its influence on bursting (Fernandez et al. 2005a). It then appears that regardless of whether IA is directly reduced or if the activation curve is shifted, mAChR modulation may produce the alterations in bursting described in this study through the activation of a PKC pathway.
Cholinergic input pathway
Cholinergic input unto pyramidal cells most likely originates from eurydendroid cells within the caudal lobe of the cerebellum (Berman and Maler 1999; Maler et al. 1981; Mathieson and Maler 1988; Phan and Maler 1983). The circuitry within the caudal lobe of the cerebellum is similar to that of the deep cerebellar nuclei in mammalian systems (Finger 1978). Although the general morphology (Guest 1983) and some of the cellular projections (Carr et al. 1986) of eurydendroid cells have been investigated, the sensory responsiveness of this cell type is unknown and may be activated by input from higher brain centers similar to the cholinergic pathways in mammalian systems. The results presented here make the analysis of this input pathway a critical component required to understand its effects on sensory processing.
A recent review has highlighted the similarities between ELL pyramidal neurons and LGN relay cells in terms of burst firing (Krahe and Gabbiani 2004). LGN relay cells have a well-characterized burst mechanism (Sherman 2001) and in the LGN bursts transmit information about low-frequency stimuli (Lesica and Stanley 2004). The results presented here along with previous work showing an increase in burst firing can increase a neurons response to low frequencies (Oswald et al. 2004) are consistent with the results from the LGN. Because A-type potassium currents are present in LGN relay cells (Budde et al. 1992) and because acetylcholine has been shown to increase their excitability (Kemp and Sillito 1982; Sillito et al. 1983), it is possible that regulation of burst firing by A-type channels regulates information transmission in the LGN in a manner similar to that observed in ELL.
Overall, control of frequency tuning through the regulation of A-type currents is likely to be found in other systems and may be a general mechanism by which neural responses to sensory input are regulated. Enhanced processing of specific sensory information through increases in excitability mediated by the downregulation of an IA current may thus be a general feature of sensory processing regulating higher cognitive functions such as attention in mammals.
Acknowledgments
We thank J. Bastian for useful discussions.
GRANTS
This research was supported by funding from the National Science and Engineering Research Council to R. Krahe and Canadian Institute of Health Research to C. W. Bourque, R. J. Dunn, and M. J. Chacron.
References
- Abbott LF. Where are the switches on this thing? In: van Hemmen JL, Sejnowski TJ, editors. 23 Problems in Systems Neuroscience. New York: Oxford; 2005. pp. 423–431. [Google Scholar]
- Akins PT, Surmeier DJ, Kitai ST. Muscarinic modulation of a transient K+ conductance in rat neostriatal neurons. Nature. 1990;344:240–242. doi: 10.1038/344240a0. [DOI] [PubMed] [Google Scholar]
- Bakin JS, Weinberger NM. Induction of a physiological memory in the cerebral cortex by stimulation of the nucleus basalis. Proc Natl Acad Sci USA. 1996;93:11219–11224. doi: 10.1073/pnas.93.20.11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastian J. Electrolocation. I. How the electroreceptors of Apteronotus albifrons code for moving objects and other electrical stimuli. 1981;144:465–479. [Google Scholar]
- Bastian J. The role of amino acid neurotransmitters in the descending control of electroreception. J Comp Physiol [A] 1993;172:409–423. doi: 10.1007/BF00213523. [DOI] [PubMed] [Google Scholar]
- Bastian J. Plasticity in an electrosensory system. I. General features of a dynamic sensory filter. J Neurophysiol. 1996a;76:2483–2496. doi: 10.1152/jn.1996.76.4.2483. [DOI] [PubMed] [Google Scholar]
- Bastian J. Plasticity in an electrosensory system. II. Postsynaptic events associated with a dynamic sensory filter. J Neurophysiol. 1996b;76:2497–2507. doi: 10.1152/jn.1996.76.4.2497. [DOI] [PubMed] [Google Scholar]
- Bastian J, Chacron MJ, Maler L. Receptive field organization determines pyramidal cell stimulus-encoding capability and spatial stimulus selectivity. J Neurosci. 2002;22:4577–4590. doi: 10.1523/JNEUROSCI.22-11-04577.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastian J, Chacron MJ, Maler L. Plastic and nonplastic pyramidal cells perform unique roles in a network capable of adaptive redundancy reduction. Neuron. 2004;41:767–779. doi: 10.1016/s0896-6273(04)00071-6. [DOI] [PubMed] [Google Scholar]
- Bastian J, Nguyenkim J. Dendritic modulation of burst-like firing in sensory neurons. J Neurophysiol. 2001;85:10–22. doi: 10.1152/jn.2001.85.1.10. [DOI] [PubMed] [Google Scholar]
- Berman NJ, Maler L. Distal versus proximal inhibitory shaping of feedback excitation in the electrosensory lateral line lobe: implications for sensory filtering. J Neurophysiol. 1998a;80:3214–3232. doi: 10.1152/jn.1998.80.6.3214. [DOI] [PubMed] [Google Scholar]
- Berman NJ, Maler L. Inhibition evoked from primary afferents in the electrosensory lateral line lobe of the weakly electric fish (Apteronotus leptorhynchus) J Neurophysiol. 1998b;80:3173–3196. doi: 10.1152/jn.1998.80.6.3173. [DOI] [PubMed] [Google Scholar]
- Berman NJ, Maler L. Interaction of GABAB-mediated inhibition with voltage-gated currents of pyramidal cells: computational mechanism of a sensory searchlight. J Neurophysiol. 1998c;80:3197–3213. doi: 10.1152/jn.1998.80.6.3197. [DOI] [PubMed] [Google Scholar]
- Berman NJ, Maler L. Neural architecture of the electrosensory lateral line lobe: adaptations for coincidence detection, a sensory searchlight and frequency-dependent adaptive filtering. J Exp Biol. 1999;202:1243–1253. doi: 10.1242/jeb.202.10.1243. [DOI] [PubMed] [Google Scholar]
- Borst A, Haag J. Effects of mean firing on neural information rate. J Comput Neurosci. 2001;10:213–221. doi: 10.1023/a:1011225232120. [DOI] [PubMed] [Google Scholar]
- Borst A, Theunissen FE. Information theory and neural coding. Nat Neurosci. 1999;2:947–957. doi: 10.1038/14731. [DOI] [PubMed] [Google Scholar]
- Bucci DJ, Holland PC, Gallagher M. Removal of cholinergic input to rat posterior parietal cortex disrupts incremental processing of conditioned stimuli. J Neurosci. 1998;18:8038–8046. doi: 10.1523/JNEUROSCI.18-19-08038.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budde T, Mager R, Pape HC. Different types of potassium outward current in relay neurons acutely isolated from the rat lateral geniculate nucleus. Eur J Neurosci. 1992;4:708–722. doi: 10.1111/j.1460-9568.1992.tb00180.x. [DOI] [PubMed] [Google Scholar]
- Carr CE, Maler L, Taylor B. A time-comparison circuit in the electric fish midbrain. II. Functional morphology. J Neurosci. 1986;6:1372–1383. doi: 10.1523/JNEUROSCI.06-05-01372.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacron MJ. Nonlinear information processing in a model sensory system. J Neurophysiol. 2006;95:2933–2946. doi: 10.1152/jn.01296.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacron MJ, Doiron B, Maler L, Longtin A, Bastian J. Non-classical receptive field mediates switch in a sensory neuron’s frequency tuning. Nature. 2003;423:77–81. doi: 10.1038/nature01590. [DOI] [PubMed] [Google Scholar]
- Chacron MJ, Maler L, Bastian J. Feedback and feedforward control of frequency tuning to naturalistic stimuli. J Neurosci. 2005;25:5521–5532. doi: 10.1523/JNEUROSCI.0445-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Johnston D. Properties of single voltage-dependent K+ channels in dendrites of CA1 pyramidal neurones of rat hippocampus. J Physiol. 2004;559:187–203. doi: 10.1113/jphysiol.2004.068114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, McCormack T, Moreno H, Nadal MS, Ozaita A, Pountney D, Saganich M, Vega-Saenz dM, Rudy B. Molecular diversity of K+ channels. Ann NY Acad Sci. 1999;868:233–285. doi: 10.1111/j.1749-6632.1999.tb11293.x. [DOI] [PubMed] [Google Scholar]
- Connor JA, Stevens CF. Voltage clamp studies of a transient outward membrane current in gastropod neural somata. J Physiol. 1971;213:21–30. doi: 10.1113/jphysiol.1971.sp009365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cover T, Thomas J. Elements of Information Theory. New York: Wiley; 2001. [Google Scholar]
- Delgado-Lezama R, Perrier JF, Nedergaard S, Svirskis G, Hounsgaard J. Metabotropic synaptic regulation of intrinsic response properties of turtle spinal motoneurons. J Physiol. 1997;504:97–102. doi: 10.1111/j.1469-7793.1997.097bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doiron B, Chacron MJ, Maler L, Longtin A, Bastian J. Inhibitory feedback required for network oscillatory responses to communication but not prey stimuli. Nature. 2003a;421:539–543. doi: 10.1038/nature01360. [DOI] [PubMed] [Google Scholar]
- Doiron B, Laing C, Longtin A, Maler L. Ghostbursting: a novel neuronal burst mechanism. J Comput Neurosci. 2002;12:5–25. doi: 10.1023/a:1014921628797. [DOI] [PubMed] [Google Scholar]
- Doiron B, Longtin A, Turner RW, Maler L. Model of gamma frequency burst discharge generated by conditional backpropagation. J Neurophysiol. 2001;86:1523–1545. doi: 10.1152/jn.2001.86.4.1523. [DOI] [PubMed] [Google Scholar]
- Doiron B, Noonan L, Lemon N, Turner RW. Persistent Na+ current modifies burst discharge by regulating conditional backpropagation of dendritic spikes. J Neurophysiol. 2003b;89:324–337. doi: 10.1152/jn.00729.2002. [DOI] [PubMed] [Google Scholar]
- Donoghue JP, Carroll KL. Cholinergic modulation of sensory responses in rat primary somatic sensory cortex. Brain Res. 1987;408:367–371. doi: 10.1016/0006-8993(87)90407-0. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW. Central cholinergic systems and cognition. Annu Rev Psychol. 1997;48:649–684. doi: 10.1146/annurev.psych.48.1.649. [DOI] [PubMed] [Google Scholar]
- Fernandez FR, Mehaffey WH, Molineux ML, Turner RW. High-threshold K+ current increases gain by offsetting a frequency-dependent increase in low-threshold K+ current. J Neurosci. 2005a;25:363–371. doi: 10.1523/JNEUROSCI.3950-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez FR, Mehaffey WH, Turner RW. Dendritic Na+ current inactivation can increase cell excitability by delaying a somatic depolarizing afterpotential. J Neurophysiol. 2005b;94:3836–3848. doi: 10.1152/jn.00653.2005. [DOI] [PubMed] [Google Scholar]
- Fine A, Hoyle C, Maclean CJ, Levatte TL, Baker HF, Ridley RM. Learning impairments following injection of a selective cholinergic immunotoxin, ME20.4 IgG-saporin, into the basal nucleus of Meynert in monkeys. Neuroscience. 1997;81:331–343. doi: 10.1016/s0306-4522(97)00208-x. [DOI] [PubMed] [Google Scholar]
- Finger TE. Efferent neurons of the teleost cerebellum. Brain Res. 1978;153:608–614. doi: 10.1016/0006-8993(78)90346-3. [DOI] [PubMed] [Google Scholar]
- Frank K, Becker MC. Physical Techniques in Biological Research. part A. New York: Academic; 1964. Microelectrodes for recording and stimulation; pp. 23–84. [Google Scholar]
- Gardiner CW. Handbook of Stochiastic Methods. Berlin: Springer; 1985. [Google Scholar]
- Gu Q. Contribution of acetylcholine to visual cortex plasticity. Neurobiol Learn Mem. 2003;80:291–301. doi: 10.1016/s1074-7427(03)00073-x. [DOI] [PubMed] [Google Scholar]
- Guest RM. Phd thesis. Ottawa, Ontario, Canada: University of Ottawa; 1983. Alternate Cerebellar Circuitry—The Morphology of the Caudal Cerebellar Lobe of the Weakly Electric Fish. [Google Scholar]
- Hasselmo ME, Anderson BP, Bower JM. Cholinergic modulation of cortical associative memory function. J Neurophysiol. 1992;67:1230–1246. doi: 10.1152/jn.1992.67.5.1230. [DOI] [PubMed] [Google Scholar]
- Heiligenberg W. Sensory control of behavior in electric fish. Curr Opin Neurobiol. 1991;1:633–637. doi: 10.1016/s0959-4388(05)80041-8. [DOI] [PubMed] [Google Scholar]
- Hoffman DA, Johnston D. Downregulation of transient K+ channels in dendrites of hippocampal CA1 pyramidal neurons by activation of PKA and PKC. J Neurosci. 1998;18:3521–3528. doi: 10.1523/JNEUROSCI.18-10-03521.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DA, Magee JC, Colbert CM, Johnston D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 1997;387:869–875. doi: 10.1038/43119. [DOI] [PubMed] [Google Scholar]
- Jarvis MR, Mitra PP. Sampling properties of the spectrum and coherency of sequences of action potentials. Neural Comput. 2001;13:717–749. doi: 10.1162/089976601300014312. [DOI] [PubMed] [Google Scholar]
- Kemp JA, Sillito AM. The nature of the excitatory transmitter mediating X and Y cell inputs to the cat dorsal lateral geniculate nucleus. J Physiol. 1982;323:377–391. doi: 10.1113/jphysiol.1982.sp014078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilgard MP, Merzenich MM. Cortical map reorganization enabled by nucleus basalis activity. Science. 1998;279:1714–1718. doi: 10.1126/science.279.5357.1714. [DOI] [PubMed] [Google Scholar]
- Koch C. Biophysics of Computation. New York: University Press; 1999. [Google Scholar]
- Krahe R, Gabbiani F. Burst firing in sensory systems. Nat Rev Neurosci. 2004;5:13–23. doi: 10.1038/nrn1296. [DOI] [PubMed] [Google Scholar]
- Lemon N, Turner RW. Conditional spike backpropagation generates burst discharge in a sensory neuron. J Neurophysiol. 2000;84:1519–1530. doi: 10.1152/jn.2000.84.3.1519. [DOI] [PubMed] [Google Scholar]
- Lesica NA, Stanley GB. Encoding of natural scene movies by tonic and burst spikes in the lateral geniculate nucleus. J Neurosci. 2004;24:10731–10740. doi: 10.1523/JNEUROSCI.3059-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas-Meunier E, Fossier P, Baux G, Amar M. Cholinergic modulation of the cortical neuronal network. Pfluegers. 2003;446:17–29. doi: 10.1007/s00424-002-0999-2. [DOI] [PubMed] [Google Scholar]
- Maccaferri G, Mangoni M, Lazzari A, DiFrancesco D. Properties of the hyperpolarization-activated current in rat hippocampal CA1 pyramidal cells. J Neurophysiol. 1993;69:2129–2136. doi: 10.1152/jn.1993.69.6.2129. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- Maler L, Collins M, Mathieson WB. The distribution of acetylcholinesterase and choline acetyl transferase in the cerebellum and posterior lateral line lobe of weakly electric fish (Gymnotidae) Brain Res. 1981;226:320–325. doi: 10.1016/0006-8993(81)91106-9. [DOI] [PubMed] [Google Scholar]
- Maler L, Sas E, Johnston S, Ellis W. An atlas of the brain of the electric fish Apteronotus leptorhynchus. J Chem Neuroanat. 1991;4:1–38. doi: 10.1016/0891-0618(91)90030-g. [DOI] [PubMed] [Google Scholar]
- Mathieson WB, Maler L. Morphological and electrophysiological properties of a novel in vitro preparation: the electrosensory lateral line lobe brain slice. J Comp Physiol [A] 1988;163:489–506. doi: 10.1007/BF00604903. [DOI] [PubMed] [Google Scholar]
- McCormick DA. Functional properties of a slowly inactivating potassium current in guinea pig dorsal lateral geniculate relay neurons. J Neurophysiol. 1991;66:1176–1189. doi: 10.1152/jn.1991.66.4.1176. [DOI] [PubMed] [Google Scholar]
- Mehaffey WH, Doiron B, Maler L, Turner RW. Deterministic multiplicative gain control with active dendrites. J Neurosci. 2005;25:9968–9977. doi: 10.1523/JNEUROSCI.2682-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda MI, Bermudez-Rattoni F. Reversible inactivation of the nucleus basalis magnocellularis induces disruption of cortical acetylcholine release and acquisition, but not retrieval, of aversive memories. Proc Natl Acad Sci USA. 1999;96:6478–6482. doi: 10.1073/pnas.96.11.6478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittmann T, Alzheimer C. Muscarinic inhibition of persistent Na+ current in rat neocortical pyramidal neurons. J Neurophysiol. 1998;79:1579–1582. doi: 10.1152/jn.1998.79.3.1579. [DOI] [PubMed] [Google Scholar]
- Nakajima Y, Nakajima S, Leonard RJ, Yamaguchi K. Acetylcholine raises excitability by inhibiting the fast transient potassium current in cultured hippocampal neurons. Proc Natl Acad Sci USA. 1986;83:3022–3026. doi: 10.1073/pnas.83.9.3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura TY, Coetzee WA, Vega-Saenz dM, Artman M, Rudy B. Modulation of Kv4 channels, key components of rat ventricular transient outward K+ current, by PKC. Am J Physiol Heart Circ Physiol. 1997;273:H1775–H1786. doi: 10.1152/ajpheart.1997.273.4.H1775. [DOI] [PubMed] [Google Scholar]
- Nelson ME, Maciver MA. Prey capture in the weakly electric fish Apteronotus albifrons: sensory acquisition strategies and electrosensory consequences. J Exp Biol. 1999;202:1195–1203. doi: 10.1242/jeb.202.10.1195. [DOI] [PubMed] [Google Scholar]
- Oswald AM, Chacron MJ, Doiron B, Bastian J, Maler L. Parallel processing of sensory input by bursts and isolated spikes. J Neurosci. 2004;24:4351–4362. doi: 10.1523/JNEUROSCI.0459-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan M, Maler L. Distribution of muscarinic receptors in the caudal cerebellum and electrosensory lateral line lobe of gymnotiform fish. Neurosci Lett. 1983;42:137–143. doi: 10.1016/0304-3940(83)90396-8. [DOI] [PubMed] [Google Scholar]
- Rasmusson DD, Dykes RW. Long-term enhancement of evoked potentials in cat somatosensory cortex produced by co-activation of the basal forebrain and cutaneous receptors. Exp Brain Res. 1988;70:276–286. doi: 10.1007/BF00248353. [DOI] [PubMed] [Google Scholar]
- Roddey JC, Girish B, Miller JP. Assessing the performance of neural encoding models in the presence of noise. J Comput Neurosci. 2000;8:95–112. doi: 10.1023/a:1008921114108. [DOI] [PubMed] [Google Scholar]
- Sadeghi SG, Chacron MJ, Taylor MC, Cullen KE. Neural variability, detection thresholds, and information transmission in the vestibular system. J Neurosci. 2007;27:771–781. doi: 10.1523/JNEUROSCI.4690-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Hasselmo ME, Bruno JP, Givens B. Unraveling the attentional functions of cortical cholinergic inputs: interactions between signal-driven and cognitive modulation of signal detection. Brain Res Brain Res Rev. 2005;48:98–111. doi: 10.1016/j.brainresrev.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Schoppa NE, Westbrook GL. Regulation of synaptic timing in the olfactory bulb by an A-type potassium current. Nat Neurosci. 1999;2:1106–1113. doi: 10.1038/16033. [DOI] [PubMed] [Google Scholar]
- Shannon CE. The mathematical theory of communication. Bell Systems Technical Journal. 1948:379–423. [Google Scholar]
- Sherman SM. Tonic and burst firing: dual modes of thalamocortical relay. Trends Neurosci. 2001;24:122–126. doi: 10.1016/s0166-2236(00)01714-8. [DOI] [PubMed] [Google Scholar]
- Sillito AM, Kemp JA, Berardi N. The cholinergic influence on the function of the cat dorsal lateral geniculate nucleus (dLGN) Brain Res. 1983;280:299–307. doi: 10.1016/0006-8993(83)90059-8. [DOI] [PubMed] [Google Scholar]
- Song WJ. Genes responsible for native depolarization-activated K+ currents in neurons. Neurosci Res. 2002;42:7–14. doi: 10.1016/s0168-0102(01)00305-4. [DOI] [PubMed] [Google Scholar]
- Stocker M, Krause M, Pedarzani P. An apamin-sensitive Ca2+-activated K+ current in hippocampal pyramidal neurons. Proc Natl Acad Sci USA. 1999;96:4662–4667. doi: 10.1073/pnas.96.8.4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svirskis G, Hounsgaard J. Transmitter regulation of plateau properties in turtle motoneurons. J Neurophysiol. 1998;79:45–50. doi: 10.1152/jn.1998.79.1.45. [DOI] [PubMed] [Google Scholar]
- Tai C, Kuzmiski JB, MacVicar BA. Muscarinic enhancement of R-type calcium currents in hippocampal CA1 pyramidal neurons. J Neurosci. 2006;26:6249–6258. doi: 10.1523/JNEUROSCI.1009-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Mishkin M, Aigner TG. Effects of muscarinic blockade in perirhinal cortex during visual recognition. Proc Natl Acad Sci USA. 1997;94:12667–12669. doi: 10.1073/pnas.94.23.12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RW, Maler L. Oscillatory and burst discharge in the apteronotid electrosensory lateral line lobe. J Exp Biol. 1999;202:1255–1265. doi: 10.1242/jeb.202.10.1255. [DOI] [PubMed] [Google Scholar]
- Turner RW, Maler L, Deerinck T, Levinson SR, Ellisman MH. TTX-sensitive dendritic sodium channels underlie oscillatory discharge in a vertebrate sensory neuron. J Neurosci. 1994;14:6453–6471. doi: 10.1523/JNEUROSCI.14-11-06453.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RW, Plant JR, Maler L. Oscillatory and burst discharge across electrosensory topographic maps. J Neurophysiol. 1996;76:2364–2382. doi: 10.1152/jn.1996.76.4.2364. [DOI] [PubMed] [Google Scholar]
- Voytko ML, Olton DS, Richardson RT, Gorman LK, Tobin JR, Price DL. Basal forebrain lesions in monkeys disrupt attention but not learning and memory. J Neurosci. 1994;14:167–186. doi: 10.1523/JNEUROSCI.14-01-00167.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger NM. The nucleus basalis and memory codes: auditory cortical plasticity and the induction of specific, associative behavioral memory. Neurobiol Learn Mem. 2003;80:268–284. doi: 10.1016/s1074-7427(03)00072-8. [DOI] [PubMed] [Google Scholar]