

Summary
IgA nephropathy (IgAN) is the most common primary glomerulonephritis worldwide. Lifelong mesangial deposition of IgA1 complexes subsist inflammation and nephron loss, but the complex pathogenesis in detail remains unclear. In regard to the heterogeneous course, classical immunosuppressive and specific therapeutic regimens adapted to the loss of renal function will here be discussed in addition to the essential common renal supportive therapy. Renal supportive therapy alleviates secondary, surrogate effects or sequelae on renal function and proteinuria of high intraglomerular pressure and subsequent nephrosclerosis by inhibition of the renin angiotensin system (RAASB). In patients with physiological (ΔGFR < 1·5 ml/min/year) or mild (ΔGFR 1·5–5 ml/min/year) decrease of renal function and proteinuric forms (> 1 g/day after RAASB), corticosteroids have shown a reduction of proteinuria and might protect further loss of renal function. In patients with progressive loss of renal function (ΔGFR > 3 ml/min within 3 months) or a rapidly progressive course with or without crescents in renal biopsy, cyclophosphamide with high‐dose corticosteroids as induction therapy and azathioprine maintenance has proved effective in one randomized controlled study of a homogeneous cohort in loss of renal function (ΔGFR). Mycophenolic acid provided further maintenance in non‐randomized trials. Differentiated, precise, larger, randomized, placebo‐controlled studies focused on the loss of renal function in the heterogeneous forms of IgAN are still lacking. Prospectively, fewer toxic agents will be necessary in the treatment of IgAN.
Keywords: IgA nephropathy, cyclophosphamide, mycophenolic acid, corticosteroids, high dose intravenous immunoglobulines
Introduction
Pathogenesis of the primary, idiopathic mesangioproliferative Immunoglobulin a nephropathy – Morbus Berger
In the early 1960s, Berger and Hinglais first described the entity of mesangial immunoglobulin (Ig)A deposits by immunofluorescence, frequently in concordance with IgG and complement factor 3 (C3) 1. They established the technique of immunofluorescence microscopy as a standard in renal histopathology. Primary or idiopathic IgA nephropathy (IgAN) – Morbus Berger – is the most common form of primary glomerulonephritis worldwide with heterogeneous outcome, and at least 30% of affected patients have a progressive clinical course with loss of renal function after 10–20 years 2.
During the last 50 years there has been an extensive, unresolved discussion concerning the origin and the formation of the polymeric IgA1 immune complexes, in particular, and the mechanism of mesangial deposition on a cellular and humoral basis (pIgA; IgA‐IC) (Fig. 1, Table 1). Aberrant glycosylation is the main characteristic of these mesangial IgA‐immune complexes with poor galactosylated pIgA1 15. Disease progression might be associated with the amount of aberrant IgA1 16 and circulating autoantibodies 17, 18, 19, 20. A mucosal origin was proposed by the polymeric structure, presence of IgA1 and the J secretory component in the pathogenic inflammatory mesangial IgA‐IC. Hence, systemic IgA is monomeric and mainly IgA1. After stem cell bone marrow transplantation, a decrease of IgA and remission of IgAN was described in murine models 21, 22, 23. Mesangial deposition of IgA could be mediated be IgG anti‐mesangial cell autoantibodies (IgG‐MESCA) in the sera of patients with IgA nephropathy, specific by F(ab′)(2) binding to 48‐ and 55‐kD autoantigen(s) 17, 18, 19. Soluble FcαRI (CD89) receptor was identified in the formation of IgA‐IC 24, 25, 26, 27, 28. Mesangial binding might be mediated by membrane‐bound Fc alpha receptors that could be expressed on autochthonous mesangial cells or immigrating myeloid cells. Asialoglycoprotein receptor (ASGP)‐R, CD 89 and the transferrin receptor (TfR1 or CD71) were involved and induce mesangial cell activation 29, 30, 31, 32. Mesangial deposition induces infiltration of granulocytes and macrophages and activation of the alternative complement pathway by complement factor 3 (C3). Functional nephron loss by the inflammatory response discharges into in a downstream cascade of fibrosis, high glomerular pressure and hypertension, which appears as sequelae or surrogate parameters, e.g. proteinuria. Therefore, proteinuria consists of two fractions: (i) mesangial damage by inflammation due to IgAN and (ii) conversely, high glomerular pressure by altered glomerular microdynamics due to nephron loss 33, 34, 35, 36. Therefore, the individual linear regression analysis of the time‐dependent course of estimated glomerular filtration rate (GFR) (eGFR; ΔGFR) or inverse serum creatinine is the only validated direct method in the assessment disease activity.
Figure 1.
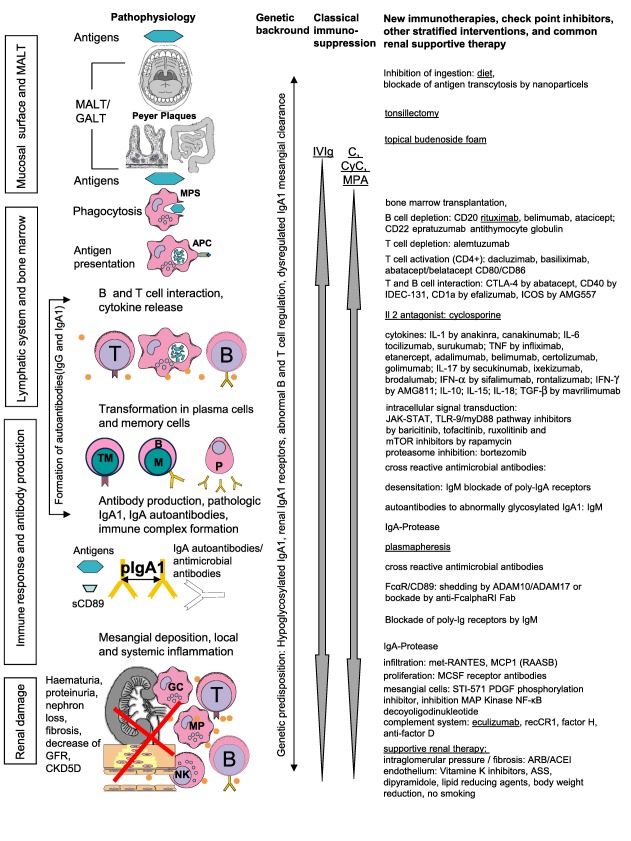
Pathophysiology, proven immunosuppressive drugs and new immunotherapies, check‐point inhibitors and other stratified interventions with their modes of action in immunoglobulin A nephropathy (IgAN). The pleiotropic effects of the classical immunosuppressive drugs are depicted in Table 1 and their clinical use in Table 2. Underlined interventions were used in therapy of primary IgAN. References are given in Table 1 and in the text. ACEI = angiotensin converting enzyme inhibitor = ADAM A disintegrin and metalloproteinase; AMG = anti‐interferon (IFN)‐γ IgG1 monoclonal antibody; APC = antigen‐presenting cells; ARB = angiotensin receptor blocker; ASS = acetylsalicylic acid; C = corticosteroids; CD = cluster of differentiation; CKD = chronic kidney disease; CTLA = cytotoxic T lymphocyte‐associated protein; CyC = cyclophosphamide; Fab = fragment antigen‐binding; GALT = gut‐associated lymphoid tissue; GFR = glomerular filtration rate; ICOS = inducible T cell co‐stimulator; IDEC = epidermal cell‐like dendritic cells; IL = interleukine IVIg = intravenous immunoglobulin; JAK‐STAT = Janus kinase–signal transducers and activators of transcription; MALT = mucosa‐associated lymphoid tissue; MAP = mitogen‐activated protein; MCSF = macrophage colony‐stimulating factor, MMF = mycophenolate mofetil = MPA mycophenolic acid; MPS = mononuclear phagocyte system; mTOR = mechanistic target of rapamycin; nuclear factor (NF) kappaB nuclear factor k‐light‐chain‐enhancer of activated B cells; NK = natural killer cell; p = pathological; PDGF = platelet‐derived growth factor; RAASB = renin angiotensin system blocker; RANTES = regulated on activation, normal T cell expressed and secreted; s = soluble; STI‐571 = Imatinib mesylate; TNF = tumour necrosis factor; TLR = Toll‐like receptor.
Table 1.
Genomic and non‐genomic effects of classical immunosuppressive drugs proven in clinical studies in progressive immunoglobulin A nephropathy (IgAN)
| Immunosuppressive drug/intervention | Therapy | Cellular Effects | Humoral Effects |
|---|---|---|---|
|
Corticosteroids Unspecific pleiotropic effects on immune system and mesenchymal cells (mesangiocytes, podocytes, angiocytes, smooth muscle cells, and fibroblasts) 3 |
Induction, Maintenance |
Genomic mechanisms anti‐inflammatory/immunosuppressive effects transcriptional
post‐transcriptional
translational
post‐translational
Non genomic specific classical GR
nonclassical GR
•apoptosis (?)
•signal transduction, Ca2+, second messengers non specific
|
Genomic mechanisms anti‐inflammatory/immunosuppressive effects transcriptional
post‐transcriptional
antiproliferative effects on non‐immune cells
Non genomic specific classical GR
nonclassical GR
|
|
IVIG Polyvalent IgG Immunomodulation by targeting on lymphocytes and elimination of antigens and antiantibodies 4, 5, 6, 7 |
Induction, less toxic, avoid osmotic nephrosis |
Regulation of the proliferation of lymphocytes modulation of T‐effector cells Fab mediated activities
Fc dependent activities
|
Fab mediated activities
|
|
Cyclophosphamide Alkylating agent inhibitor of proliferation and function 8 |
Induction, toxic, cumulative dose |
Inhibition of the proliferation of B‐, e.g. naïve B‐cells, and T‐cells and of inflammation | Decrease of antibody production and cytokines |
|
Azathioprine Antimetabolite Inhibition of Proliferation[9] |
Failed in maintenance (more T‐cell targeting?), phototoxicity, hepatotoxicity, bone marrow depression |
Non specific inhibition of the purine synthesis Mostly T‐cells blockade of antigen recognition by T‐cells |
Indirect, following T‐cell hit mild decrease of serum IgA levels |
|
Mycophenolic Acid Antimetabolit Inhibition of the proliferation of activated lymphocytes in the G1‐S phase (B‐ and T‐cells) and smooth muscle cells 8, 10, 11, 12, 13, 14 |
Maintenance, bone marrow depression |
Specific inhibition of the proliferation of B‐ and T‐cells by blockade of the de novo purine synthesis Induction of apoptosis of activated T‐lymphocytes Inhibition of antigen presentation of dendritic cells Inhibition of migration Proliferation of smooth muscle cells Expression of adhesion molecules (VLA‐4, E‐ and P‐ Selectin, VCAM‐1) Expression of CD 154 and CD 28 |
Decrease of antibody production, cytokines and anti‐inflammatory effects |
The unanswered burning question is the pathological explanation of the heterogeneous courses of IgAN: (i) the structure of the IgA1, (ii) the secondary, surrogate effects and comorbidity and (iii) the complement system. Clinically, mesangial inflammation and subsequent renal injury appears in haematuria, mixed tubular casts, proteinuria, reflecting intraglomerular pressure and damage of the glomerular filtration barrier and progredient nephron loss, with increase of serum creatinine, arterial hypertension and secondary nephrosclerosis. These findings, called nephritic syndrome 37, 38, require renal biopsy.
IgAN, a polygenetic disease with different incidences worldwide
Genetic predisposition in polygenetic IgAN remains uncertain, and familiar forms are extremely rare 39. Some genetic factors (6q21, 1q32, 22q12, 17q13, 8q32, 1q13, 9q34, 16q11) have been proposed as influencing renal prognosis 40. A recent publication identified, in a genomewide scan, a copy number variable region at 3p21.1 that might influence the TLR9 expression levels in IgA nephropathy patients with worse prognosis 41. Differences in patients with several ethnicities might be detected 42, but without therapeutic consequences, while pharmacogenetic studies have not been conducted. However, the worldwide differences in the incidence in IgAN from 0·8 (Germany 43, Spain 44) to 10·5 (Australia 45) per 100 000 patients per year seems to depend upon different referral to renal biopsy more than on ethnicity or genetic factors 46.
Is the prognosis and response on the therapy predictable by initial histological findings?
Histological grading systems were established in IgAN by Lee et al. 47. Histological grading of IgA nephropathy predicting renal outcome: revisiting H. S. Lee's glomerular grading system and Haas et al. 48. In 2009, the ‘new’ Oxford Classification was proposed 49, 50. Unfortunately, this classification did not include important histological findings, such as crescents with extracapillary proliferation and arterial hyalinosis 51, 52, 53, thrombotic microangiopathy 54 or techniques such as immune staining and electronmicroscopy 51, which were relevant and indispensable in the diagnosis 1 and prognosis 1, 48, 51, 52, 55, 56, 57, 58, 59. In order of the deficiencies, also confirmed by the authors themselves 51, we do not recommend the Oxford Classification. Clearly, no histological grading correlated with clinical outcomes after therapy in most studies 4, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70. However, therapeutic interventions should be based not only or decided upon isolated histological findings 71. Decrease of kidney function, decline of GFR (> 12 ml/min/year) and/or refractory proteinuria (> 1·0 g/day) with age‐related normal macroscopic morphology, normal kidney size and parenchyma/pyelon ratio and specific findings in renal histology (mesangial hypercellularity, crescents or adhesions) 47, 48, 51, 54, 71, 72, 73 require further immunosuppressive interventions after symptomatic therapy.
Assessment of progression and subsequent treatment decision
Due to the heterogeneity of progression the of IgAN, risk‐adjusted precise homogeneous patient selection for treatment decisions in study inclusion, or interpretation of the outcomes of progressive patients, are the most important criteria in study design (Table 2, Fig. 2) 33, 34, 64, 74. In all kidney diseases, the recommended standard in the assessment of renal function is the individual linear regression analysis of the time‐dependent course of estimated GFR (eGFR; ΔGFR) or inverse serum creatinine before and after therapy, especially in IgAN 5, 35, 60, 61, 62, 64, 69, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83 (Table 2, Fig. 2). Doubling of serum creatinine is not recommended in the determination of the loss of renal function (ΔGFR), because the loss of renal function calculated by serum creatinine (eGFR) is not linear between 110 and 40 ml/min 84, 85, 86. The reason for this is that a linear decline in ‘true’ GFR does not result in a straight line as a reciprocal of serum creatinine analysis. This is because the filtration of serum creatinine is influenced, in proportion, by increased tubular secretion of serum creatinine as renal function declines. Thus, basing outcome analysis on reciprocal serum creatinines only provides a false picture of changes in ‘true’ GFR. Furthermore, doubling of eGFR as an end‐point is not recommended regarding the interindividual differences in the time dependent decline of renal function in such a heterogeneous disease.
Table 2.
Synopsis of treatment strategies in immunoglobulin A nephropathy (IgAN): imunosuppressive therapy, tonsillectomy and supportive therapy in IgAN stratified by risk factors [stage of renal chronic kidney disease, loss renal function (ΔGFR), arterial hypertension and proteinuria], evidence‐based level, study design and inclusion criteria
| Therapy | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CKD | Proteinuria | RR | Immunsuppression | |||||||
| Stage | (g/day) | (mm Hg) | CTX | C | MPA/MMF | CsA | IVIG | ACEI or ARB | Tonsillectomy | Fish oil |
| 1 | <1 | Normal | ||||||||
| >1 | Normal | C+Fish oil: Hogg et al. 2006 179 | ACEI: Praga 2003 180 | TE+ C: Hotta et al. 2001 110 | +K: Hogg et al. 1995 2006 2010 179, 181, 182 | |||||
| Katafuchi et al. 2003 183 | ACEI+ARB: Tanaka et al. 2004 184 | |||||||||
| Kuriki et al. 2003 185 | ACEI+ARB: Russo et al. 2001 186 | |||||||||
| C+TE: Hotta et al. 2001 110 | ACEI: Maschio et al. 1994 1996 100, 187 | |||||||||
| Shoji et al. 2000 188 | ||||||||||
| C+Aza: Yoshikawa and Ito 1999 189 | ||||||||||
| >1 | >125/75 | Manno et al. 2009 115 | ARB+HCT: Uzu et al. 2005 190 | Xie et al. 2003 191 | ||||||
| 1–2 | > 1 | CTX+C+Aza/HDC+C+Aza versus RAASB Rauen et al. 2015 36 | Pozzi et al. 1999 67 C+Aza: Pozzi et al. 2010 66 | Lai et al. 1987 120 | ||||||
| >3 | Tesar et al . 2015 35 | ±C: Liang et al . 2014 81 | ||||||||
| 1–3 | >0·75 | |||||||||
| 2 | n.n. | >125/75 | Kobayashi et al. 1986, 1988, 1989, 1996 192, 193, 194, 195, 196 | |||||||
| 2–3 | >1 | CTX±Aza±C: Ballardie and Roberts 2002 64 | Maes et al. 2004 70 | Rostoker et al . 1994 5 | ARB: Li et al . 2006 82 | Iino et al. 1993 197 | ||||
| >2 | ||||||||||
| 3 | >1 | Woo et al. 1987 198 | C+TE: Sato et al. 2003 199 | +C: Liu et al. 2014 200 | ±C Rasche et al . 2006a 4 | TE+C: Sato et al. 2003 199 | Donadio et al . 1994 1999 2000 2001 201, 202, 203, 204 | |||
| C+Aza: Goumenos et al. 1995, 2003 205, 206 | Tang et al. 2005, 2010 68, 69 | Pettersson et al.1994 207 | ||||||||
| C+CTX: Roccatello et al. 2000 208 | ||||||||||
| C+CTX: Tsuruya et al. 2000 209 | ||||||||||
| Peters 2015 210 | ||||||||||
| 4 | >1 | >125/75 | Frisch et al. 2005 142 | ACEI+ARB: Nakao et al. 2003 211 | TE+CTX: Rasche et al. 1999 109 | |||||
| CTX±MPA±C: Rasche et al . 2006b/2015 61, 63 | ||||||||||
| C±CTX: Tumlin et al . 2003 79 | ||||||||||
| Rasche et al . 2003 62 | ||||||||||
| CTX+TE: Rasche et al. 1999 109 | ||||||||||
| >2 | >125/75 | Tamura et al . 2003 80 | ||||||||
| n.n. | >2 | n.n. | Chen et al. 2002 143 | |||||||
| n.n. | nephritic syndrom | n.n. | +C: Liu et al. 2014 200 | |||||||
Evidence‐based level grade 1b is marked in grey and all other studies have been classified as less than 1b. With regard to the heterogeneity of the disease, studies with linear regression analysis (ΔGFR) as the most accurate, standard method in the assessment of the loss of renal function before and during the study period are underlined. AZA, azathioprine; ACEI, angiotensin converting enzyme inhibitors; ARB, angiotensin receptor blocker; CKD, chronic kidney disease; C, corticosteroids; CTX, cyclophosphamide; HCT, hydrochlorothiazide; HD, high dose IVIg, high‐dose immunoglobulin therapy; MMF, mycophenolate mofetil; MPA, mycophenolic acid; RAASB, renin angiotensin system blocker; RR, blood pressure; TE, tonsillectomy.
Figure 2.
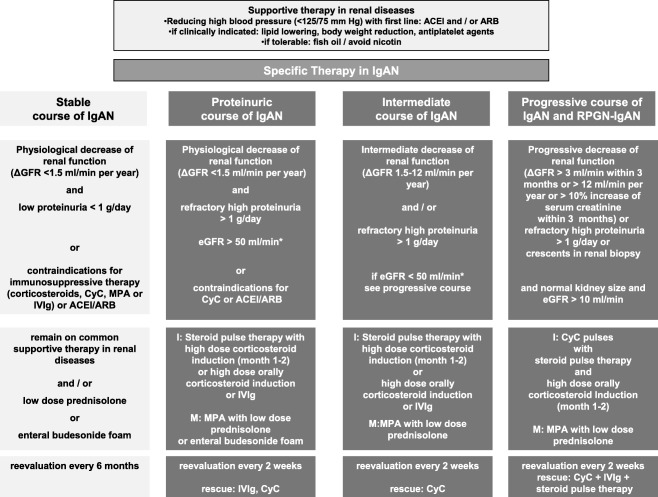
Risk‐adapted treatment strategy in order of degree of proteinuria and loss of renal function (ΔGFR) based on clinical studies (Table 2). Normal kidney size and morphology in ultrasound has to be evaluated before specific immunosuppressive treatment is initiated. Corticosteroids monotherapy showed proven benefit only in patients with mild to moderate impaired renal function (*, Table 2) and the progressive treatment strategy is recommended in patients with severely impaired renal function. In patients with nephrotic syndrome, the proteinuric course treatment regimen will be recommended.
Clearly, only progressive patients should be treated with high‐risk immunosuppressive therapy in a professional setting, e.g. cyclophosphamide 36.
Therefore, progressive loss of renal function or the decrease of estimated GFR (eGFR; ΔGFR) could be differentiated in four grades or stages: (i) approximately 20–30% or less of the patients: stable disease with physiological decrease of renal function (ΔGFR < 1·5 ml/min/year) and low‐grade proteinuria below 1 g/g day with renin angiotensin system blocker (RAASB); (ii) approximately 50% of the patients with intermediate and progressive disease ΔGFR > 1·5–30 ml/min/year or more than 3 ml/min within 3 months; (iii) approximately 10% of the patients with rapidly progressive forms and the presence of crescents in renal histology (RPGN) and a ΔGFR > 3 ml/min/month or > 30 ml/min/year; and (iv) approximately 7% of the patients with a nephrotic syndrome (proteinuria > 3·5 g/g, plus hypoalbuminaemia and oedema).
However, in most meta‐analyses and randomized controlled studies (RCTs) this criterion, focusing on the heterogeneity of IgAN, was neglected (Table 1) 33, 35, 64, 74, 83, 87, 88, 89, 90, 91. Table 1 presents an extensive overview of all publications regarding several immunosuppressive therapies of progressive IgAN concerning evidence‐based medicine (EBM) level (≥ 1b) and is stratified by renal risk factors: stage of chronic kidney disease (CKD) and the assessment of the loss of renal function before and during the study period (ΔGFR) with linear regression analysis (ΔGFR). Risk‐adapted treatment strategy in order of degree of proteinuria and loss of renal function (ΔGFR) are shown in Fig. 2.
Unfortunately, the low incidence and the heterogeneity of IgAN limit larger trials with results that apply EBM criteria in level 1. Therefore, only one trial in progressive IgAN fulfilled the criteria of EBM level 1 (RCT) and, conversely, a precise, detailed, homogeneous selection of patients with ΔGFR before and during therapy 64.
Point of no return and limitations of immunosuppressive therapy
It has been suggested that untreated patients with a serum creatinine exceeding 2·5–2·7 mg/dl (‘the point of no return’) will develop end‐stage renal disease within a period of approximately 1 year 92, 93, but this judgement should be revised with immunosuppressive therapy 4, 61, 62, 63, 64, 77, 94, 95.
Contraindications for immunosuppressive treatment will be: serum creatinine > 4·5 mg/dl (> 480 µmol/l), small kidney size (< 9 cm) in ultrasound, acute or chronic infections (including human immunodeficiency virus, hepatitis B and C virus), carcinoma, leucocyte counts < 3·0/nl, platelet counts < 80/nl, gastrointestinal bleeding, haemolytic anaemia, pregnancy, lactation or women with childbearing potential. In all drug regimens, even in supportive therapy, current information on the contraindications of applicable regulatory documents (summary of product characteristics) should be considered.
Treatment in non‐progressive disease and supportive therapy
Symptomatic or common supportive renal therapy, reducing blood pressure and hyperlipidaemia in order to avoid nephrosclerosis, renin–angiotensin–aldosterone blockade and others
Downstream effects, namely glomerular and interstitial fibrosis, may be mediated by monocyte chemotactic protein‐1 (MCP‐1), interleukin (IL)−6, IL‐8, transforming growth factor (TGF)‐beta, regulated on activation, normal T cell expressed and secreted (RANTES) and CCR1‐ and CCR5‐positive cells 96, 97 and may be reduced by RAASB 98 (Fig. 1).
In patients with stable disease without progression in linear regression analysis of serum creatinine (or eGFR) only supportive therapy with ACE inhibitors (ACEI), angiotensin receptor blockers (ARB), cholesterol lowering and fish oil were proposed in the former KDIGO clinical best practice guidelines for glomerulonephritis by Eckardt et al. from 2012 99. However, corticosteroids have also demonstrated proven benefit in all studies in stable disease (Table 2). Further, in progressive disease, taking into account the risk of worsening renal function and hyperkalaemia, ACEI and/or ARB should be prescribed in lowering intraglomerular pressure and avoiding interstitial sclerosis 100 with a secondary effect on the loss of renal function, e.g. proteinuria as a surrogate parameter, due to nephrosclerosis 101.
Only 7% of the patients with IgAN have proteinuria in the nephrotic range and normal renal function. High‐dose corticosteroid induction is beneficial, and in refractory cases or in maintenance therapy, mycophenolic acid or leflunomide 102, 103, 104 and in RAASB‐resistant disease adrenocorticotrophic hormone 105 will also be beneficial.
Tonsillectomy and mucosa‐associated lymphoid tissue (MALT)
Due to the proposed mucosal origin, tonsillectomy was performed in reducing the contact of potential antigens with the mucosa‐associated lymphoid tissue (MALT), inhibition of the migration of B and T cells in the locoregional lymphatic nodes and decrease of the total systemic amount of pIgA. Furthermore, tonsilla palatina has been mentioned as a potential trigger for the systemic response (Fig. 1). After tonsillectomy the episodes of macrohaematuria might have been reduced 106, 107, 108, but there was no clear benefit in reducing the disease progression 77, 90, 109. However, tonsillectomy affected only a part of the widespread MALT. Hence, in combination with steroid pulses 110, 111, 112, possible effects have been described in patients with mild renal impairment and proteinuria < 1 g/g (CKD G1‐2 A3) 71. In patients with progressive disease or renal impairment, tonsillectomy could provoke acute renal injury with irreversible detoriation of renal function, and should be avoided 77, 109, 110.
Topical corticosteroids for suppression of the MALT system
Topical application of enteric budesonide foam targeted to the ileocecal region had a significant effect on urine albumin excretion, accompanied by a minor reduction of serum creatinine and a modest improvement of eGFR 113. However, systemic budesonide absorption is approximately 10–20% 114.
High‐dose corticosteroid induction therapy and long‐term application of systemic corticosteroids decreases proteinuria and risk of renal failure
In two large randomized trials, corticosteroids, high‐dose intravenous pulses and low‐dose corticosteroid maintenance reduced proteinuria and the risk of renal failure in proteinuric patients with mild impaired renal function significantly (serum creatinine/creatinine clearance in control versus study group: 88 versus 98 µmol/l/87 versus 93 ml/min) and proteinuria (1·8 versus 2·0 g/day; Table 2) 65, 66, 67. These results were confirmed in a large RCT trial (97 patients, follow‐up 8 years) with high‐dose corticosteroid induction over 6 months and ramipril versus ramipril monotherapy in patients without progression and mild renal impairment, but without maintenance therapy 115. Recently, a large retrospective cohort study demonstrated that continuous application of corticosteroids in addition to ACEI or ARB prolongs renal survival time significantly, in contrast to RAASB monotherapy 35, 116.
Cyclophosphamide and high‐dose corticosteroid pulses with azathioprine maintenance in patients with non‐progressive disease
In the STOP‐IgAN trial, patients with mild renal impairment, no sign of progression and persistent low‐degree proteinuria were treated after a 6‐month run‐in phase of maximum intensified renal supportive therapy (RAASB) in an RCT with cyclophosphamide orally, corticosteroid pulses and only supportive therapy stratified by proteinuria (supportive care versus supportive care plus CyP or high‐dose corticosteroid pulses: serum creatinine 76/76 µmol/l; GFR 57/61 ml/min, absolute change in eGFR over 36 months −4·7/–4·2 ml/min per 1·73 m2 body surface area (BSA) per year, estimated loss of renal function 1·6 versus 1·4 ml/min per 1·73 m2 BSA per year, mean annual change in the slope of the reciprocal of serum creatinine concentration −0·02 versus −0·01 mg/dl, proteinuria 1·6/1·8 g/day, protein creatinine ratio 1·0–1·1. g/g). However, in this heterogeneous cohort, cyclophosphamide or high corticosteroid pulses demonstrated significant effects on the primary end‐point (full clinical remission). Full clinical remission was defined as proteinuria with a protein‐to‐creatinine ratio of < 0·2 and stable renal function with a decrease in the eGFR of < 5 ml/min per 1·73 m2 from the baseline eGFR at the end of the 3‐year trial phase. The secondary end‐point, defined as a decrease in the eGFR of at least 15 ml/min per 1·73 m2 from the baseline eGFR, was not significant 36. This trial is discussed controversially, even in study design and statistical power (δ value < 0·3), patient selection (inhomogeneous, no information of ΔGFR before therapy and after therapy), inclusion criteria (proteinuria as a sequelae or surrogate parameter of nephron loss), observation time (only 3 years) and the lack of any kind of renal histology 33, 74, 83, 117, 118, 119. Clearly, cyclophosphamide and corticosteroids will not ameliorate the physiological loss of renal function, even in patients with IgAN, and should be limited in IgAN to patients with a defined decrease of renal function (ΔGFR).
Treatment of progressive disease – classical systemic immunosuppressive therapy
Classical immunomodulatory drugs and interventions: intravenous immunoglobulin (IVIg), corticosteroids, cyclophosphamide and mycophenolate maintenance
In light of the autoimmune pathogenesis, cyclophosphamide, corticosteroids, mycophenolic acid and the less toxic alternative of IVIg are effective in the reduction of the systemic amount of IgA antibodies and local response in the mesangium (Fig. 1). Therefore, in patients with rapid loss of renal function, immunosuppressive therapy is necessary and has to be continued lifelong for kidney survival. The pleiotrophic effects of these drugs promises advantages because several checkpoints of the cascade will be inhibited but toxic effects, except IVIg, must be considered.
T cell‐targeted drugs, e.g. cyclosporin, can reduce proteinuria in combination with corticosteroids mediated by glomerular vasoconstriction, but a worsened glomerular filtration rate 120.
Plasmapheresis, in combination with cyclophosphamide and corticosteroids as a short‐term intervention, is restricted to rapidly progressive forms, mainly in secondary IgAN, e.g. Henoch–Schoenlein purpura or in p‐ or c‐anti‐neutrophil cytoplasmic antibodies (ANCA)‐positive vasculitis. Stem cell transplantation is used only in mice 21, 22, 23.
IVIg – non‐toxic, limited immunomodulation
In patients with pregnancy, childbearing potential or high cumulative doses with cyclophosphamide, IVIg is a less toxic alternative with comparable effects in the reduction of ΔGFR from −1·05 ml/min/month to −0·15 ml/min/month (P = 0·024) and proteinuria (from 2·4 g/l to 1·0 g/l, P = 0·015) 4, 5, 60, 61, 62, 63. In Kaplan–Meier analysis median survival time was only 4·7 years with IVIg versus 10·5 with cyclophosphamide pulse therapy/mycophenolic acid (CyP/MPA). Therefore, IVIg is an option as induction therapy for 6 months. However, 3 years after IVIg, further loss of renal function was observed 4, 60, 63, and further maintenance therapy will be needed with prednisolone and/or mycophenolate mofetil (MMF)/MPA plus prednisolone 60, 61, 62, 63, 77.
Corticosteroids are essential in induction and maintenance therapy
Corticosteroids display anti‐inflammatory effects and induce apoptosis and show proven benefit in both long‐term use and pulse therapy 3, 35, 65, 66, 67, 116 (Table 1, Fig. 1). Corticosteroids will be a necessary standard in addition to mycophenolate 60, 61, 62, 63, even in other autoimmune diseases 121. These effects may be responsible for a reduction of the proliferative lesions, glomerular sclerosis and tubular fibrosis in IgAN with a superior renal survival compared with patients receiving only ARB/ACEI 35, 36, 67, 122.
Cyclophosphamide in combination with corticosteroids – advantages of CyP and intensified immunosuppression
Treatment with cyclophosphamide plus steroids has been used with great success for more than 30 years in several publications regarding IgA nephropathy with progressive loss of renal function (Table 2). Cyclophosphamide is a highly potent cytotoxic agent used frequently for cytoreductive induction therapy in autoimmune disease by depletion and inhibition of T and B lymphocytes, but its long‐term use is limited due to the high cumulative toxicity 123, therefore further maintenance therapy is needed 64 (Fig. 1, Table 1). In an RCT, Ballardie et al. used cyclophosphamide orally 1·5 mg/kg/day adjusted to the nearest 50 mg for 3 months in order to avoid severe leucopenia, anaemia and thrombocytopenia or other side effects with an estimated cumulative dose of 9 g 64. Continuous oral application of cyclophosphamide has to be monitored weekly by experts, and severe leucopenia with adverse events has been described 36. Intravenously CyP pulses have shown superiority regarding safety 124 and less toxicity 123, 124, 125 by short‐term acrolein bladder exposure with fewer cumulative doses 62 with equal 126 or better 124, 125 efficacy, as also proved in other autoimmune diseases compared with oral cyclophosphamide.
Intensified and escalated immunosuppression adapted to leucocytes or neutrophils have demonstrated a better outcome in autoimmune diseases 123, 127 and in IgAN 62. We have adjusted the doses to leucocyte count nadir 2 weeks after CyP with a remarkable depression of leucocytes close to 3·5/ml 61, 62, 63, 77 accompanied by low‐dose corticosteroids (5–7·5 mg prednisone/day). However, the majority of our patients (67%, 31 of 47) showed further disease activity 4 months on average after CyP, and further administration of MPA was necessary 61, 62, 63, 77.
Concept of sequential therapy – safety maintenance with low risk and toxicity
The concept of sequential therapy, induction and maintenance has already been introduced in the treatment of lupus nephritis 10, ANCA‐associated glomerulonephritis 128, 129, 130, 131 and other autoimmune diseases. In IgAN, sequential therapy was used in two RCTs and several other studies 60, 61, 62, 63, 64, 66 (Table 2). In both RCTs, high‐dose prednisolone medication orally accompanied the induction therapy for 3 months (Ballardie et al. 40 mg for 3 months 64 and Pozzi 0·5 mg/kg/day, alternate‐day regimen 66). Serum creatinine/creatinine clearance in the patients in the Pozzi et al. study was almost normal (control versus study group: 88 versus 98 µmol/l/87 versus 93 ml/min); proteinuria (1·8 versus 2·0 g/day) was the focus of the treatment and no information was given before therapy regarding the decline of renal function (ΔGFR) 67. Contrary to Pozzi et al. 67, Ballardie et al. included patients with moderate to severe impaired renal function (serum creatinine > 130 µmol/l) and a homogeneous progression in linear regression analysis with the equivalent slopes, as in our patients (Ballardie −16·8 to −3·8 ml/min/year and controls −15·6 to −16·5; proteinuria 4·6–4·2 g/day and controls 3·9–0·8 g/day) 64. We started with a low‐dose corticosteroid regimen (20 mg/day prednisolone) and we reduced every 2 weeks to 5 mg/ day until the end of the study 4, 60, 61, 62, 63, 77, 109.
In non‐progressive IgAN patients after steroid pulses azathioprine (1·5 mg/kg/day), limited for 6 months, provided no additional benefit to steroids alone after a 5‐year follow‐up, but more side effects have been described 36, 66. However, after cyclophosphamide and corticosteroid induction, maintenance with azathioprine (1·5 mg/kg/day) was continued favourably to the study end. The study by Pozzi et al. started in 1987 65, 66, 67 and the study of Ballardie et al. in 1991 64. The more specific drug, mycophenolic acid, was not available at that time, because MMF has been approved for transplantation since 1996 and free mycophenolic acid (ecMPS) since 2004. Clearly, mycophenolic acid had demonstrated superiority in transplant medicine in preventing acute and chronic rejection to azathioprine 132, 133 and in safety 134, 135, 136, 137.
Mycophenolic acid
MPA acts by reversible uncompetitive inhibition of inosine 5′‐monophosphate dehydrogenase (IMPDH), which is essential for de‐novo biosynthesis of guanine nucleotides and lymphocyte proliferation 138, 139, 140. Specifically by MPA, the proliferation of B and T cells is inhibited and the Ig cytokine secretion of B cells is suppressed 11 and the apoptosis of activated T lymphocytes is induced 12. MPA inhibits the migration of lymphocytes and antigen presentation by dendritic cells 13 (Table 1). However, other forms of important inflammatory response were not influenced by MPA as the expression of activation markers of inflammation, including CD25 and CD69 14.
MPA was used with different outcomes in several studies with 60, 61, 62, 63 and without cytotoxic induction therapy, but in some studies without low‐dose prednisolone 68, 70, 141, 142. However, the patients in these studies were not comparable regarding risk factors, e.g. progression of renal failure in linear regression analysis (ΔGFR), proteinuria and age 68, 70, 141, 142. This might explain why an RCT in 21 IgAN patients with mild renal impairment without induction therapy and without prednisolone showed no significant effect on the loss of renal function 70, 142. However, a decrease of proteinuria was observed in two RCTs in 31 patients 143 and in 16 patients 68. Clearly, MPA monotherapy is less effective and corticosteroids are needed additionally for long‐term maintenance 35, 60, 61, 62, 63, 64, 65, 66, 67, 121.
Genetic or pharmacogenetic aspects have not been explored in published studies of IgAN patients with supportive or immunosuppressive studies. Hence, only in one study has detailed information of ethnicity been given 61. Genetic variants of the uridine diphosphate–glucoronyltransferases may enlarge the drug exposition with MMF in the area under the curve and will be responsible for more side effects 144, but larger trials in IgAN patients with MPA/MMF with worldwide scan of this defect have not been published. No signs of less exposure or inosine 5′‐monophosphate dehydrogenase suppression between MMF and MPA in our patients were demonstrated in concordance with other studies in a pharmacokinetic/pharmacodynamic study 145.
In our study, sequential MPA maintenance therapy was effective in patients with progressive IgAN in reducing further loss of renal function (ΔGFR) from −0·4 to −0·1 ml/min/month with a trend in reduction of the proteinuria from 1·0 to 0·6 g/l 63. The full effect of MPA on renal function was observed after an average time lag of 6 months and in reduction of proteinuria after 5 months. Maintenance therapy with MPA and low‐dose corticosteroids consolidates the clinical outcome after induction therapy with CyP or steroids over 6 years, while reducing side effects and cumulative toxicity of cyclophosphamide and corticosteroids 10, 14, 60, 61, 62, 63, 146, 147. The strengths of our non‐randomized study were the homogeneous cohort, the long observation time, the number of treated patients, intraindividual course with ΔGFR before and under therapy and the proven concept of sequential therapy in another RCT with the same criteria, but with azathioprine instead of MPA 64. However, an RCT with cyclophosphamide and MPA will be needed, but the concept and the realization will be limited by the small number of patients with an incidence of < 0·6/100 000/year, decline of resources and support in the health systems for monitoring or study preparations, from governments and pharmaceutical industries and to deny patients a proven and save therapy.
Targeted inhibition of the generation, formation and deposition of the pathogenic IgA immune complexes and the downstream cascade with new immunotherapies, checkpoint inhibitors and other stratified interventions
Because of the still‐unclear pathophysiology, the genetic susceptibility in the production of aberrant glycosylated IgA1 and the multiple targets on several locations, e.g. MALT, systemic immune system, bone marrow and mesangium, and the development of therapeutic strategies in the specific inhibition of the pernicious cascade, are our future challenges (Fig. 1).
Hypothetical strategies – in‐vitro or animal models
Nanoparticles may protect from the ingestion of potential triggering antigens in the mucosal area 148.
Cleavage of the pathological IgA1 and the IgA1 IC with a specific IgA protease is a controversial topic in casual‐specific therapy for IgAN 30, 149, 150, 151, 152, 153, 154. However, IgA is one of the most frequent immunoglobulins and is extremely important for the defence and integrity of the surfaces. Therefore, the loss of integrity could be induced 155, 156.
Desensitizing may be helpful in reducing pathological IgA1 by shifting plasma cells to IgG‐producing cells 157. Blockade of aberrant glycosylated IgA1 by autoantibodies 16, 20, 158 or poly‐Ig receptors Fcalpha/muR 31 on mesangial cells may attenuate the influence of aberrant glycosylated IgA1 159.
Hyperexpression of nuclear factor kappa B (NF‐kB) is found in IgAN and proteasome inhibitors; e.g. bortezomib will be a therapeutic option in progressive glomerular diseases limited by neurotoxicity 160. In IgAN the Akt/mTOR/p70S6K pathway and Toll‐like receptor (TLR)−9 161, 162 is activated and the inhibitor rapamycin might be an option in the treatment of IgAN 163.
The deposition of IgA in the mesangium may be mediated by the soluble type I IgA receptor (FcalphaRI or CD89) and transferrin receptor (TfR) on mesangial cells 164, 165, 166. This might be inhibited by anti‐FcalphaRI Fab 167. Inhibition of factor Xa by DX‐9065a may reduce mesangial proliferation 168.
Secondary IgAN – cytokine inhibition by biologicals
Proinflammatory cytokines play a pivotal role in the inflammatory downstream events and might be a possible target for intervention. Beneficial effects of biologicals were reported in secondary IgA by inhibition: IL‐1 with anakinra 169; I:‐6 with tocilizumab 170, 171; TNF with infliximab 172, 173 and adalimumab 174.
Biologicals – B cell and complement inhibition in human case reports
Recently, case reports have been published after B cell depletion with rituximab with beneficial response in patients with nephrotic syndrome 175 and RPGN‐IgAN after kidney transplantation 176. Anti‐thymocyte globulin (ATG) induction therapy reduces disease recurrence in renal transplant recipients with primary IgA nephropathy 177. B and T cell interaction will be inhibited by biologicals, e.g. cytotoxic T lymphocyte (CTLA)−4 abatacept, in one patient with rheumatoid arthritis 174. In a patient with RPGN‐IgAN refractory to CyP, corticosteroids and plasmapheresis 178 will offer new aspects in the inhibition of the complement system by humanized anti‐C5 monoclonal antibody eculizimab.
However, all these specific interventions will interfere with other important ongoing parallel immune processes and probably inhibit crucial functions of the immune system. Contrary to these specific inhibitions, the advantages of the classical drugs were the multi‐modal types of action, long‐term experience and cost‐effectiveness.
Concluding remarks
καιρὸς δ' ἐπὶ πᾶσιν ἄριστος: Hesiod, ΕΡΓΑ ΚΑΙ ΗΜΕΡΑΙ, 694
Approximately 3000 years ago, Hesiod taught ‘why, when and how’ interventional therapy in a heterogeneous disease such as IgAN has to be started (καιρὸς). Supportive therapy might be effective in reducing renal fibrosis by lowering intraglomerular pressure. However, in an autoimmune disease with lifelong deposition of altered IgA1 immune complexes, corticosteroid therapy will promise benefit even for mild progressive course and in patients with proteinuria. The right moment (καιρòς) for immunosuppressive intervention with cyclophosphamide is defined by an accelerated decrease of renal function in linear regression analysis (ΔGFR). Immunosuppressive therapy with cyclophosphamide has been proved (δ' ἐπὶ πᾶσιν ἄριστος) in several trials. Mycophenolate maintenance therapy with low‐dose steroids provides a reduction of further progress after cyclophosphamide induction in concordance with other autoimmune diseases. However, the heterogeneity of the disease needs further sufficiently powered, larger, randomized, placebo‐controlled clinical studies (δ' ἐπὶ πᾶσιν ἄριστος), with the focus on maintenance therapy with corticosteroids and MMF regarding the loss of GFR in linear regression analysis and the optimal duration of therapy. In future, less toxic and more specific drugs will be needed to prevent prolonged mesangial deposition of the altered IgA and prevent loss of renal function.
Disclosure
None to declare.
References
- 1. Berger J, Hinglais N. Les dépôts intercapillaires d'IgA‐IgG [Intercapillary deposits of IgA]. J Urol Nephrol (Paris) 1968; 74:694–5. [PubMed] [Google Scholar]
- 2. Lee H, Hwang JH, Paik JH et al Long‐term prognosis of clinically early IgA nephropathy is not always favorable. BMC Nephrol 2014; 15:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Czock D, Keller F, Rasche FM et al Pharmacokinetics and pharmacodynamics of systemically administered glucocorticoids. Clin Pharmacokinet 2005; 44:61–98. [DOI] [PubMed] [Google Scholar]
- 4. Rasche FM, Keller F, Lepper PM et al High‐dose intravenous immunoglobulin pulse therapy in patients with progressive immunoglobulin A nephropathy: a long‐term follow‐up. Clin Exp Immunol 2006; 146:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rostoker G, Desvaux‐Belghiti D, Pilatte Y et al High‐dose immunoglobulin therapy for severe IgA nephropathy and Henoch–Schonlein purpura. Ann Intern Med 1994; 120:476–84. [DOI] [PubMed] [Google Scholar]
- 6. Gelfand EW. Intravenous immune globulin in autoimmune and inflammatory diseases. N Engl J Med 2012; 367:2015–25. [DOI] [PubMed] [Google Scholar]
- 7. Yu Z, Lennon VA. Mechanism of intravenous immune globulin therapy in antibody‐mediated autoimmune diseases. N Engl J Med 1999; 340:227–8. [DOI] [PubMed] [Google Scholar]
- 8. Fassbinder T, Saunders U, Mickholz E et al Differential effects of cyclophosphamide and mycophenolate mofetil on cellular and serological parameters in patients with systemic lupus erythematosus. Arthritis Res Ther 2015; 17:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lacki JK, Klama K, Michels H et al The effect of methotrexate and azathioprine on the serum levels of IgA‐alpha(1)‐antitrypsin complex in juvenile chronic arthritis. Braz J Med Biol Res 1997; 30:763–7. [DOI] [PubMed] [Google Scholar]
- 10. Contreras G, Pardo V, Leclercq B et al Sequential therapies for proliferative lupus nephritis. N Engl J Med 2004; 350:971–80. [DOI] [PubMed] [Google Scholar]
- 11. Jonsson CA, Carlsten H. Mycophenolic acid inhibits inosine 5'‐monophosphate dehydrogenase and suppresses immunoglobulin and cytokine production of B cells. Int Immunopharmacol 2003; 3:31–7. [DOI] [PubMed] [Google Scholar]
- 12. Allison AC, Eugui EM. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology 2000; 47:85–118. [DOI] [PubMed] [Google Scholar]
- 13. Colic M, Stojic‐Vukanic Z, Pavlovic B et al Mycophenolate mofetil inhibits differentiation, maturation and allostimulatory function of human monocyte‐derived dendritic cells. Clin Exp Immunol 2003; 134:63–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Izeradjene K, Revillard JP. Apoptosis of superantigen‐activated T cells induced by mycophenolate mofetil treatment. Transplantation 2001; 71:118–25. [DOI] [PubMed] [Google Scholar]
- 15. Novak J, Julian BA, Tomana M et al IgA glycosylation and IgA immune complexes in the pathogenesis of IgA nephropathy. Semin Nephrol 2008; 28:78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhao N, Hou P, Lv J et al The level of galactose‐deficient IgA1 in the sera of patients with IgA nephropathy is associated with disease progression. Kidney Int 2012; 82:790–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Darvill AM, Ballardie FW. Mesangial autoantigens in IgA nephropathy: matrix synthesis and localization. J Lab Clin Med 2006; 147:301–9. [DOI] [PubMed] [Google Scholar]
- 18. O'Donoghue DJ, Darvill A, Ballardie FW. Mesangial cell autoantigens in immunoglobulin A nephropathy and Henoch–Schonlein purpura. J Clin Invest 1991; 88:1522–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ballardie FW, Brenchley PE, Williams S et al Autoimmunity in IgA nephropathy. Lancet 1988; 2:588–92. [DOI] [PubMed] [Google Scholar]
- 20. Berthoux F, Suzuki H, Thibaudin L et al Autoantibodies targeting galactose‐deficient IgA1 associate with progression of IgA nephropathy. J Am Soc Nephrol 2012; 23:1579–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Imasawa T, Utsunomiya Y. Stem cells in renal biology: bone marrow transplantation for the treatment of IgA nephropathy. Exp Nephrol 2002; 10:51–8. [DOI] [PubMed] [Google Scholar]
- 22. Imasawa T, Nagasawa R, Utsunomiya Y et al Bone marrow transplantation attenuates murine IgA nephropathy: role of a stem cell disorder. Kidney Int 1999; 56:1809–17. [DOI] [PubMed] [Google Scholar]
- 23. Imasawa T, Utsunomiya Y, Kawamura T et al Evidence suggesting the involvement of hematopoietic stem cells in the pathogenesis of IgA nephropathy. Biochem Biophys Res Commun 1998; 249:605–11. [DOI] [PubMed] [Google Scholar]
- 24. van Zandbergen G, Westerhuis R, Mohamad NK et al Crosslinking of the human Fc receptor for IgA (FcalphaRI/CD89) triggers FcR gamma‐chain‐dependent shedding of soluble CD89. J Immunol 1999; 163:5806–12. [PubMed] [Google Scholar]
- 25. Moresco RN, Speeckaert MM, Zmonarski SC et al Urinary myeloid IgA Fc alpha receptor (CD89) and transglutaminase‐2 as new biomarkers for active IgA nephropathy and Henoch–Schonlein purpura nephritis. BBA Clin 2016; 5:79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Narita I, Goto S, Saito N et al Genetic polymorphisms in the promoter and 5' UTR region of the Fc alpha receptor (CD89) are not associated with a risk of IgA nephropathy. J Hum Genet 2001; 46:694–8. [DOI] [PubMed] [Google Scholar]
- 27. Launay P, Grossetete B, Arcos‐Fajardo M et al Fcalpha receptor (CD89) mediates the development of immunoglobulin A (IgA) nephropathy (Berger's disease). Evidence for pathogenic soluble receptor–Iga complexes in patients and CD89 transgenic mice. J Exp Med 2000; 191:1999–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. van Zandbergen G, van Kooten C, Mohamad NK et al Reduced binding of immunoglobulin A (IgA) from patients with primary IgA nephropathy to the myeloid IgA Fc‐receptor, CD89. Nephrol Dial Transplant 1998; 13:3058–64. [DOI] [PubMed] [Google Scholar]
- 29. Leung JC, Tsang AW, Chan DT et al Absence of CD89, polymeric immunoglobulin receptor, and asialoglycoprotein receptor on human mesangial cells. J Am Soc Nephrol 2000; 11:241–9. [DOI] [PubMed] [Google Scholar]
- 30. Moura IC, Arcos‐Fajardo M, Sadaka C et al Glycosylation and size of IgA1 are essential for interaction with mesangial transferrin receptor in IgA nephropathy. J Am Soc Nephrol 2004; 15:622–34. [DOI] [PubMed] [Google Scholar]
- 31. Moura IC, Centelles MN, Arcos‐Fajardo M et al Identification of the transferrin receptor as a novel immunoglobulin (Ig)A1 receptor and its enhanced expression on mesangial cells in IgA nephropathy. J Exp Med 2001; 194:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Haddad E, Moura IC, Arcos‐Fajardo M et al Enhanced expression of the CD71 mesangial IgA1 receptor in Berger disease and Henoch–Schonlein nephritis: association between CD71 expression and IgA deposits. J Am Soc Nephrol 2003; 14:327–37. [DOI] [PubMed] [Google Scholar]
- 33. Ballardie FW. IgA nephropathy treatment 25 years on: can we halt progression? The evidence base. Nephrol Dial Transplant 2004; 19:1041–6. [DOI] [PubMed] [Google Scholar]
- 34. Ballardie FW, Cowley RD. Prognostic indices and therapy in IgA nephropathy: toward a solution. Kidney Int 2008; 73:249–51. [DOI] [PubMed] [Google Scholar]
- 35. Tesar V, Troyanov S, Bellur S et al Corticosteroids in IgA nephropathy: a retrospective analysis from the VALIGA study. J Am Soc Nephrol 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rauen T, Eitner F, Fitzner C et al Intensive supportive care plus immunosuppression in IgA nephropathy. N Engl J Med 2015; 373:2225–36. [DOI] [PubMed] [Google Scholar]
- 37. Bright R. Reports of medical cases. London: Longman, Rees, Orme, Brown, and Green, 1827. [Google Scholar]
- 38. Volhard F, Fahr T. Die Brightsche Nierenkrankheit. Berlin: Springer, 1914. [Google Scholar]
- 39. Novak J, Renfrow MB, Gharavi AG et al Pathogenesis of immunoglobulin A nephropathy. Curr Opin Nephrol Hypertens 2013; 22:287–94. [DOI] [PubMed] [Google Scholar]
- 40. Salvadori M, Rosso G. Update on immunoglobulin A nephropathy, part I: pathophysiology. World J Nephrol 2015; 4:455–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sallustio F, Cox SN, Serino G et al Genome‐wide scan identifies a copy number variable region at 3p21.1 that influences the TLR9 expression levels in IgA nephropathy patients. Eur J Hum Genet 2015; 23:940–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kiryluk K, Li Y, Sanna‐Cherchi S et al Geographic differences in genetic susceptibility to IgA nephropathy: GWAS replication study and geospatial risk analysis. PLOS Genet 2012; 8:e1002765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Werner T, Brodersen HP, Janssen U. [Analysis of the spectrum of nephropathies over 24 years in a West German center based on native kidney biopsies]. Med Klin (Munich) 2009; 104:753–9. [DOI] [PubMed] [Google Scholar]
- 44. Rivera F, Lopez‐Gomez JM, Perez‐Garcia R. Frequency of renal pathology in Spain 1994–1999. Nephrol Dial Transplant 2002; 17:1594–602. [DOI] [PubMed] [Google Scholar]
- 45. Briganti EM, Dowling J, Finlay M et al The incidence of biopsy‐proven glomerulonephritis in Australia. Nephrol Dial Transplant 2001; 16:1364–7. [DOI] [PubMed] [Google Scholar]
- 46. Geddes CC, Rauta V, Gronhagen‐Riska C et al A tricontinental view of IgA nephropathy. Nephrol Dial Transplant 2003; 18:1541–8. [DOI] [PubMed] [Google Scholar]
- 47. Lee HS, Lee MS, Lee SM et al Histological grading of IgA nephropathy predicting renal outcome: revisiting H. S. Lee's glomerular grading system. Nephrol Dial Transplant 2005; 20:342–8. [DOI] [PubMed] [Google Scholar]
- 48. Haas M. Histologic subclassification of IgA nephropathy: a clinicopathologic study of 244 cases. Am J Kidney Dis 1997; 29:829–42. [DOI] [PubMed] [Google Scholar]
- 49. Cattran DC, Coppo R, Cook HT et al The Oxford classification of IgA nephropathy: rationale, clinicopathological correlations, and classification. Kidney Int 2009; 76:534–45. [DOI] [PubMed] [Google Scholar]
- 50. Roberts IS, Cook HT, Troyanov S et al The Oxford classification of IgA nephropathy: pathology definitions, correlations, and reproducibility. Kidney Int 2009; 76:546–56. [DOI] [PubMed] [Google Scholar]
- 51. Roberts IS. Oxford classification of immunoglobulin A nephropathy: an update. Curr Opin Nephrol Hypertens 2013; 22:281–6. [DOI] [PubMed] [Google Scholar]
- 52. Serriello I, Polci R, Feriozzi S et al Extracapillary proliferation is an independent predictive factor in IgA nephropathy. Nephrology (Carlton) 2015; 20:654–9. [DOI] [PubMed] [Google Scholar]
- 53. Kaneko Y, Yoshita K, Kono E et al Extracapillary proliferation and arteriolar hyalinosis are associated with long‐term kidney survival in IgA nephropathy. Clin Exp Nephrol 2016; 20:569–77. [DOI] [PubMed] [Google Scholar]
- 54. El Karoui K, Hill GS, Karras A et al A clinicopathologic study of thrombotic microangiopathy in IgA nephropathy. J Am Soc Nephrol 2012; 23:137–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lee H, Yi SH, Seo MS et al Validation of the Oxford classification of IgA nephropathy: a single‐center study in Korean adults. Korean J Intern Med 2012; 27:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lee MJ, Kim SJ, Oh HJ et al Clinical implication of crescentic lesions in immunoglobulin A nephropathy. Nephrol Dial Transplant 2014; 29:356–64. [DOI] [PubMed] [Google Scholar]
- 57. Lee SM, Rao VM, Franklin WA et al IgA nephropathy: morphologic predictors of progressive renal disease. Hum Pathol 1982; 13:314–22. [DOI] [PubMed] [Google Scholar]
- 58. Ibels LS, Gyory AZ. IgA nephropathy: analysis of the natural history, important factors in the progression of renal disease, and a review of the literature. Medicine (Balt) 1994; 73:79–102. [PubMed] [Google Scholar]
- 59. Peters CD, Ring T. Validation of the Oxford classification of IgA nephropathy: valid or invalid? Kidney Int 2015; 87:661–2. [DOI] [PubMed] [Google Scholar]
- 60. Rasche FM, Keller F, von Muller L et al Sequential immunosuppressive therapy in progressive IgA nephropathy. Contrib Nephrol 2007; 157:109–13. [DOI] [PubMed] [Google Scholar]
- 61. Rasche FM, Keller F, von Muller L et al Mycophenolic acid therapy after cyclophosphamide pulses in progressive IgA nephropathy. J Nephrol 2006; 19:465–72. [PubMed] [Google Scholar]
- 62. Rasche FM, Klotz CH, Czock D et al Cyclophosphamide pulse therapy in advanced progressive IgA nephropathy. Nephron Clin Pract 2003; 93:131–6. [DOI] [PubMed] [Google Scholar]
- 63. Rasche FM, Keller F, Rasche WG et al Sequential therapy with cyclophosphamide and mycophenolic acid in patients with progressive IgA nephropathy – a long‐term follow‐up. Clin Exp Immunol 2016; 183:307–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ballardie FW, Roberts IS. Controlled prospective trial of prednisolone and cytotoxics in progressive IgA nephropathy. J Am Soc Nephrol 2002; 13:142–8. [DOI] [PubMed] [Google Scholar]
- 65. Pozzi C, Andrulli S, Del Vecchio L et al Corticosteroid effectiveness in IgA nephropathy: long‐term results of a randomized, controlled trial. J Am Soc Nephrol 2004; 15:157–63. [DOI] [PubMed] [Google Scholar]
- 66. Pozzi C, Andrulli S, Pani A et al Addition of azathioprine to corticosteroids does not benefit patients with IgA nephropathy. J Am Soc Nephrol 2010; 21:1783–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pozzi C, Bolasco PG, Fogazzi GB et al Corticosteroids in IgA nephropathy: a randomised controlled trial. Lancet 1999; 353:883–7. [DOI] [PubMed] [Google Scholar]
- 68. Tang S, Leung JC, Chan LY et al Mycophenolate mofetil alleviates persistent proteinuria in IgA nephropathy. Kidney Int 2005; 68:802–12. [DOI] [PubMed] [Google Scholar]
- 69. Tang SC, Tang AW, Wong SS et al Long‐term study of mycophenolate mofetil treatment in IgA nephropathy. Kidney Int 2010; 77:543–9. [DOI] [PubMed] [Google Scholar]
- 70. Maes BD, Oyen R, Claes K et al Mycophenolate mofetil in IgA nephropathy: results of a 3‐year prospective placebo‐controlled randomized study. Kidney Int 2004; 65:1842–9. [DOI] [PubMed] [Google Scholar]
- 71. Hoshino Y, Kaga T, Abe Y et al Renal biopsy findings and clinical indicators of patients with hematuria without overt proteinuria. Clin Exp Nephrol 2015; 19:918–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Roberts IS. Pathology of IgA nephropathy. Nat Rev Nephrol 2014; 10:445–54. [DOI] [PubMed] [Google Scholar]
- 73. El Karoui K, Hill GS, Karras A et al Focal segmental glomerulosclerosis plays a major role in the progression of IgA nephropathy. II. Light microscopic and clinical studies. Kidney Int 2011; 79:643–54. [DOI] [PubMed] [Google Scholar]
- 74. Ballardie FW, Gartside S, Mallick NP. Computer prediction of the need for dialysis and transplantation using calculated creatinine clearance. BMJ (Clin Res Ed) 1983; 286:1328–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mitch WE, Walser M, Buffington GA et al A simple method of estimating progression of chronic renal failure. Lancet 1976; 2:1326–1328. [DOI] [PubMed] [Google Scholar]
- 76. Fellin G, Gentile MG, Duca G et al Renal function in IgA nephropathy with established renal failure. Nephrol Dial Transplant 1988; 3:17–23. [PubMed] [Google Scholar]
- 77. Rasche FM, Sailer LC, Czock D et al Tonsillectomy, high dose immunoglobulins, and cyclophosphamide in progressive IgA‐nephropathy. Acta Otolaryngol Suppl 2004; 32–7. [DOI] [PubMed] [Google Scholar]
- 78. Tumlin JA, Hennigar RA. Clinical presentation, natural history, and treatment of crescentic proliferative IgA nephropathy. Semin Nephrol 2004; 24:256–68. [DOI] [PubMed] [Google Scholar]
- 79. Tumlin JA, Lohavichan V, Hennigar R. Crescentic, proliferative IgA nephropathy: clinical and histological response to methylprednisolone and intravenous cyclophosphamide. Nephrol Dial Transplant 2003; 18:1321–9. [DOI] [PubMed] [Google Scholar]
- 80. Tamura S, Ueki K, Ideura H et al Corticosteroid therapy in patients with IgA nephropathy and impaired renal function. Clin Nephrol 2001; 55:192–5. [PubMed] [Google Scholar]
- 81. Liang Y, Zhang J, Liu D et al Retrospective study of mycophenolate mofetil treatment in IgA nephropathy with proliferative pathological phenotype. Chin Med J (Engl) 2014; 127:102–8. [PubMed] [Google Scholar]
- 82. Li PK, Leung CB, Chow KM et al Hong Kong study using valsartan in IgA nephropathy (HKVIN): a double‐blind, randomized, placebo‐controlled study. Am J Kidney Dis 2006; 47:751–60. [DOI] [PubMed] [Google Scholar]
- 83. Ballardie FW. Quantitative appraisal of treatment options for IgA nephropathy. J Am Soc Nephrol 2007; 18:2806–9. [DOI] [PubMed] [Google Scholar]
- 84. Peralta CA, Vittinghoff E, Bansal N et al Trajectories of kidney function decline in young black and white adults with preserved GFR: results from the Coronary Artery Risk Development in Young Adults (CARDIA) study. Am J Kidney Dis 2013; 62:261–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bolignano D, Mattace‐Raso F, Sijbrands EJ et al The aging kidney revisited: a systematic review. Ageing Res Rev 2014; 14:65–80. [DOI] [PubMed] [Google Scholar]
- 86. Andrews PA, Burnapp L, Manas D et al Summary of the British Transplantation Society/Renal Association U.K. guidelines for living donor kidney transplantation. Transplantation 2012; 93:666–73. [DOI] [PubMed] [Google Scholar]
- 87. Xu G, Tu W, Jiang D et al Mycophenolate mofetil treatment for IgA nephropathy: a meta‐analysis. Am J Nephrol 2009; 29:362–7. [DOI] [PubMed] [Google Scholar]
- 88. Cheng J, Zhang X, Zhang W et al Efficacy and safety of glucocorticoids therapy for IgA nephropathy: a meta‐analysis of randomized controlled trials. Am J Nephrol 2009; 30:315–22. [DOI] [PubMed] [Google Scholar]
- 89. Cheng J, Zhang W, Zhang XH et al ACEI/ARB therapy for IgA nephropathy: a meta analysis of randomised controlled trials. Int J Clin Pract 2009; 63:880–8. [DOI] [PubMed] [Google Scholar]
- 90. Feehally J, Coppo R, Troyanov S et al Tonsillectomy in a European cohort of 1,147 patients with IgA nephropathy. Nephron 2015; 132:15–24. [DOI] [PubMed] [Google Scholar]
- 91. Vecchio M, Bonerba B, Palmer SC et al Immunosuppressive agents for treating IgA nephropathy. Cochrane Database Syst Rev 2015; 8:CD003965. [DOI] [PubMed] [Google Scholar]
- 92. Scholl U, Wastl U, Risler T et al The ‘point of no return’ and the rate of progression in the natural history of IgA nephritis. Clin Nephrol 1999; 52:285–92. [PubMed] [Google Scholar]
- 93. D'Amico G, Minetti L, Ponticelli C et al Prognostic indicators in idiopathic IgA mesangial nephropathy. Q J Med 1986; 59:363–78. [PubMed] [Google Scholar]
- 94. Pozzi C, Del Vecchio L, Locatelli F. Can immunosuppressive therapy be useful in IgA nephropathy when the ‘point of no return’ has already been exceeded? Nephron 2002; 92:699–701. [DOI] [PubMed] [Google Scholar]
- 95. Rivera GM, Merino Rivas JL, Alarcon Garcelan MC et al Outcome of HIV‐infected patients of peritoneal dialysis: experience in a center and literature review. Nefrologia 2008; 28:505–10. [PubMed] [Google Scholar]
- 96. Kim MJ, McDaid JP, McAdoo SP et al Spleen tyrosine kinase is important in the production of proinflammatory cytokines and cell proliferation in human mesangial cells following stimulation with IgA1 isolated from IgA nephropathy patients. J Immunol 2012; 189:3751–8. [DOI] [PubMed] [Google Scholar]
- 97. Wagrowska‐Danilewicz M, Danilewicz M, Stasikowska O. CC chemokines and chemokine receptors in IgA nephropathy (IgAN) and in non‐IgA mesangial proliferative glomerulonephritis (MesProGN). The immunohistochemical comparative study. Pol J Pathol 2005; 56:121–6. [PubMed] [Google Scholar]
- 98. Ihm CG, Jeong KW, Lee SH et al Effects of therapeutic agents on the inflammatory and fibrogenic factors in IgA nephropathy. Nephrology (Carlton) 2007; 12 Suppl 3:S25–6. [DOI] [PubMed] [Google Scholar]
- 99.KDIGO Clinical Practice Guideline for Glomerulonephritis. Chapter 10: immunoglobulin A nephropathy. Kidney Int Suppl 2012; 2:209–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Maschio G, Cagnoli L, Claroni F et al ACE inhibition reduces proteinuria in normotensive patients with IgA nephropathy: a multicentre, randomized, placebo‐controlled study. Nephrol Dial Transplant 1994; 9:265–9. [PubMed] [Google Scholar]
- 101. Rekola S, Bergstrand A, Bucht H. Deterioration rate in hypertensive IgA nephropathy: comparison of a converting enzyme inhibitor and beta‐blocking agents. Nephron 1991; 59:57–60. [DOI] [PubMed] [Google Scholar]
- 102. Kang Z, Li Z, Duan C et al Mycophenolate mofetil therapy for steroid‐resistant IgA nephropathy with the nephrotic syndrome in children. Pediatr Nephrol 2015; 30:1121–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Yaginuma T, Yamamoto H, Mitome J et al Successful treatment of nephrotic syndrome caused by recurrent IgA nephropathy with chronic active antibody‐mediated rejection three years after kidney transplantation. Clin Transplant 2011; 25:28–33. [DOI] [PubMed] [Google Scholar]
- 104. Liu XW, Li DM, Xu GS et al Comparison of the therapeutic effects of leflunomide and mycophenolate mofetil in the treatment of immunoglobulin A nephropathy manifesting with nephrotic syndrome. Int J Clin Pharmacol Ther 2010; 48:509–13. [DOI] [PubMed] [Google Scholar]
- 105. Bomback AS, Canetta PA, Beck LH Jr et al Treatment of resistant glomerular diseases with adrenocorticotropic hormone gel: a prospective trial. Am J Nephrol 2012; 36:58–67. [DOI] [PubMed] [Google Scholar]
- 106. Masuda Y, Tamura S, Sugiyama N. The effect of tonsillectomy and its postoperative clinical course in IgA nephropathy with chronic tonsillitis. Adv Otorhinolaryngol 1992; 47:203–7. [DOI] [PubMed] [Google Scholar]
- 107. Akagi H, Kosaka M, Hattori K et al Long‐term results of tonsillectomy as a treatment for IgA nephropathy. Acta Otolaryngol Suppl 2004; 38–42. [DOI] [PubMed] [Google Scholar]
- 108. Sugiyama N, Shimizu J, Nakamura M et al Clinicopathological study of the effectiveness of tonsillectomy in IgA nephropathy accompanied by chronic tonsillitis. Acta Otolaryngol Suppl 1993; 508:43–8. [DOI] [PubMed] [Google Scholar]
- 109. Rasche FM, Schwarz A, Keller F. Tonsillectomy does not prevent a progressive course in IgA nephropathy. Clin Nephrol 1999; 51:147–52. [PubMed] [Google Scholar]
- 110. Hotta O, Miyazaki M, Furuta T et al Tonsillectomy and steroid pulse therapy significantly impact on clinical remission in patients with IgA nephropathy. Am J Kidney Dis 2001; 38:736–43. [DOI] [PubMed] [Google Scholar]
- 111. Hotta O, Taguma Y, Kurosawa K et al Early intensive therapy for clinical remission of active IgA nephropathy: a three‐year follow‐up study. 1993; 35:967–73. [PubMed] [Google Scholar]
- 112. Hotta O, Taguma Y, Yoshizawa N et al Long‐term effects of intensive therapy combined with tonsillectomy in patients with IgA nephropathy. Acta Otolaryngol Suppl 1996; 523:165–8. [PubMed] [Google Scholar]
- 113. Smerud HK, Barany P, Lindstrom K et al New treatment for IgA nephropathy: enteric budesonide targeted to the ileocecal region ameliorates proteinuria. Nephrol Dial Transplant 2011; 26:3237–42. [DOI] [PubMed] [Google Scholar]
- 114. Brunner M, Vogelsang H, Greinwald R et al Colonic spread and serum pharmacokinetics of budesonide foam in patients with mildly to moderately active ulcerative colitis. Aliment Pharmacol Ther 2005; 22:463–70. [DOI] [PubMed] [Google Scholar]
- 115. Manno C, Torres DD, Rossini M et al Randomized controlled clinical trial of corticosteroids plus ACE‐inhibitors with long‐term follow‐up in proteinuric IgA nephropathy. Nephrol Dial Transplant 2009; 24:3694–701. [DOI] [PubMed] [Google Scholar]
- 116. Zhou YH, Tang LG, Guo SL et al Steroids in the treatment of IgA nephropathy to the improvement of renal survival: a systematic review and meta‐analysis. PLOS ONE 2011; 6:e18788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Schena FP, Manno C. Intensive supportive care plus immunosuppression in IgA nephropathy. N Engl J Med 2016; 374:992. [DOI] [PubMed] [Google Scholar]
- 118. Ayoub I, Hebert L, Rovin BH. Intensive supportive care plus immunosuppression in IgA nephropathy. N Engl J Med 2016; 374:991–2. [DOI] [PubMed] [Google Scholar]
- 119. Robert T, Cambier A, Hertig A. Intensive supportive care plus immunosuppression in IgA nephropathy. N Engl J Med 2016; 374:991. [DOI] [PubMed] [Google Scholar]
- 120. Lai KN, Lai FM, Li PK et al Cyclosporin treatment of IgA nephropathy: a short term controlled trial. BMJ (Clin Res Ed) 1987; 295:1165–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Hahn BH, McMahon MA, Wilkinson A et al American College of Rheumatology guidelines for screening, treatment, and management of lupus nephritis. Arthritis Care Res (Hoboken) 2012; 64:797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Lv J, Xu D, Perkovic V et al Corticosteroid therapy in IgA nephropathy. J Am Soc Nephrol 2012; 23:1108–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Austin HA III, Klippel JH, Balow JE et al Therapy of lupus nephritis. Controlled trial of prednisone and cytotoxic drugs. N Engl J Med 1986; 314:614–9. [DOI] [PubMed] [Google Scholar]
- 124. Haubitz M, Schellong S, Gobel U et al Intravenous pulse administration of cyclophosphamide versus daily oral treatment in patients with antineutrophil cytoplasmic antibody‐associated vasculitis and renal involvement: a prospective, randomized study. Arthritis Rheum 1998; 41:1835–44. [DOI] [PubMed] [Google Scholar]
- 125. Guillevin L, Cordier JF, Lhote F et al A prospective, multicenter, randomized trial comparing steroids and pulse cyclophosphamide versus steroids and oral cyclophosphamide in the treatment of generalized Wegener's granulomatosis. Arthritis Rheum 1997; 40:2187–98. [DOI] [PubMed] [Google Scholar]
- 126. Boumpas DT, Austin HA III, Vaughn EM et al Controlled trial of pulse methylprednisolone versus two regimens of pulse cyclophosphamide in severe lupus nephritis. Lancet 1992; 340:741–5. [DOI] [PubMed] [Google Scholar]
- 127. Mok CC, Ho CT, Siu YP et al Treatment of diffuse proliferative lupus glomerulonephritis: a comparison of two cyclophosphamide‐containing regimens. Am J Kidney Dis 2001; 38:256–64. [DOI] [PubMed] [Google Scholar]
- 128. Schaier M, Scholl C, Scharpf D et al High interpatient variability in response to mycophenolic acid maintenance therapy in patients with ANCA‐associated vasculitis. Nephrol Dial Transplant 2015; 30 Suppl 1:i138–45. [DOI] [PubMed] [Google Scholar]
- 129. Han F, Liu G, Zhang X et al Effects of mycophenolate mofetil combined with corticosteroids for induction therapy of microscopic polyangiitis. Am J Nephrol 2011; 33:185–92. [DOI] [PubMed] [Google Scholar]
- 130. Hiemstra TF, Walsh M, Mahr A et al Mycophenolate mofetil vs azathioprine for remission maintenance in antineutrophil cytoplasmic antibody‐associated vasculitis: a randomized controlled trial. JAMA 2010; 304:2381–8. [DOI] [PubMed] [Google Scholar]
- 131. Iatrou C, Zerbala S, Revela I et al Mycophenolate mofetil as maintenance therapy in patients with vasculitis and renal involvement. Clin Nephrol 2009; 72:31–7. [DOI] [PubMed] [Google Scholar]
- 132. A blinded randomized clinical trial of mycophenolate mofetil for the prevention of acute rejection in cadaveric renal transplantation . The Tricontinental Mycophenolate Mofetil Renal Transplantation Study Group. Transplantation 1996; 61:1029–37. [PubMed] [Google Scholar]
- 133. Clayton PA, McDonald SP, Chapman JR et al Mycophenolate versus azathioprine for kidney transplantation: a 15‐year follow‐up of a randomized trial. Transplantation 2012; 94:152–8. [DOI] [PubMed] [Google Scholar]
- 134. Bullingham R, Monroe S, Nicholls A et al Pharmacokinetics and bioavailability of mycophenolate mofetil in healthy subjects after single‐dose oral and intravenous administration. J.Clin.Pharmacol 1996; 36:315–24. [DOI] [PubMed] [Google Scholar]
- 135. Budde K, Braun KP, Glander P et al Pharmacodynamic monitoring of mycophenolate mofetil in stable renal allograft recipients. Transplant Proc 2002; 34:1748–50. [DOI] [PubMed] [Google Scholar]
- 136. Sollinger H. Enteric‐coated mycophenolate sodium: therapeutic equivalence to mycophenolate mofetil in de novo renal transplant patients. Transplant Proc 2004; 36:517S–20S. [DOI] [PubMed] [Google Scholar]
- 137. Salvadori M, Holzer H, de Mattos A et al Enteric‐coated mycophenolate sodium is therapeutically equivalent to mycophenolate mofetil in de novo renal transplant patients. Am J Transplant 2004; 4:231–6. [DOI] [PubMed] [Google Scholar]
- 138. Langman LJ, LeGatt DF, Halloran PF et al Pharmacodynamic assessment of mycophenolic acid‐induced immunosuppression in renal transplant recipients. Transplantation 1996; 62:666–72. [DOI] [PubMed] [Google Scholar]
- 139. Langman LJ, Shapiro AM, Lakey JR et al Pharmacodynamic assessment of mycophenolic acid‐induced immunosuppression by measurement of inosine monophosphate dehydrogenase activity in a canine model. Transplantation 1996; 61:87–92. [DOI] [PubMed] [Google Scholar]
- 140. Glander P, Hambach P, Braun KP et al Effect of mycophenolate mofetil on IMP dehydrogenase after the first dose and after long‐term treatment in renal transplant recipients. Int J Clin Pharmacol Ther 2003; 41:470–6. [DOI] [PubMed] [Google Scholar]
- 141. Maes B, Evenepoel P, Kuypers D et al A prospective placebo‐controlled randomized single centre study of mycophenolate mofetil treatment for IgA nephropathy: lack of clinical efficacy after two years [Abstract]. J Am Soc Nephrol 2001; 12:114A. 11134257 [Google Scholar]
- 142. Frisch G, Lin J, Rosenstock J et al Mycophenolate mofetil (MMF) vs placebo in patients with moderately advanced IgA nephropathy: a double‐blind randomized controlled trial. Nephrol Dial Transplant 2005; 20:2139–45. [DOI] [PubMed] [Google Scholar]
- 143. Chen X, Chen P, Cai G et al [A randomized control trial of mycophenolate mofetil treatment in severe IgA nephropathy] in Chinese. Zhonghua Yi Xue Za Zhi 2002; 82:796–801. [PubMed] [Google Scholar]
- 144. Kuypers DR, de Jonge H, Naesens M et al Effects of CYP3A5 and MDR1 single nucleotide polymorphisms on drug interactions between tacrolimus and fluconazole in renal allograft recipients. Pharmacogenet Genomics 2008; 18:861–8. [DOI] [PubMed] [Google Scholar]
- 145. Czock D, Rasche FM, Carius A et al Pharmacokinetics and pharmacodynamics of mycophenolic acid after enteric‐coated mycophenolate versus mycophenolate mofetil in patients with progressive IgA nephritis. J Clin Pharmacol 2007; 47:850–9. [DOI] [PubMed] [Google Scholar]
- 146. Langford CA, Talar‐Williams C, Sneller MC. Mycophenolate mofetil for remission maintenance in the treatment of Wegener's granulomatosis. Arthritis Rheum 2004; 51:278–83. [DOI] [PubMed] [Google Scholar]
- 147. Pesavento TE, Bay WH, Agarwal G et al Mycophenolate therapy in frequently relapsing minimal change disease that has failed cyclophosphamide therapy. Am J Kidney Dis 2004; 43:3–6. [DOI] [PubMed] [Google Scholar]
- 148. Ensign LM, Cone R, Hanes J. Oral drug delivery with polymeric nanoparticles: the gastrointestinal mucus barriers. Adv Drug Deliv Rev 2012; 64:557–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Janoff EN, Rubins JB, Fasching C et al Pneumococcal IgA1 protease subverts specific protection by human IgA1. Mucosal Immunol 2016. Feb 5. pii: ASN.2015080856. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Lechner SM, Abbad L, Boedec E et al IgA1 protease treatment reverses mesangial deposits and hematuria in a model of IgA nephropathy. J Am Soc Nephrol 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Yin G, Wu Y, Zeng CH et al Coexistence of Fabry disease and IgA nephropathy: a report of two cases. Ir J Med Sci 2014; 183:671–5. [DOI] [PubMed] [Google Scholar]
- 152. Inoue T, Sugiyama H, Kitagawa M et al Suppression of adiponectin by aberrantly glycosylated IgA1 in glomerular mesangial cells in vitro and in vivo . PLOS ONE 2012; 7:e33965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Kokubo T, Hiki Y, Iwase H et al Protective role of IgA1 glycans against IgA1 self‐aggregation and adhesion to extracellular matrix proteins. J Am Soc Nephrol 1998; 9:2048–54. [DOI] [PubMed] [Google Scholar]
- 154. Lamm ME, Emancipator SN, Robinson JK et al Microbial IgA protease removes IgA immune complexes from mouse glomeruli in vivo: potential therapy for IgA nephropathy. Am J Pathol 2008; 172:31–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Xie LS, Huang J, Qin W et al Immunoglobulin A1 protease: a new therapeutic candidate for immunoglobulin A nephropathy. Nephrology (Carlton) 2010; 15:584–6. [DOI] [PubMed] [Google Scholar]
- 156. Mistry D, Stockley RA. IgA1 protease. Int J Biochem Cell Biol 2006; 38:1244–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Larche M, Akdis CA, Valenta R. Immunological mechanisms of allergen‐specific immunotherapy. Nat Rev Immunol 2006; 6:761–71 [DOI] [PubMed] [Google Scholar]
- 158. Suzuki Y, Suzuki H, Yasutake J et al Paradigm shift in activity assessment of IgA nephropathy – optimizing the next generation of diagnostic and therapeutic maneuvers via glycan targeting. Expert Opin Biol Ther 2015; 15:583–93. [DOI] [PubMed] [Google Scholar]
- 159. Heybeli C, Unlu M, Yildiz S et al IgA nephropathy: association of C4d with clinical and histopathological findings and possible role of IgM. Ren Fail 2015; 37:1464–9. [DOI] [PubMed] [Google Scholar]
- 160. Coppo R. Proteasome inhibitors in progressive renal diseases. Nephrol Dial Transplant 2014; 29 Suppl 1:i25–30. [DOI] [PubMed] [Google Scholar]
- 161. Nakata J, Suzuki Y, Suzuki H et al Changes in nephritogenic serum galactose‐deficient IgA1 in IgA nephropathy following tonsillectomy and steroid therapy. PLOS ONE 2014; 9:e89707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Suzuki H, Suzuki Y, Narita I et al Toll‐like receptor 9 affects severity of IgA nephropathy. J Am Soc Nephrol 2008; 19:2384–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Tian J, Wang Y, Guo H et al The Akt/mTOR/p70S6K pathway is activated in IgA nephropathy and rapamycin may represent a viable treatment option. Exp Mol Pathol 2015; 99:435–40. [DOI] [PubMed] [Google Scholar]
- 164. Moura IC, Benhamou M, Launay P et al The glomerular response to IgA deposition in IgA nephropathy. Semin Nephrol 2008; 28:88–95. [DOI] [PubMed] [Google Scholar]
- 165. Tamouza H, Vende F, Tiwari M et al Transferrin receptor engagement by polymeric IgA1 induces receptor expression and mesangial cell proliferation: role in IgA nephropathy. Contrib Nephrol 2007; 157:144–7. [DOI] [PubMed] [Google Scholar]
- 166. Moura IC, Arcos‐Fajardo M, Gdoura A et al Engagement of transferrin receptor by polymeric IgA1: evidence for a positive feedback loop involving increased receptor expression and mesangial cell proliferation in IgA nephropathy. J Am Soc Nephrol 2005; 16:2667–76. [DOI] [PubMed] [Google Scholar]
- 167. Kanamaru Y, Pfirsch S, Aloulou M et al Inhibitory ITAM signaling by Fc alpha RI‐FcR gamma chain controls multiple activating responses and prevents renal inflammation. J Immunol 2008; 180:2669–78. [DOI] [PubMed] [Google Scholar]
- 168. Tanaka M, Arai H, Liu N et al Role of coagulation factor Xa and protease‐activated receptor 2 in human mesangial cell proliferation. Kidney Int 2005; 67:2123–33. [DOI] [PubMed] [Google Scholar]
- 169. Liu DJ, Liu Y, Ran LM et al Genetic variants in interleukin genes and susceptibility to IgA nephropathy: a meta‐analysis. DNA Cell Biol 2014; 33:345–54. [DOI] [PubMed] [Google Scholar]
- 170. Sumida K, Ubara Y, Suwabe T et al Complete remission of myeloperoxidase‐anti‐neutrophil cytoplasmic antibody‐associated crescentic glomerulonephritis complicated with rheumatoid arthritis using a humanized anti‐interleukin 6 receptor antibody. Rheumatology (Oxf) 2011; 50:1928–30. [DOI] [PubMed] [Google Scholar]
- 171. Komatsuda A, Wakui H, Togashi M et al IgA nephropathy associated with Castleman disease with cutaneous involvement. Am J Med Sci 2010; 339:486–90. [DOI] [PubMed] [Google Scholar]
- 172. Kluger N, Du‐Thanh A, Bessis D et al Psoriasis‐associated IgA nephropathy under infliximab therapy. Int J Dermatol 2015; 54:e79–80. [DOI] [PubMed] [Google Scholar]
- 173. Ueno Y, Tanaka S, Onitake T et al Infliximab treatment for Crohn's disease in a patient with IgA nephropathy. Clin J Gastroenterol 2009; 2:380–3. [DOI] [PubMed] [Google Scholar]
- 174. Michel M, Henri P, Vincent FB et al Mesangial immunoglobulin (Ig)A glomerulonephritis in a patient with rheumatoid arthritis treated with abatacept. Joint Bone Spine 2013; 80:660–3. [DOI] [PubMed] [Google Scholar]
- 175. Sugiura H, Takei T, Itabashi M et al Effect of single‐dose rituximab on primary glomerular diseases. Nephron Clin Pract 2011; 117:c98–105. [DOI] [PubMed] [Google Scholar]
- 176. Chancharoenthana W, Townamchai N, Leelahavanichkul A et al Rituximab for recurrent IgA nephropathy in kidney transplantation: a report of 3 cases and proposed mechanisms. Nephrology (Carlton) 2016. Jan 12. doi: 10.1111/nep.12722. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 177. Berthoux F, El Deeb S, Mariat C et al Antithymocyte globulin (ATG) induction therapy and disease recurrence in renal transplant recipients with primary IgA nephropathy. Transplantation 2008; 85:1505–7. [DOI] [PubMed] [Google Scholar]
- 178. Ring T, Pedersen BB, Salkus G et al Use of eculizumab in crescentic IgA nephropathy: proof of principle and conundrum? Clin Kidney J 2015; 8:489–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Hogg RJ, Lee J, Nardelli N et al Clinical trial to evaluate omega‐3 fatty acids and alternate day prednisone in patients with IgA nephropathy: report from the Southwest Pediatric Nephrology Study Group. Clin J Am Soc Nephrol 2006; 1:467–74. [DOI] [PubMed] [Google Scholar]
- 180. Praga M, Gutierrez E, Gonzalez E et al Treatment of IgA nephropathy with ACE inhibitors: a randomized and controlled trial. J Am Soc Nephrol 2003; 14:1578–83. [DOI] [PubMed] [Google Scholar]
- 181. Hogg RJ. A randomized, placebo‐controlled, multicenter trial evaluating alternate‐day prednisone and fish oil supplements in young patients with immunoglobulin A nephropathy. Scientific Planning Committee of the IgA Nephropathy Study. Am J Kidney Dis 1995; 26:792–6. [DOI] [PubMed] [Google Scholar]
- 182. Hogg RJ, Fitzgibbons L, Atkins C et al Efficacy of omega‐3 fatty acids in children and adults with IgA nephropathy is dosage‐ and size‐dependent. Clin J Am Soc Nephrol 2006; 1:1167–72. [DOI] [PubMed] [Google Scholar]
- 183. Katafuchi R, Ikeda K, Mizumasa T et al Controlled, prospective trial of steroid treatment in IgA nephropathy: a limitation of low‐dose prednisolone therapy. Am J Kidney Dis 2003; 41:972–83. [DOI] [PubMed] [Google Scholar]
- 184. Tanaka H, Suzuki K, Nakahata T et al Combined therapy of enalapril and losartan attenuates histologic progression in immunoglobulin A nephropathy. Pediatr Int 2004; 46:576–9. [DOI] [PubMed] [Google Scholar]
- 185. Kuriki M, Asahi K, Asano K et al Steroid therapy reduces mesangial matrix accumulation in advanced IgA nephropathy. Nephrol Dial Transplant 2003; 18:1311–5. [DOI] [PubMed] [Google Scholar]
- 186. Russo D, Minutolo R, Pisani A et al Coadministration of losartan and enalapril exerts additive antiproteinuric effect in IgA nephropathy. Am J Kidney Dis 2001; 38:18–25. [DOI] [PubMed] [Google Scholar]
- 187. Maschio G, Alberti D, Janin G et al Effect of the angiotensin‐converting‐enzyme inhibitor benazepril on the progression of chronic renal insufficiency. The Angiotensin‐Converting‐Enzyme Inhibition in Progressive Renal Insufficiency Study Group. N Engl J Med 1996; 334:939–45. [DOI] [PubMed] [Google Scholar]
- 188. Shoji T, Nakanishi I, Suzuki A et al Early treatment with corticosteroids ameliorates proteinuria, proliferative lesions, and mesangial phenotypic modulation in adult diffuse proliferative IgA nephropathy. Am J Kidney Dis 2000; 35:194–201. [DOI] [PubMed] [Google Scholar]
- 189. Yoshikawa N, Ito H. Combined therapy with prednisolone, azathioprine, heparin–warfarin, and dipyridamole for paediatric patients with severe IgA nephropathy – is it relevant for adult patients? Nephrol Dial Transplant 1999; 14:1097–9. [DOI] [PubMed] [Google Scholar]
- 190. Uzu T, Harada T, Namba T et al Thiazide diuretics enhance nocturnal blood pressure fall and reduce proteinuria in immunoglobulin A nephropathy treated with angiotensin II modulators. J Hypertens 2005; 23:861–5. [DOI] [PubMed] [Google Scholar]
- 191. Xie Y, Nishi S, Ueno M et al The efficacy of tonsillectomy on long‐term renal survival in patients with IgA nephropathy. Kidney Int 2003; 63:1861–7. [DOI] [PubMed] [Google Scholar]
- 192. Kobayashi Y, Fujii K, Hiki Y et al Steroid therapy in IgA nephropathy: a prospective pilot study in moderate proteinuric cases. Q J Med 1986; 61:935–43. [PubMed] [Google Scholar]
- 193. Kobayashi Y, Fujii K, Hiki Y et al Steroid therapy in IgA nephropathy: a retrospective study in heavy proteinuric cases. Nephron 1988; 48:12–7. [DOI] [PubMed] [Google Scholar]
- 194. Kobayashi Y, Hiki Y, Fujii K et al Effect of corticosteroids on renal function in progressive IgA nephropathy – long‐term follow‐up study. Nippon Jinzo Gakkai Shi 1988; 30:1135–42. [PubMed] [Google Scholar]
- 195. Kobayashi Y, Hiki Y, Fujii K et al Moderately proteinuric IgA nephropathy: prognostic prediction of individual clinical courses and steroid therapy in progressive cases. Nephron 1989; 53:250–6. [DOI] [PubMed] [Google Scholar]
- 196. Kobayashi Y, Hiki Y, Kokubo T et al Steroid therapy during the early stage of progressive IgA nephropathy. A 10‐year follow‐up study. Nephron 1996; 72:237–42. [DOI] [PubMed] [Google Scholar]
- 197. Iino Y, Ambe K, Kato Y et al Chronic tonsillitis and IgA nephropathy. Clinical study of patients with and without tonsillectomy. Acta Otolaryngol Suppl 1993; 508:29–35. [DOI] [PubMed] [Google Scholar]
- 198. Woo KT, Chiang GS, Lau YK et al IgA nephritis in Singapore: clinical, prognostic indices, and treatment. Semin Nephrol 1987; 7:379–81. [PubMed] [Google Scholar]
- 199. Sato M, Hotta O, Tomioka S et al Cohort study of advanced IgA nephropathy: efficacy and limitations of corticosteroids with tonsillectomy. Nephron Clin Pract 2003; 93:c137–45. [DOI] [PubMed] [Google Scholar]
- 200. Liu X, Dewei D, Sun S et al Treatment of severe IgA nephropathy: mycophenolate mofetil/prednisone compared to cyclophosphamide/prednisone. Int J Clin Pharmacol Ther 2014; 52:95–102. [DOI] [PubMed] [Google Scholar]
- 201. Donadio JV Jr. Use of fish oil to treat patients with immunoglobulin a nephropathy. Am J Clin Nutr 2000; 71:373–5. [DOI] [PubMed] [Google Scholar]
- 202. Donadio JV, Bergstralh EJ, Offord KP et al A controlled trial of fish oil in IgA nephropathy. Mayo Nephrology Collaborative Group. N Engl J Med 1994; 331:1194–9. [DOI] [PubMed] [Google Scholar]
- 203. Donadio JV Jr, Grande JP, Bergstralh EJ et al The long‐term outcome of patients with IgA nephropathy treated with fish oil in a controlled trial. Mayo Nephrology Collaborative Group. J Am Soc Nephrol 1999; 10:1772–7. [DOI] [PubMed] [Google Scholar]
- 204. Donadio JV Jr, Larson TS, Bergstralh EJ et al A randomized trial of high‐dose compared with low‐dose omega‐3 fatty acids in severe IgA nephropathy. J Am Soc Nephrol 2001; 12:791–9. [DOI] [PubMed] [Google Scholar]
- 205. Goumenos D, Ahuja M, Shortland JR et al Can immunosuppressive drugs slow the progression of IgA nephropathy? Nephrol Dial Transplant 1995; 10:1173–81. [PubMed] [Google Scholar]
- 206. Goumenos DS, Davlouros P, El Nahas AM et al Prednisolone and azathioprine in IgA nephropathy ‐ a ten‐year follow‐up study. Nephron Clin Pract 2003; 93:58–68. [DOI] [PubMed] [Google Scholar]
- 207. Pettersson EE, Rekola S, Berglund L et al Treatment of IgA nephropathy with omega‐3‐polyunsaturated fatty acids: a prospective, double‐blind, randomized study. Clin Nephrol 1994; 41:183–90. [PubMed] [Google Scholar]
- 208. Roccatello D, Ferro M, Cesano G et al Steroid and cyclophosphamide in IgA nephropathy. Nephrol Dial Transplant 2000; 15:833–5. [DOI] [PubMed] [Google Scholar]
- 209. Tsuruya K, Harada A, Hirakata H et al Combination therapy using prednisolone and cyclophosphamide slows the progression of moderately advanced IgA nephropathy. Clin Nephrol 2000; 53:1–9. [PubMed] [Google Scholar]
- 210. Peters HP, van den Brand JA, Berger SP et al Immunosuppressive therapy in patients with IgA nephropathy. Neth J Med 2015; 73:284–9. [PubMed] [Google Scholar]
- 211. Nakao N, Yoshimura A, Morita H et al Combination treatment of angiotensin‐II receptor blocker and angiotensin‐converting‐enzyme inhibitor in non‐diabetic renal disease (COOPERATE): a randomised controlled trial. Lancet 2003; 361:117–24. [DOI] [PubMed] [Google Scholar]