

Summary
Hematopoietic stem cells (HSCs) sustain long-term reconstitution of hematopoiesis in transplantation recipients, yet their role in the endogenous steady-state hematopoiesis remains unclear. In particular, recent studies suggested that HSCs provide a relatively minor contribution to immune cell development in the adults. We directed transgene expression in a fraction of HSCs that maintained reconstituting activity during serial transplantations. Inducible genetic labeling showed that transgene-expressing HSCs gave rise to other phenotypic HSCs, confirming their top position in the differentiation hierarchy. The labeled HSCs rapidly contributed to committed progenitors of all lineages and to mature myeloid cells and lymphocytes, but not to B-1a cells or tissue macrophages. Importantly, labeled HSCs gave rise to more than two-thirds of all myeloid cells and platelets in adult mice, and this contribution could be accelerated by an induced interferon response. Thus, classically defined HSCs maintain immune cell development in the steady state and during systemic cytokine responses.
eTOC/In Brief
The role of hematopoietic stem cells (HSCs) in steady-state hematopoiesis remains controversial. Here, Sawai et al. use lineage tracing to reveal a major contribution of HSCs to all blood cell lineages, including myeloid cells and lymphocytes, throughout the adult life.
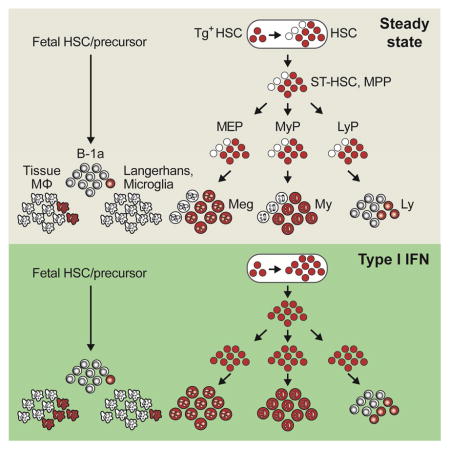
Introduction
Throughout the adult life, all blood cell types including platelets, erythrocytes and leukocytes undergo constant replenishment from the bone marrow (BM). Continuous blood development (hematopoiesis) is particularly important for the immune system, in which myeloid cells such as granulocytes, monocytes and dendritic cells (DCs) are very short-lived (Geering et al., 2013; Geissmann et al., 2010). Peripheral T and B lymphocytes are relatively long-lived and may be sustained in part by homeostatic proliferation, yet they require the input of new cells to maintain a diverse polyclonal repertoire of antigen specificities (Montecino-Rodriguez et al., 2013). The only exceptions are cell types that develop primarily during embryogenesis, such as tissue macrophages (MΦ) (Ginhoux and Jung, 2014) and innate-like lymphocytes such as B-1a cells (Montecino-Rodriguez and Dorshkind, 2012). Dysregulation of hematopoiesis is associated with immunodeficiencies and age-related immune dysfunction, and is a direct cause of myelodysplastic syndromes and leukemia.
The BM of adult mammals maintains a rare population of hematopoietic stem cells (HSCs) that can permanently reconstitute blood formation when transferred into lethally irradiated or otherwise myelosuppressed hosts (Eaves, 2015; Shizuru et al., 2005). The long-term reconstituting HSCs are thought to give rise to phenotypically and transcriptionally distinct short-term HSCs (ST-HSCs) that lack durable self-renewal and can produce only a transient wave of reconstitution. ST-HSCs and the partially overlapping multipotent progenitors (MPPs) progressively generate lineage-restricted progenitors and mature cells of the myeloid (e.g. granulocytes and monocytes), lymphoid (T, B and NK cells) and megaerythroid (platelets and erythrocytes) lineages (Wilson et al., 2015). Transplantation-competent HSCs are less proliferative than downstream progenitors (Cheshier et al., 1999; Passegue et al., 2005; Wilson et al., 2008), and are localized in specific BM microenvironment(s) termed the HSC niche (Boulais and Frenette, 2015). The durable reconstitution by HSCs underlies the success of clinical hematopoietic transplantation and gene therapy protocols, the life-saving therapies for many diseases including leukemia and primary immunodeficiencies.
It has long been assumed that the paradigm of hierarchical HSC differentiation, which was derived from transplantation studies, also applies to steady-state hematopoiesis. In other words, all hematopoietic cells, including all major immune cell types such as myeloid cells and lymphocytes, are continuously generated from endogenous HSCs in the BM. This notion, however, has been challenged by recent studies. Thus, clonal cell analysis using transposon-mediated “barcoding” suggested that granulocyte development is completely uncoupled from the HSC activity (Sun et al., 2014). A subsequent study using lineage tracing showed that genetically labeled HSCs did contribute to myeloid and lymphoid cells. However, the contribution was infrequent, and it was therefore proposed that most mature cells are derived primarily from ST-HSCs (Busch et al., 2015). Collectively these studies raise the possibility that steady-state hematopoiesis is sustained primarily by progenitors rather than by HSCs, with the latter predominantly activated only during hematopoietic stress such as transplantation. This model therefore poses a fundamental question in developmental immunology, namely: what is the ultimate source of continuous lymphopoiesis and myelopoiesis in adult mammals?
The HSC activity in the BM of normal adult mice is contained in a small subset of Lineage marker (Lin)− Sca-1+ c-Kit+ (LSK) cells (Ikuta and Weissman, 1992; Morrison and Weissman, 1994) that express high levels of CD150 (Slamf1) and endothelial protein C receptor (EPCR) but low levels of CD135 (Flt3), CD48 (Slamf2) and CD34 (Balazs et al., 2006; Forsberg et al., 2005; Kent et al., 2009; Kiel et al., 2005). However, these phenotypically defined HSCs are heterogeneous in their differentiation potential and self-renewal capacity, as revealed by single-cell transplantation (Benz et al., 2012; Challen et al., 2010; Dykstra et al., 2007; Morita et al., 2010) and genetic reporters (Chen et al., 2016; Sanjuan-Pla et al., 2013). Importantly, only a minor fraction of HSCs can support long-term multilineage reconstitution in serial transplantations, as opposed to primary transplantations (Oguro et al., 2013; Yamamoto et al., 2013). These cells presumably comprise the top of hematopoietic hierarchy, yet their contribution to hematopoiesis has not been studied. Indeed, genetic labeling in (Sun et al., 2014) was not HSC-specific, and in (Busch et al., 2015) involved a small (~1%) random fraction of the HSC population. Therefore, an efficient genetic labeling of HSCs, particularly of the most potent HSCs with superior self-renewal capacity, is required to elucidate their contribution to endogenous hematopoiesis.
Here we developed genetic systems to prospectively label HSCs with serial reconstitution capacity, and to test their fundamental properties in unperturbed animals. The labeled HSCs gave rise to other cells within the HSC population, confirming their highest position in the hematopoietic hierarchy. Importantly, these HSCs provided a major sustained contribution to hematopoiesis in the steady state and during interferon response. Our results therefore favor a view in which HSCs provide an ultimate source of adult immune cell development.
Results
Pdzk1ip1-GFP transgene is expressed in a subset of HSCs
To genetically mark and trace HSCs, we used bacterial artificial chromosome (BAC) clones to drive transgene expression from the Pdzk1ip1 locus. Pdzk1ip1 (also known as Map17) is expressed in the murine HSC population but is strongly reduced in more differentiated cells or in mobilized HSCs (Cabezas-Wallscheid et al., 2014; Forsberg et al., 2010; Wilson et al., 2015). We used a BAC clone containing the entire Tal1-Pdzk1ip1 locus to direct the expression of green fluorescent protein (GFP) from the Pdzk1ip1 gene (Fig. S1A). The resulting GFP transgene was expressed at high levels in all LSK CD150+ CD48− HSCs; however, it was also detectable in some LSK CD150− CD48+ multipotent progenitors (MPPs), and the majority of Lin− Sca1− c-Kit+ CD150+ megakaryocyte-erythrocyte progenitors (MEPs) (Fig. S1B–D).
To improve the specificity of Pdzk1ip1 transgene expression, we dissociated Pdzk1ip1 and Tal1 loci by using a 5′ truncated BAC clone lacking the promoter and upstream region of Tal1 (Fig. S1A). The resulting Pdzk1ip1-GFP transgene was expressed in a small population of Lin− cells, most of which had the HSC phenotype (Fig. 1A). Conversely, the HSC population contained ~27% GFP+ cells, whereas progenitors including MPPs, MEPs and myeloid progenitors (Lin− Sca1− c-Kit+ CD150−, MyPs) contained <1% of GFP+ cells (Fig. 1B,C). Some Lin+ cells with lower GFP levels were also detected and found to represent mature granulocytes (3–4% of these) (Fig. 1A–C). By several phenotypic definitions currently used in the field (Fig. S1E), GFP-expressing cells were enriched among the HSCs that were rigorously defined as CD34−/lo (Wilson et al., 2008) or CD229− (HSC-1; Oguro et al., 2013) (Fig. 1D,E). Conversely, GFP+ cells expressed lower levels of CD34 and CD229 and higher levels of EPCR than the GFP− cells (Fig 1F). Finally, we tested the proliferation of HSC populations by measuring the incorporation of nucleoside analogue 5-bromodeoxyuridine (BrdU) in vivo. As shown in Fig. 1G, GFP+ HSCs showed the lowest rate of BrdU incorporation compared to GFP− HSCs or progenitors. Thus, the 5′ truncated Pdzk1ip1 transgene was preferentially expressed in a subset of HSCs with the most undifferentiated phenotype and the lowest proliferation rate.
Fig. 1. Pdzk1ip1-GFP is expressed in a fraction of adult bone marrow HSCs.

A. The expression of GFP within the total BM cells of adult Pdzk1ip1-GFP mice and the phenotype of gated GFP+ cells. The phenotypes of total or Lin− cells from control non-transgenic BM are shown for comparison. Note that GFP+ Lin+ cells correspond to SSChi granulocytes and GFP+ Lin− cells correspond to HSCs.
B. Representative histograms of GFP expression in HSCs (LSK CD48− CD150+), MPPs (LSK CD48+ CD150−), MyPs (Lin− Sca1− cKit+ CD150−), and MEPs (Lin− Sca1− cKit+ CD150+) from Pdzk1ip1-GFP and control non-transgenic animals.
C. The fraction of GFP+ cells within the indicated BM populations (MkP defined as Lin− Sca1− cKit+ CD150+ CD41+; B, B220+; T, TCRβ+; Gran, Gr1+ CD11b+ SSChi). Bars represent medians of individual animals shown (n=13 for all cells except lymphocytes, for which n=5).
D. The expression of GFP in the HSC and progenitor (MPP1–4) populations defined according to (Cabezas-Wallscheid et al., 2014; Wilson et al., 2008) and the fraction of GFP+ cells within each population (bars represent medians of individual animals shown, n=7).
E. The expression of GFP in the HSC (HSC-1 and HSC-2) and progenitor (MPP and HPC-1) populations defined according to (Oguro et al., 2013). The expression of CD229 in the HPC-1 population is shown as a control (orange shadow histogram). Bars represent medians of individual animals shown (n=8).
F. Expression of surface markers in GFP+ and GFP− HSCs. Control populations showing the expression range of each marker are shown as grey shadow histograms. Representative of 3 individual animals.
G. The fraction of GFP+ and GFP− HSCs and progenitors (MPPs and MyPs) that incorporated BrdU after a 3-day pulse. Shown are values from 6 individual animals. Statistical significance: *p ≤ 0.05; **p ≤ 0.01.
See also Fig. S1.
Transgene-expressing HSCs have an undifferentiated expression profile and serial reconstitution capacity
We performed global mRNA sequencing (RNA-Seq) of sorted GFP+ and GFP− HSCs, along with the MPP population from the same pooled sample as a control. Clustering and principal component analysis (PCA) of the resulting expression profiles confirmed the expected differences between HSCs and MPPs, and the high concordance between GFP+ and GFP− HSCs (Fig. 2A and Dataset S1). The expression of Pdzk1ip1 was only slightly enriched in GFP+ HSCs, confirming that the truncated Pdzk1ip1-GFP transgene has a more restricted expression than the endogenous gene. Consistent with their immature phenotype, GFP+ HSCs showed a slight decrease of differentiation markers (e.g. Cd34, Itga2b and Cd48) and of multiple genes associated with proliferation (e.g. Mki67, Ccne2). (Dataset S1). Direct comparison with RNA-Seq profiles of wild-type HSCs and progenitor populations (Cabezas-Wallscheid et al., 2014) showed that genes enriched in GFP+ compared to GFP− HSCs were also highly enriched in wild-type HSCs compared to their immediate progeny (MPP1), and vice versa (Fig. 2B). Furthermore, combined PCA of all samples showed that GFP+ HSCs segregated closest to the HSC samples of (Cabezas-Wallscheid et al., 2014) (data not shown). We conclude that the expression profile of GFP+ HSCs is consistent with the least differentiated status, including the reduction of differentiation markers and proliferation signature genes.
Fig. 2. Pdzk1ip1-GFP+ HSCs have an undifferentiated expression profile and serial reconstitution capacity.

A–B. RNA-Seq analysis of GFP+ and GFP− HSCs and MPPs from Pdzk1ip1-GFP mice. A. Unsupervised clustering dendrogram of the three samples.
B. Comparison with RNA-Seq profiles of wild-type HSCs and their immediate progeny (MPP1) (Cabezas-Wallscheid et al., 2014). Shown is pairwise comparison of transcript frequencies in HSCs and MPP1 for all genes (total) or for genes enriched in the GFP+ or GFP− HSCs from Pdzk1ip1-GFP mice (Dataset S1). Genes increased >2-fold in HSCs or MPP1 are highlighted in green or red, respectively.
C. The fraction of GFP+ cells in donor-derived HSCs and MPPs of primary recipients reconstituted with either GFP+ or GFP− HSCs six months post-transplant (mean ± S.D. of 5 recipients).
D. The design of experiment to assess the ability of 20 sorted GFP+ (green) or GFP− (grey) CD45+ EPCR+ CD150+ CD48− (ESLAM) HSCs to reconstitute hematopoiesis in the primary (1°) and secondary (2°) recipients.
E. The fraction of donor-derived cells among different blood cell lineages in recipients assessed at least 16 weeks post-transplant in primary and secondary recipients of 20 ESLAM HSCs. Shown are values for individual recipient mice pooled from two independent experiments.
See also Fig. S2 and Dataset S1.
We tested the reconstitution capacity of Pdzk1ip1 transgene-expressing HSCs in serial transplantations. Primary transplantation of GFP+ or GFP− HSCs yielded comparably robust and lineage-balanced reconstitution of myeloid (granulocytes, monocytes), lymphoid (T and B cells) and megakaryocytic (platelets) lineages when HSCs were isolated as LSK CD48− CD150+ (Fig S2A) or as LSK CD135− cells (Fig S2B). Notably, GFP+ HSCs regenerated both GFP+ and GFP− HSCs in transplanted hosts, whereas recipients of GFP− HSCs contained only GFP− cells (Fig. 2C). Thus, GFP+ HSCs could give rise to GFP− HSCs but not vice versa. Moreover, GFP− HSCs generated from the original GFP+ cells in primary recipients showed poor reconstitution of secondary recipients, in contrast to the robust activity of the HSCs that retained GFP expression (Fig S2C).
We then transplanted recipient mice with 20 GFP+ or GFP− HSCs defined as CD45+ EPCR+ CD48− CD150+ (ESLAM) (Kent et al., 2009) (Fig. 2D). Both subsets gave robust reconstitution of all primary recipients at 8–12 weeks (data not shown) and at 16 weeks (Fig. 2E). However, again only the GFP+ progeny of these GFP+ ESLAM HSCs reconstituted secondary recipients (Fig. 2E). Of note, the majority of GFP+ ESLAM HSCs recipients showed multilineage reconstitution of granulocytes, T and B cells (6/6 and 6/8 in primary and secondary transplantations, respectively). In single-cell transplantations, 5 out of 14 primary recipients showed multilineage reconstitution (Fig. S2D), whereas 8/14 primarily reconstituted granulocytes (corresponding to α- and β-HSCs, respectively (Dykstra et al., 2007)). Thus, the Pdzk1ip1 transgene expression selectively marks HSCs that are uniquely capable of reconstituting secondary recipients. Collectively, the transgene-expressing HSCs display the most undifferentiated phenotype and expression profile, serial multilineage reconstitution capacity and the ability to regenerate themselves and the transgene-negative subset.
Pdzk1ip1-CreER transgene specifically labels undifferentiated HSCs
To inducibly label and trace the Pdzk1ip1 transgene-expressing HSCs, we created mice expressing a tamoxifen-inducible Cre recombinase-estrogen receptor fusion protein (CreER) under the control of the same 5′ truncated Pdzk1ip1 BAC clone. The resulting Pdzk1ip1-CreER transgenic animals were crossed to the Rosa26tdTomato (R26Tom) strain, in which Cre recombination permanently activates expression of the red fluorescent protein tdTomato (Tom). Three days after a single administration of tamoxifen, Tom expression was observed in a small fraction of BM cells, the majority of which represented HSC (Fig. 3A). The labeling was observed in a fraction of HSCs (mean, 32.6%; median 30%, Fig. 3B,C) that was comparable to the Pdzk1ip1-GFP mice. The labeling was selective for HSCs, as it was observed in only ~3% of MPPs and <0.6% of all downstream progenitors (Fig. 3B,C). In addition to HSCs, the expression of Pdzk1ip1 transgenes was detected in the kidney, reproductive system, and skin (data not shown). Unlike in the Pdzk1ip1-GFP transgene, no “leaky” labeling of granulocytes was observed, likely because of lower transgene expression levels in these cells.
Fig. 3. Pdzk1ip1-CreER transgene inducibly labels undifferentiated HSCs.

A. The expression of tdTomato (Tom) in Pdzk1ip1-CreER R26Tom/Tom mice or Cre-negative R26Tom/Tom controls three days after a single tamoxifen administration. Shown is representative expression of Tom within the total BM cells and the phenotype of gated Tom+ cells.
B,C. Representative histograms of Tom expression in the indicated cell populations (panel B) and the fraction of Tom+ cells within each population defined as in Fig. 1B,C (panel C, bars indicate median of individual animals, n=6).
D–E. Histograms and fractions of Tom expression in HSC/progenitor populations defined as in Fig. 1D and E, respectively (bars represent medians of individual animals shown, n=6).
F. Expression of surface markers in the Tom+ and Tom− subsets of HSCs. Indicated control populations showing the expression range of each marker are shown as grey shadow histograms. Representative of 3 individual animals.
G. The retention of H2B-GFP marker protein in the HSC/progenitor subsets. Pdzk1ip1-CreER R26Tom R26rtTA Col1a1tetO-H2BGFP mice were pulsed for 6 weeks to express H2B-GFP, chased for 14 weeks, induced with tamoxifen and analyzed 3 days later. Shown are representative histograms of H2B-GFP expression in Tom+ (+) or Tom− (−) HSCs, MPPs and MyPs, and the fractions of GFP+ cells in these populations from individual animals.
H. Labeled HSCs in the BM sections of Pdzk1ip1-CreER R26Tom/Tom mice 3 days post-tamoxifen were detected by endogenous tdTomato fluorescence (arrowhead). Shown is representative localization of Tom+ HSCs in sections stained for the sinusoid endothelial marker endomucin. Statistical significance: *p ≤ 0.05; **p ≤ 0.01; ns, not significant.
See also Fig. S3.
As in the case of Pdzk1ip1-GFP+ cells, the labeled Tom+ cells were significantly enriched among the CD34−/lo HSCs (Wilson et al., 2008) (Fig. 3D). No significant difference was observed between CD229− HSC-1 and CD229+ HSC-2 (Oguro et al., 2013), yet both phenotypes were highly enriched for Tom+ cells compared to ST-HSCs or progenitors (Fig. 3E). Moreover, Tom+ HSCs had slightly lower CD34 and CD229 and higher EPCR levels than the Tom− cells (Fig. 3F). To directly compare the HSC subsets labeled by GFP and CreER transgenes, we crossed the two transgenic strains and analyzed GFP and Tom expression 3 days after tamoxifen treatment. Despite a lower GFP expression in double-transgenic compared to single-transgenic mice, the fraction of GFP+ HSCs was significantly enriched in the Tom+ population, and vice versa (Fig. S3A–C). Thus, similar to the Pdzk1ip1-GFP transgene, Pdzk1ip1-CreER preferentially labels a subset of HSCs with the most immature phenotype.
HSCs are thought to turn over slowly and therefore to retain labeled DNA or chromatin (Foudi et al., 2009; Wilson et al., 2008). To analyze label retention, the Pdzk1ip1-CreER mice were crossed with the mice that allow global doxycycline (Dox)-inducible expression of the histone 2B-GFP (H2B-GFP) fusion protein (Foudi et al., 2009). The resulting Pdzk1ip1-CreER R26Tom R26rtTA Col1a1tetO-H2BGFP mice were administered Dox for 6 weeks to induce H2B-GFP and then kept without Dox for 14 weeks to allow label dilution. The mice were then induced with tamoxifen and analyzed 3 days later for the expression of GFP in the labeled Tom+ HSC subset. Up to 50% of Tom+ HSCs retained H2B-GFP, whereas the H2B-GFP+ fraction was significantly lower in Tom− HSCs and virtually absent in progenitors (Fig. 3G). These data suggest that the labeled HSCs are heterogeneous with respect to label retention but are enriched in label-retaining cells. The intensity of H2B-GFP signal in Tom+ HSCs was progressively decreasing with time (data not shown), suggesting that they still undergo substantial proliferation.
The labeled Tom+ HSCs could be detected in frozen BM sections by their native red fluorescent signal, and were confirmed to express CD150 but not lineage markers, CD48 or CD41 (Fig. S3D,E). Simultaneous staining for endothelium showed that most Tom+ HSCs were located adjacent to the endomucin-positive endothelium of the sinusoids (Fig. 3H), consistent with recent observations (Acar et al., 2015; Ding and Morrison, 2013). Given the broad distribution of sinusoids in the BM (Kunisaki et al., 2013), the identity of cell types and vessels adjacent to Tom+ HSCs still remains to be determined. Overall, the labeled HSCs demonstrate preferential label retention and perivascular localization, the expected attributes of a bona fide HSC.
Labeled HSCs give rise to other phenotypic HSCs
We used lineage tracing to analyze the progeny of labeled HSCs. If labeled cells represent a random sampling of the HSC population, their frequency would remain constant over time, as observed previously (Busch et al., 2015). On the other hand, if labeled HSCs give rise to other cells within the HSC population, the fraction of labeled HSCs would increase with time. Indeed, the initial fraction of labeled HSCs increased ~2-fold after several weeks (Fig. 4A). Serial BM biopsies of the same tamoxifen-induced animals confirmed the rapid increase in labeled HSCs by 12 weeks, eventually reaching 80–90% Tom+ by 46 weeks (Fig. 4B). As expected, the labeling of immediate HSC progeny (ST-HSCs and MPPs, Fig. S4A) increased >5-fold by 11–12 weeks, and approached (but did not equal) that of HSCs by 28–40 weeks (Fig. 4A,B). The fluorescence intensity of Tom in HSCs increased as well, likely reflecting its accrual in these slowly dividing cells (Fig. S4B). Thus, similar to transplantation settings, the labeled HSCs give rise to other cells within the HSC population, suggesting that they represent the highest level of hematopoietic hierarchy. Furthermore, the actual initial labeling of this “top-level” HSC subset must have been high (80–90%) to allow the similarly high endpoint labeling of the entire HSC compartment (Fig. 4C).
Fig. 4. Labeled HSCs give rise to other cells within the HSC population.

A. The fraction of labeled Tom+ cells in the HSC/progenitor compartment of Pdzk1ip1-CreER R26Tom/Tom reporter mice. The HSC and progenitor subsets within the LSK population were defined on the basis of CD150/CD48 expression as shown in Fig. S4A. Shown are the median (bars) and individual values (circles) for animals sacrificed at 3 days, 3–4 weeks or 11 weeks after tamoxifen treatment.
B. Continuous analysis of Tom+ cells in the HSC/progenitor compartment of tamoxifen-treated reporter mice by serial BM biopsy. Shown is the fraction of Tom+ cells in HSC/progenitor subsets in the same animals over time (median ± interquartile range, n= 5).
C. The dynamics of Tom+ label accumulation in the HSC compartment (left) and its decomposition into two-subset model (right). The modeled composition of the HSC compartment is shown as a pie diagram. The labeled fraction of “top-level” HSCs is assumed to be constant since they are fully self-renewing while the labeled fraction of their progeny is steadily growing.
See also Fig. S4.
Labeled HSCs contribute to all hematopoietic lineages
We analyzed the spectrum of top-HSC differentiation into hematopoietic progenitors (defined as in Fig. S5A,B). Because minor fractions of ST-HSCs and MPPs were labeled at the outset of lineage tracing (>10%, Fig. 4A,B), the origin of Tom+ cells below these frequencies cannot be ascertained in our tracing experiments. However, any Tom+ cells above these frequencies must necessarily have arisen from an upstream population with the highest labeling, i.e. the HSCs. We found that the labeling of lineage-restricted erythroid (EryPs), megakaryocytic (MkPs), myeloid (MyPs), and monocyte and dendritic cell (MDPs, CDPs) progenitors reached >30% 11 weeks post-induction (Fig. 5A). Common lymphoid progenitors (CLPs), early thymic T cell progenitors (ETPs) and immature thymocytes showed lower labeling at 4–11 weeks, consistent with the slower kinetics of HSC differentiation into lymphoid progenitors as observed before (Busch et al., 2015). However, by 36 weeks the labeling of all progenitors reached or exceeded ~60%, revealing a major contribution of labeled HSCs to all lineages.
Fig. 5. Labeled HSCs provide a major contribution to all lineage-committed progenitors.

A–B. The fraction of labeled Tom+ cells in lineage-committed progenitors from Pdzk1ip1-CreER R26Tom/Tom reporter mice. Cell populations in the BM and thymus were defined as shown in Fig. S5A. Shown are the median (bars) and values from individual animals (circles) at the indicated time points after tamoxifen treatment in the BM (panel A) and thymus (panel B). The median fractions of Tom+ HSCs at the respective time points are indicated by dashed lines.
C. The fraction of labeled Tom+ cells in myeloid progenitors and granulocytes (Grans) from Pdzk1ip1-CreER R26Tom/Tom reporter mice analyzed by serial BM biopsy. Shown is the fraction of Tom+ cells in the same animals over time (median ± interquartile range, n= 5).
D. A linear model of granulopoiesis based on serial BM biopsy data. Estimated values of differentiation and regeneration for each population are shown in the units of cells per week. Each population is shown by a circle with an area proportional to the number of cells, the width of each differentiation arrow (linear, blue) is proportional to the differentiation rate, the width of the regeneration arrow (circular, purple) is proportional to the regeneration rate, except the rates for MyPs that are ~100 times fold higher that for other populations.
See also Fig. S5.
We used serial BM biopsy to examine the differentiation of labeled HSCs into mature granulocytes (Gr), asking in particular how very few HSCs could sustain the massive continuous production of these cells. The biopsy itself did not affect the rate of HSC differentiation as judged by the emergence of labeled Gr (Fig. S5C). The labeling of common progenitors (MPPs), committed progenitors (MyPs) and the terminal differentiated progeny (Gr) followed a nearly identical kinetics from <3% at the outset to ~70% by 46 weeks (Fig. 5C). These data suggest that the rate-limiting HSC-to-MPP transition is followed by the rapid and linear differentiation of MPPs to granulocytes. Fig. 5D shows a mathematical model of this process with estimated values of differentiation and regeneration for each cell population. For the labeled HSCs at the top of the differentiation sequence, these rates are equal as expected for a fully self-renewing compartment. For the downstream compartments, these rates appear very close, i.e. all inputs are amplified and converted into the output. Notably, the absolute values of these rates progressively increase by several orders of magnitude, reflecting the increasing proliferation rates and cell numbers in each progenitor population. Thus, the few differentiating HSCs can continuously produce large numbers of granulocytes through massive amplification at progenitor stages.
The kinetics and spectrum of HSC differentiation into peripheral cells
We examined tamoxifen-induced Pdzk1ip1-CreER R26Tom mice by peripheral blood (PB) sampling (Fig. S6A). The procedure itself did not affect the rate of HSC differentiation compared to the endpoint analysis (Fig. S5C). Labeled platelets and myeloid cells became detectable at 2 and 4 wks, respectively, and thereafter increased in parallel to reach ~66% by 32 wks (Fig. 6A). Peripheral NK, T and B cells were labeled with a longer lag period (8–12 weeks) and respectively lower rates (Fig. 6A). Endpoint analysis confirmed that myeloid cells including granulocytes, monocytes and DCs (Fig. S6B) became labeled up to 60–70% at 36–46 wks (Fig. 6B). Similarly, high labeling frequencies were observed in NK cells, thymocytes and immature B cells in the BM, confirming that the slow labeling of peripheral lymphocytes reflects their slow turnover (Fig. 6B). The endpoint labeling of mature cells (60–70%) approached that of HSCs (80–90%) but did not fully equilibrate with it (Fig. 6B), suggesting that a few HSCs did not differentiate within the analyzed period.
Fig. 6. Labeled HSCs provide a major contribution to mature hematopoietic cells.

A. The fraction of labeled Tom+ cells in peripheral blood cells of Pdzk1ip1-CreER R26Tom/Tom reporter mice at the indicated time points after tamoxifen administration. (median ± range of 4 animals, representative of two experiments).
B–D. The fraction of labeled Tom+ cells in mature cell types of reporter mice. Shown are the median (bars) and values from individual animals (circles) at the indicated time points after tamoxifen treatment.
B. Major immune cell types in the spleen or thymus. The median fractions of Tom+ HSCs at the respective time points are indicated by dashed lines.
C. Conventional B cells in the spleen or peritoneal cavity (PC) and B-1a cells in PC.
D. Resident macrophages from the indicated tissues.
See also Fig. S6.
We analyzed the cell types that are thought to emerge during embryonic hematopoiesis (Fig. S6C). Very few Tom+ cells (<5%) were detected at 36–46 wks among B-1a cells in the peritoneal cavity (PC), compared to conventional B cells in the spleen or PC (Fig. 6C). Similarly, hardly any labeling was detected in the MΦ in the lung and the brain (microglia) or in the resident DCs in the epidermis (Langerhans cells) (Fig. 6D). A higher fraction of Tom+ cells (20–30%) was found among MΦ in the PC, spleen and liver (Kupffer cells) (Fig. 6D). Thus, the contribution of HSCs to embryo-derived immune cell types is limited but varies substantially between tissues.
We treated tamoxifen-induced mice with the double-stranded RNA polymer poly-I:C, an inducer of type I interferon that increases HSC proliferation (Essers et al., 2009; Pietras et al., 2014). We found that the pattern of HSC contribution to peripheral cells was similar to that in the steady state (Fig. 7A). However, the kinetics was greatly accelerated, so that the same ~70% labeling of myeloid cells and platelets was reached in only half the time (Fig. 7A). Moreover, at the 40 wk endpoint the fraction of Tom+ progenitors and mature cells (~90%, Fig. 7B) approached that of Tom+ HSCs (~95%, shown in the figure as a dashed line). These results suggest that poly-I:C mobilized nearly all labeled HSCs into differentiation. The contribution to B-1a cells, Langerhans cells and microglia remained very low (<10%, Fig. 7C,D), but increased in MΦ in the spleen (~50%), PC and liver (30–40%). Collectively, these results show that HSCs provide a major sustained contribution to steady-state hematopoiesis, except for the few embryo-derived immune cell types. This contribution could be accelerated by an acute inflammatory stimulus, revealing a feedback control of HSC differentiation by innate immune responses.
Fig. 7. Interferon response accelerates HSC differentiation.

Pdzk1ip1-CreER R26Tom/Tom reporter mice were induced with tamoxifen and then treated with poly-I:C on days 6, 8 and 10.
A. The fraction of Tom+ cells in the peripheral blood (presented as in Fig. 6A; median ± range of 4 animals).
B–D. The fraction of Tom+ cells at the 40 week endpoint; shown are the median (bars) and values from individual animals (circles).
B. Major immune cell types in the spleen or thymus. The median fraction of Tom+ HSCs at this time point is indicated for comparison (dashed line).
C. Conventional B cells in the spleen or PC and B-1a cells in PC.
D. Resident macrophages from the indicated tissues.
Discussion
We used the murine Pdzk1ip1 gene as a driver to target the expression of transgenes to the HSC population. Though the function of Pdzk1ip1 is not well studied, the gene, unlike in mice, is not expressed in human HSCs (Bagger et al., 2016), and consequently it is unlikely to have a major role in HSC function. Indeed, the lack of an HSC-intrinsic function is characteristic of multiple phenotypic (CD150, EPCR) and genetic (α-catulin) HSC markers. The full-length BAC clone recapitulated the expression of endogenous Pdzk1ip1 and targeted transgene expression to all HSCs and to some progenitors, similar to several other HSC-specific genes (Gazit et al., 2014; Kataoka et al., 2011). In contrast, the artificially 5′ truncated Pdzk1ip1 locus targeted transgene expression to a subset of HSC, with only minimal expression in progenitors. The artificial 5′ truncation removed the shared regulatory regions of Tal1 and Pdzk1ip1, apparently reducing the transgene’s epigenetic stability. Indeed, the ectopic expression in a fraction of granulocytes was observed in several founder lines of the truncated transgene, but not with the full-length transgene. On the other hand, the truncation also likely accounted for the transgene’s unique specificity for immature HSCs and the sensitivity to the loss of self-renewal potential.
Whereas the original definition of long-term HSCs reflected their capacity to reconstitute primary recipients (Morrison and Weissman, 1994; Spangrude et al., 1988), only a fraction of those HSCs were capable of reconstituting secondary recipients (Oguro et al., 2013; Yamamoto et al., 2013). Several genetic markers were recently developed to identify HSC subsets with a particular lineage bias (Vwf, (Sanjuan-Pla et al., 2013)) or a superior reconstitution capacity (Hoxb5, (Chen et al., 2016)) during primary transplantations. However, the Pdzk1ip1-based transgene appears distinct in identifying HSCs that excel in secondary transplantations. Accordingly, previously used genetic markers showed only slight (Hoxb5) or no (Vwf) enrichment in Pdzk1ip1-GFP+ HSCs (Dataset S1). Transgene-expressing HSCs exhibited the most undifferentiated surface phenotype, relatively slow proliferation (as measured by label dilution) and localization in the perivascular niche in the BM. Transplanted transgene-expressing HSCs regenerated both themselves and the transgene-negative HSCs, whereas by lineage tracing the transgene-expressing HSCs give rise to other HSCs in the steady state. Collectively, these data suggest that our system identifies the least differentiated subset of HSCs with the highest self-renewal capacity and ability to produce other phenotypic HSCs.
The analysis of HSC differentiation by lineage tracing revealed their major sustained input to all major hematopoietic lineages. Indeed, HSCs produced the majority of conventional myeloid cells and platelets within ~8 months, i.e. less than half of the lifespan of a laboratory mouse. Of note, the labeling of progenitors and mature cells (60–70%) did not fully equilibrate with that of HSCs (>80%), suggesting that some HSCs do not differentiate within this time frame. However, this lack of contribution applies to a minor fraction of labeled HSCs, and therefore likely represents reversible quiescence (Wilson et al., 2008) rather than the primary HSC state. Our mathematical model estimates that 3–8% of labeled HSCs entered differentiation per day, followed by massive flux amplification at the downstream stages. This agrees with a relatively low but nevertheless considerable proliferation of all HSCs in the steady state (Cheshier et al., 1999; Kiel et al., 2007; Passegue et al., 2005; Wilson et al., 2008) and with the substantial label dilution observed in our experiments. The relatively slow but constant HSC input appears to be amplified through massive proliferation of progenitors, thereby maintaining continuous development of immune cells such as granulocytes.
The observed robust contribution of HSCs to steady-state hematopoiesis contrasts with the near-absent contribution of HSCs to granulopoiesis reported by (Sun et al., 2014). This study used an ingenious clonal labeling by inducible transposon mobilization, albeit without any cell type specificity. More importantly, potential caveats of this approach include secondary transpositions (e.g. due to the “leaky” transposase expression after the initial induction), and/or false positive integration sites due to multiple rounds of PCR cloning. These possibilities were not ruled out, and may have contributed to the observed “uncoupling” of barcodes between HSCs and granulocytes. A subsequent steady-state lineage tracing of HSCs estimated that ~30% of HSCs contribute to differentiated cells (Busch et al., 2015). This study used a CreER strain that labeled only 0.5–1% of the total HSC population 1–3 weeks after a 5-day tamoxifen treatment. Because of the low labeling efficiency, no continuous sampling was used, and the mature cell labeling was estimated relatively to the average HSC labeling. Moreover, the low efficiency likely reduced the probability of observable HSC differentiation, whereas the long lag before the start of tracing may have selected for the most quiescent HSCs. In contrast, our system efficiently labeled a distinct subset of “top-level” HSCs (rather than a small random HSC sample) within days after a single tamoxifen treatment. This efficient labeling i) facilitated lineage tracing of the majority of HSCs with precise timing and continuous sampling; ii) allowed the definition of lineage labeling in absolute terms, rather than relative to the fraction of labeled HSCs; iii) minimized the adverse effects of tamoxifen on HSCs (Nakada et al., 2014; Sanchez-Aguilera et al., 2014). The resulting data are generally consistent with those by Busch et al., but suggest a much higher and faster contribution of HSCs to adult hematopoiesis.
With respect to the immune system, two aspects of HSC differentiation appear notable. The first is the absent or limited HSC contribution to several immune cell types such as B-1a cells and tissue MΦ. These results not only confirmed the primary embryonic origin of these cells, but also revealed a marked variability in their maintenance. Thus, microglia and Langerhans cells received virtually no HSC input, consistent with their proposed earliest derivation in the embryo (Ginhoux and Jung, 2014). On the other hand, splenic MΦ and Kupffer cells showed a detectable HSC contribution in the steady state and particularly after interferon stimulation. These results further suggest that tissue MΦ represent a spectrum showing different degrees of self-renewal versus hematopoietic contribution (Gentek et al., 2014). Second, we found that type I interferon, a known inducer of HSC proliferation (Essers et al., 2009; Walter et al., 2015), accelerated multilineage HSC differentiation. Moreover, it caused a nearly-complete equilibration of labeling in HSCs and mature cells, revealing that the few fully quiescent HSCs have been induced to differentiate. These data suggest that cytokine-induced HSC proliferation can be directly coupled to increased differentiation without affecting its lineage pattern.
In conclusion, our results suggest that HSCs represent the major source of endogenous hematopoiesis, even though downstream progenitors may show homeostatic capacities and independent contributions. Our data are consistent with the recent clonal analysis in humans following hematopoietic reconsititution, revealing a major sustained contribution of multipotent HSCs over many years (Biasco et al., 2016). They also help explain the essential biology of HSCs, such as their active role in the aging of hematopoietic system (Adams et al., 2015) and the frequent origin of leukemogenic mutations in the HSC pool (Elias et al., 2014). The genetic platform described herein should facilitate further studies of HSC differentiation in the steady state, during immune responses and in pathological conditions.
Experimental Procedures
Animals
The generation of BAC transgenic mouse strains is described in the Supplemental Experimental Procedures. Transgenic Pdzk1ip1-CreER animals were crossed to Cre-inducible reporter strain R26Tom/Tom (Madisen et al., 2010) and backcrossed to achieve homozygosity for the reporter allele. For label retention studies, Pdzk1ip1-CreER R26Tom/Tom mice were crossed with the R26rtTA/rtTA Col1a1tetO-H2BGFP/tetO-H2BGFP mice (Foudi et al., 2009) to generate Pdzk1ip1-CreER R26Tom/+ R26rtTA/+ Col1a1tetO-H2BGFP/+ F1 mice. Adult 8–10 week old mice were used in all experiments.
Animal procedures
All experiments with mice were performed according to protocols approved for use by the investigators (B.R. and C.J.E.) by their corresponding Institutional Animal Care and Use Committees. For cell proliferation analysis, adult Pdzk1ip1-GFP mice were injected i.p. with 1 mg BrdU (Sigma-Aldrich) and kept on water containing 0.8 mg/ml BrdU for 3 days. To induce recombination in the Pdzk1ip1-CreER R26Tom/Tom mice, a single dose of 50 mg/kg tamoxifen (Sigma-Aldrich) in sunflower oil was administered to 8–10 week-old mice by gavage. To induce an interferon response, Pdzk1ip1-CreER R26Tom/Tom mice were treated with tamoxifen and then injected i.p. with 0.15 mg of polyinosinic:polycytidylic acid (poly-I:C, GE Life Sciences) 6, 8 and 10 days later. For label retention analyses, Pdzk1ip1-CreER R26Tom/+ R26rtTA/+ Col1a1tetO-H2BGFP/+ F1 mice were given doxycycline (2 mg/ml in drinking water containing 10 mg/ml sucrose) for 6 weeks to induce H2B-GFP expression. The mice were then kept without doxycycline for 14 weeks, treated with tamoxifen and analyzed 3 days later.
Peripheral blood (~0.1 ml) was collected by submandibular vein puncture with a sterile disposable lancet. BM biopsy was done as described (Verlinden et al., 1998) on isoflurane-anesthetized animals by inserting a 28½ G insulin syringe needle into the joint surface of the femur through the patellar tendon and then into the bone cavity. Up to 20 μl of the bone marrow suspension (~106 cells) was collected.
Hematopoietic reconstitution
Sorted HSCs from Tg(Pdzk1ip1-GFP) animals (pure C57BL/6 background) were injected i.v. into lethally irradiated C57BL/6 recipients (B6.SJL) congenic for the CD45.1 isoform of pan-hematopoietic marker CD45. Total BM (105 cells) from B6.SJL mice were injected in parallel to ensure recipient survival. The transplantation of ESLAM cells was done essentially as described (Dykstra et al., 2007; Kent et al., 2009). Briefly, sorted GFP+ and GFP− ESLAM HSCs were injected i.v. into sublethally irradiated B6.SJL-W41/W41 mice, which permit BM engraftment in the absence of lethal irradiation or the “rescue” BM. Donor-derived leukocytes in the blood and BM of recipient mice were defined as CD45.2+ CD45.1−.
Cell isolation and analysis
Cells from the peripheral blood and BM were subjected to red blood cell lysis and washed in PBS. Single-cell suspensions were stained for multicolor analysis with the indicated fluorochrome-conjugated antibodies (listed in the Supplemental Experimental Procedures, BD Biosciences or eBioscience). For BrdU incorporation, cells were stained for surface markers, fixed, permeabilized, and stained with an allophycocyanin-conjugated anti-BrdU antibody according to the manufacturer’s protocol (APC BrdU Flow Kit, BD Biosciences). The samples were acquired using LSR II or Fortessa flow cytometers or sorted on an Influx flow sorter (BD Biosciences) equipped with 561 nm lasers for the detection of red fluorescent proteins. The data were analyzed using FlowJo software (TreeStar). Gene expression analysis by RNA-Seq and the staining of bone sections are described in Supplemental Experimental Procedures.
Statistics
Unless indicated otherwise, normal distribution was not assumed and significance was estimated by non-parametric Wilcoxon matched-pairs signed-rank test.
Supplementary Material
Highlights.
a subset of self-renewing HSC in the adult bone marrow was genetically marked
HSC provide a major sustained contribution to endogenous hematopoiesis
HSC give rise to all immune cell types except B-1a cells and tissue macrophages
Multilineage contribution of HSC is accelerated by interferon response
Acknowledgments
The authors thank Drs. C. Park, W. Hu and C. Borsotti for instruction on obtaining BM biopsies. Supported by the NIH grants AG049074, AI115382, AI072571 and HL113678 (B.R.), Leona M. and Harry B. Helmsley Charitable Trust (B.R.), NIH training grant HD055165 (C.M.S.) and AI100853 (C.M.L.), and a Terry Fox Program Project grant (C.J.E.). S.B. and D.H.J.K. were recipients of Vanier Scholarships from the Canadian Institutes of Health Research.
Footnotes
Accession numbers
The results of RNA-Seq analysis have been deposited in the NCBI Gene Expression Omnibus (GEO) database under the accession no. GSE78855.
Author contributions
C.M.S., S.B., S.U., D.J.H.F.K., Y.L., C.M.L., and Ju.F. performed and interpreted experiments. A.G. and L.A.M. performed data analysis and interpretation. M.M., C.J.E. and B.R. supervised and interpreted experiments. Jo.F. and L.D. provided advice and feedback. B.R. conceived and supervised the project. C.M.S. and B.R. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acar M, Kocherlakota KS, Murphy MM, Peyer JG, Oguro H, Inra CN, Jaiyeola C, Zhao Z, Luby-Phelps K, Morrison SJ. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature. 2015;526:126–130. doi: 10.1038/nature15250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Jasper H, Rudolph KL. Aging-Induced Stem Cell Mutations as Drivers for Disease and Cancer. Cell Stem Cell. 2015;16:601–612. doi: 10.1016/j.stem.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagger FO, Sasivarevic D, Sohi SH, Laursen LG, Pundhir S, Sonderby CK, Winther O, Rapin N, Porse BT. BloodSpot: a database of gene expression profiles and transcriptional programs for healthy and malignant haematopoiesis. Nucleic Acids Res. 2016;44:D917–924. doi: 10.1093/nar/gkv1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balazs AB, Fabian AJ, Esmon CT, Mulligan RC. Endothelial protein C receptor (CD201) explicitly identifies hematopoietic stem cells in murine bone marrow. Blood. 2006;107:2317–2321. doi: 10.1182/blood-2005-06-2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benz C, Copley MR, Kent DG, Wohrer S, Cortes A, Aghaeepour N, Ma E, Mader H, Rowe K, Day C, et al. Hematopoietic stem cell subtypes expand differentially during development and display distinct lymphopoietic programs. Cell Stem Cell. 2012;10:273–283. doi: 10.1016/j.stem.2012.02.007. [DOI] [PubMed] [Google Scholar]
- Biasco L, Pellin D, Scala S, Dionisio F, Basso-Ricci L, Leonardelli L, Scaramuzza S, Baricordi C, Ferrua F, Cicalese MP, et al. In Vivo Tracking of Human Hematopoiesis Reveals Patterns of Clonal Dynamics during Early and Steady-State Reconstitution Phases. Cell Stem Cell. 2016 doi: 10.1016/j.stem.2016.04.016. e-pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulais PE, Frenette PS. Making sense of hematopoietic stem cell niches. Blood. 2015;125:2621–2629. doi: 10.1182/blood-2014-09-570192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch K, Klapproth K, Barile M, Flossdorf M, Holland-Letz T, Schlenner SM, Reth M, Hofer T, Rodewald HR. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature. 2015;518:542–546. doi: 10.1038/nature14242. [DOI] [PubMed] [Google Scholar]
- Cabezas-Wallscheid N, Klimmeck D, Hansson J, Lipka DB, Reyes A, Wang Q, Weichenhan D, Lier A, von Paleske L, Renders S, et al. Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA Methylome analysis. Cell Stem Cell. 2014;15:507–522. doi: 10.1016/j.stem.2014.07.005. [DOI] [PubMed] [Google Scholar]
- Challen GA, Boles NC, Chambers SM, Goodell MA. Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-beta1. Cell Stem Cell. 2010;6:265–278. doi: 10.1016/j.stem.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JY, Miyanishi M, Wang SK, Yamazaki S, Sinha R, Kao KS, Seita J, Sahoo D, Nakauchi H, Weissman IL. Hoxb5 marks long-term haematopoietic stem cells and reveals a homogenous perivascular niche. Nature. 2016;530:223–227. doi: 10.1038/nature16943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheshier SH, Morrison SJ, Liao X, Weissman IL. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc Natl Acad Sci USA. 1999;96:3120–3125. doi: 10.1073/pnas.96.6.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. 2013;495:231–235. doi: 10.1038/nature11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykstra B, Kent D, Bowie M, McCaffrey L, Hamilton M, Lyons K, Lee SJ, Brinkman R, Eaves C. Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell Stem Cell. 2007;1:218–229. doi: 10.1016/j.stem.2007.05.015. [DOI] [PubMed] [Google Scholar]
- Eaves CJ. Hematopoietic stem cells: concepts, definitions, and the new reality. Blood. 2015;125:2605–2613. doi: 10.1182/blood-2014-12-570200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias HK, Schinke C, Bhattacharyya S, Will B, Verma A, Steidl U. Stem cell origin of myelodysplastic syndromes. Oncogene. 2014;33:5139–5150. doi: 10.1038/onc.2013.520. [DOI] [PubMed] [Google Scholar]
- Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, Trumpp A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458:904–908. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- Forsberg EC, Passegue E, Prohaska SS, Wagers AJ, Koeva M, Stuart JM, Weissman IL. Molecular signatures of quiescent, mobilized and leukemia-initiating hematopoietic stem cells. PLoS One. 2010;5:e8785. doi: 10.1371/journal.pone.0008785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg EC, Prohaska SS, Katzman S, Heffner GC, Stuart JM, Weissman IL. Differential expression of novel potential regulators in hematopoietic stem cells. PLoS Genet. 2005;1:e28. doi: 10.1371/journal.pgen.0010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foudi A, Hochedlinger K, Van Buren D, Schindler JW, Jaenisch R, Carey V, Hock H. Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nat Biotechnol. 2009;27:84–90. doi: 10.1038/nbt.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazit R, Mandal PK, Ebina W, Ben-Zvi A, Nombela-Arrieta C, Silberstein LE, Rossi DJ. Fgd5 identifies hematopoietic stem cells in the murine bone marrow. J Exp Med. 2014;211:1315–1331. doi: 10.1084/jem.20130428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geering B, Stoeckle C, Conus S, Simon HU. Living and dying for inflammation: neutrophils, eosinophils, basophils. Trends Immunol. 2013;34:398–409. doi: 10.1016/j.it.2013.04.002. [DOI] [PubMed] [Google Scholar]
- Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentek R, Molawi K, Sieweke MH. Tissue macrophage identity and self-renewal. Immunol Rev. 2014;262:56–73. doi: 10.1111/imr.12224. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. 2014;14:392–404. doi: 10.1038/nri3671. [DOI] [PubMed] [Google Scholar]
- Ikuta K, Weissman IL. Evidence that hematopoietic stem cells express mouse c-kit but do not depend on steel factor for their generation. Proc Natl Acad Sci USA. 1992;89:1502–1506. doi: 10.1073/pnas.89.4.1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka K, Sato T, Yoshimi A, Goyama S, Tsuruta T, Kobayashi H, Shimabe M, Arai S, Nakagawa M, Imai Y, et al. Evi1 is essential for hematopoietic stem cell self-renewal, and its expression marks hematopoietic cells with long-term multilineage repopulating activity. J Exp Med. 2011;208:2403–2416. doi: 10.1084/jem.20110447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent DG, Copley MR, Benz C, Wohrer S, Dykstra BJ, Ma E, Cheyne J, Zhao Y, Bowie MB, Gasparetto M, et al. Prospective isolation and molecular characterization of hematopoietic stem cells with durable self-renewal potential. Blood. 2009;113:6342–6350. doi: 10.1182/blood-2008-12-192054. [DOI] [PubMed] [Google Scholar]
- Kiel MJ, He S, Ashkenazi R, Gentry SN, Teta M, Kushner JA, Jackson TL, Morrison SJ. Haematopoietic stem cells do not asymmetrically segregate chromosomes or retain BrdU. Nature. 2007;449:238–242. doi: 10.1038/nature06115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel MJ, Yilmaz OH, Iwashita T, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, Mizoguchi T, Wei Q, Lucas D, Ito K, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. 2013;502:637–643. doi: 10.1038/nature12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecino-Rodriguez E, Berent-Maoz B, Dorshkind K. Causes, consequences, and reversal of immune system aging. J Clin Invest. 2013;123:958–965. doi: 10.1172/JCI64096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecino-Rodriguez E, Dorshkind K. B-1 B cell development in the fetus and adult. Immunity. 2012;36:13–21. doi: 10.1016/j.immuni.2011.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita Y, Ema H, Nakauchi H. Heterogeneity and hierarchy within the most primitive hematopoietic stem cell compartment. J Exp Med. 2010;207:1173–1182. doi: 10.1084/jem.20091318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SJ, Weissman IL. The long-term repopulating subset of hematopoietic stem cells is deterministic and isolatable by phenotype. Immunity. 1994;1:661–673. doi: 10.1016/1074-7613(94)90037-x. [DOI] [PubMed] [Google Scholar]
- Nakada D, Oguro H, Levi BP, Ryan N, Kitano A, Saitoh Y, Takeichi M, Wendt GR, Morrison SJ. Oestrogen increases haematopoietic stem-cell self-renewal in females and during pregnancy. Nature. 2014;505:555–558. doi: 10.1038/nature12932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguro H, Ding L, Morrison SJ. SLAM Family Markers Resolve Functionally Distinct Subpopulations of Hematopoietic Stem Cells and Multipotent Progenitors. Cell Stem Cell. 2013;13:102–116. doi: 10.1016/j.stem.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passegue E, Wagers AJ, Giuriato S, Anderson WC, Weissman IL. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med. 2005;202:1599–1611. doi: 10.1084/jem.20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras EM, Lakshminarasimhan R, Techner JM, Fong S, Flach J, Binnewies M, Passegue E. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J Exp Med. 2014;211:245–262. doi: 10.1084/jem.20131043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Aguilera A, Arranz L, Martin-Perez D, Garcia-Garcia A, Stavropoulou V, Kubovcakova L, Isern J, Martin-Salamanca S, Langa X, Skoda RC, et al. Estrogen Signaling Selectively Induces Apoptosis of Hematopoietic Progenitors and Myeloid Neoplasms without Harming Steady-State Hematopoiesis. Cell Stem Cell. 2014;15:791–804. doi: 10.1016/j.stem.2014.11.002. [DOI] [PubMed] [Google Scholar]
- Sanjuan-Pla A, Macaulay IC, Jensen CT, Woll PS, Luis TC, Mead A, Moore S, Carella C, Matsuoka S, Bouriez Jones T, et al. Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature. 2013;502:232–236. doi: 10.1038/nature12495. [DOI] [PubMed] [Google Scholar]
- Shizuru JA, Negrin RS, Weissman IL. Hematopoietic stem and progenitor cells: clinical and preclinical regeneration of the hematolymphoid system. Annu Rev Med. 2005;56:509–538. doi: 10.1146/annurev.med.54.101601.152334. [DOI] [PubMed] [Google Scholar]
- Spangrude GJ, Heimfeld S, Weissman IL. Purification and characterization of mouse hematopoietic stem cells. Science. 1988;241:58–62. doi: 10.1126/science.2898810. [DOI] [PubMed] [Google Scholar]
- Sun J, Ramos A, Chapman B, Johnnidis JB, Le L, Ho YJ, Klein A, Hofmann O, Camargo FD. Clonal dynamics of native haematopoiesis. Nature. 2014;514:322–327. doi: 10.1038/nature13824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verlinden SF, van Es HH, van Bekkum DW. Serial bone marrow sampling for long-term follow up of human hematopoiesis in NOD/SCID mice. Exp Hematol. 1998;26:627–630. [PubMed] [Google Scholar]
- Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, Moehrle B, Brocks D, Bayindir I, Kaschutnig P, et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature. 2015;520:549–552. doi: 10.1038/nature14131. [DOI] [PubMed] [Google Scholar]
- Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135:1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- Wilson NK, Kent DG, Buettner F, Shehata M, Macaulay IC, Calero-Nieto FJ, Sanchez Castillo M, Oedekoven CA, Diamanti E, Schulte R, et al. Combined Single-Cell Functional and Gene Expression Analysis Resolves Heterogeneity within Stem Cell Populations. Cell Stem Cell. 2015;16:712–724. doi: 10.1016/j.stem.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto R, Morita Y, Ooehara J, Hamanaka S, Onodera M, Rudolph KL, Ema H, Nakauchi H. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell. 2013;154:1112–1126. doi: 10.1016/j.cell.2013.08.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.