

Abstract
Objective
To evaluate the efficacy and safety of switching from intravenous (IV) tocilizumab (TCZ) to subcutaneous (SC) TCZ monotherapy in rheumatoid arthritis patients.
Methods
Patients who had completed 24 weeks of TCZ‐SC (162 mg/2 weeks) or TCZ‐IV (8 mg/kg/4 weeks) monotherapy in the double‐blind period of the MUSASHI study were enrolled in an 84‐week open‐label extension period. All received TCZ‐SC (162 mg/2 weeks) monotherapy. Effects of the IV to SC switch were evaluated at week 36 (12 weeks after switching).
Results
Overall, 319 patients received ≥1 dose of TCZ‐SC during the open‐label extension period; 160 switched from TCZ‐IV to TCZ‐SC (TCZ IV/SC) and 159 continued TCZ‐SC (TCZ SC/SC). Disease Activity Score in 28 joints using the erythrocyte sedimentation rate clinical remission rates were 62.5% (100 of 160) for TCZ IV/SC and 50.0% (79 of 158) for TCZ SC/SC at week 24, and were maintained at 62.5% (100 of 160) and 57.0% (90 of 158), respectively, at week 36. In the TCZ IV/SC group, 9% of patients (9 of 100) who had achieved remission at week 24 could not maintain remission at week 36. In TCZ IV/SC patients weighing ≥70 kg, the percentage with a sufficient serum TCZ concentration (≥1 μg/ml) decreased from 90.9% (10 of 11) at week 24 to 45.5% (5 of 11) at week 36. Overall safety profiles were similar in TCZ IV/SC and TCZ SC/SC except for mild injection site reactions in TCZ IV/SC.
Conclusion
Efficacy is adequately maintained in most patients switching from TCZ‐IV (8 mg/kg/4 weeks) to TCZ‐SC (162 mg/2 weeks) monotherapy. Patients receiving TCZ‐IV can switch to TCZ‐SC without serious safety concerns. Clinical efficacy may be reduced after switching in some patients with high body weight.
INTRODUCTION
Tocilizumab (TCZ) is an antibody that targets the interleukin‐6 (IL‐6) receptor, which plays a pivotal role in the generation of pathogenic Th17 cells in rheumatoid arthritis (RA) 1. As an intravenous (IV) formulation, TCZ has already been approved in Japan for the indications of RA, systemic juvenile idiopathic arthritis, polyarticular juvenile idiopathic arthritis, and Castleman's disease. In monotherapy and combination therapy with methotrexate or other disease‐modifying antirheumatic drugs (DMARDs), TCZ‐IV is clinically effective, prevents structural joint damage, and improves physical function 2, 3, 4, 5, 6, 7, 8.
Box 1. Significance & Innovations.
Understanding the clinical impact of changing formulations from monthly weight‐tiered–dose intravenous administration of tocilizumab (TCZ) to biweekly fixed‐dose subcutaneous injection is important with regard to efficacy and safety.
Switching from intravenous to subcutaneous TCZ monotherapy demonstrated the efficacy and safety of TCZ to be maintained in most patients with rheumatoid arthritis, although efficacy was reduced in a few patients with high body weights. Subcutaneous administration of TCZ monotherapy is a potential new therapeutic option for rheumatoid arthritis.
The phase III multicenter double‐blind noninferiority (MUSASHI) study, which compared subcutaneous (SC) with TCZ‐IV monotherapy, was performed in Japan from April 2010 to April 2013. This study verified the noninferiority of TCZ‐SC 162 mg/2 weeks versus TCZ‐IV 8 mg/kg/4 weeks 9. Furthermore, 2 international phase III studies, SUMMACTA and BREVACTA 10, 11, which compared TCZ‐SC with TCZ‐IV or placebo with DMARDs, were performed. These studies verified not only the noninferiority of TCZ‐SC 162 mg/2 weeks to TCZ‐IV 8 mg/kg/4 weeks, but also compared the superiority of TCZ‐SC 162 mg/2 weeks to placebo. Based on these studies, SC formulations have already been approved in Japan, the US, and European countries for the indication of RA.
Understanding the clinical impact of changing formulations from monthly IV administration to biweekly SC injection of TCZ is important with regard to efficacy, safety, and tolerability. To our knowledge, this is the first study to evaluate efficacy, safety, and tolerability in patients switching from weight‐tiered–dose TCZ‐IV monotherapy to fixed‐dose TCZ‐SC monotherapy.
PATIENTS AND METHODS
Study design
This study was a phase III randomized, parallel‐group, double‐dummy trial with a double‐blind period of 24 weeks (MUSASHI) followed by an open‐label extension period of 84 weeks in Japanese patients with RA. The protocol was approved by the Japanese Ministry of Health, Labour and Welfare and the institutional review boards at all study sites. The study was performed in accordance with the ethical standards of the current version of the 1964 Declaration of Helsinki. At enrollment in the double‐blind study, signed informed consent was received from participating patients.
Participating patients had shown an inadequate response to methotrexate, DMARDs, or anti–tumor necrosis factor (anti‐TNF) inhibitors with active disease at randomization, as previously described 9. At the start of the double‐blind period, patients were randomly assigned to a dosage of 8 mg/kg/4 weeks of TCZ‐IV or 162 mg/2 weeks of TCZ‐SC. After observations had been completed at the end of the 24‐week double‐blind period, patients received TCZ‐SC 162 mg/2 weeks under open‐label conditions for 84 weeks (see Supplementary Figure 1, available in the online version of this article at http://onlinelibrary.wiley.com/doi/10.1002/acr.22598/abstract). The eligibility criteria for participation in this extension study were described previously 9. A total of 322 patients completed the double‐blind period of the study. Efficacy of the switch from TCZ‐IV to TCZ‐SC was evaluated at week 36 (at 12 weeks after switching). Safety and immunogenicity were evaluated and reported throughout the open‐label extension period.
Efficacy
Disease activity at baseline was monitored every 4 weeks before the first dose in the open‐label extension period (the last observation in the double‐blind period). Disease activity was evaluated by the Disease Activity Score in 28 joints using the erythrocyte sedimentation rate (DAS28‐ESR), Clinical Disease Activity Index (CDAI) scores, and the American College of Rheumatology 20% improvement criteria (ACR20) response rate 12, for comparison against the baseline, defined as the values at the start of the double‐blind period. For the DAS28‐ESR, the percentages of patients with low disease activity (DAS28‐ESR ≤3.2) and remission (DAS28‐ESR <2.6) were also calculated. For the CDAI, only the percentage of patients with remission (CDAI ≤2.8) was calculated.
Safety and immunogenicity
Adverse events and serious adverse events were classified using the Medical Dictionary for Regulatory Activities, version 13.0. All events that were judged by the investigators to have occurred at the injection sites were classified as injection site reactions.
Anti‐TCZ antibodies were measured every 4 weeks during the double‐blind period, then at week 12 in the extension period. Anti‐TCZ antibodies were measured using the enzyme‐linked immunosorbent assay method described previously 13.
Pharmacokinetics
Serum TCZ concentrations were measured using the enzyme‐linked immunosorbent assay every 4 weeks throughout the double‐blind period and extension period 14.
Statistical analysis
Efficacy was evaluated for patients who completed the double‐blind period and received at least 1 dose of TCZ during the extension period. Missing values in the double‐blind period were imputed up to week 24 using the last observation carried forward (LOCF) method, and missing values in the extension period were imputed up to week 36 using the LOCF method. Safety was evaluated for all patients who received at least 1 dose of TCZ during the double‐blind period and the extension period.
RESULTS
Patient disposition and baseline characteristics
A total of 322 patients completed the double‐blind period of the study. Table 1 shows the baseline characteristics of 319 patients, including 1 patient in the TCZ‐SC group who had not completed the double‐blind period, who received at least 1 dose of TCZ‐SC during the open‐label extension period. Of these, 160 patients received TCZ‐IV in the double‐blind period (TCZ IV/SC group) and 159 received TCZ‐SC in the double‐blind period (TCZ SC/SC group); 158 patients (98.8%) in the TCZ IV/SC group and 157 (98.7%) in the TCZ SC/SC group continued treatment and completed the observations at week 36 (at 12 weeks after first dosing in the open‐label extension period).
Table 1.
Demographics and baseline characteristics of study participants (n = 319)a
| TCZ IV/SC (n = 160) | TCZ SC/SC (n = 159) | |
|---|---|---|
| Age, mean ± SD years | 51.5 ± 12.1 | 52.5 ± 12.5 |
| Body weight, mean ± SD kg | 54.3 ± 10.1 | 53.9 ± 8.8 |
| Women, % | 83.1 | 83.0 |
| Disease duration, mean ± SD years | 8.0 ± 7.3 | 7.4 ± 7.4 |
| Functional class, no. | ||
| I | 19 | 26 |
| II | 123 | 111 |
| III | 18 | 22 |
| Rheumatoid arthritis stage, no. | ||
| I | 8 | 19 |
| II | 63 | 54 |
| III | 43 | 48 |
| IV | 46 | 38 |
| DAS28‐ESR, mean ± SD | 6.2 ± 0.9 | 6.1 ± 0.9 |
| C‐reactive protein level, mean ± SD mg/dl | 2.1 ± 2.0 | 2.1 ± 2.3 |
| Interleukin‐6 level, mean ± SD pg/ml | 32.7 ± 42.8 | 39.3 ± 47.0 |
| Rheumatoid factor positive, % | 83.1 | 79.2 |
| Anti‐CCP antibody positive, % | 90.6 | 88.7 |
| Previous treatment with anti‐TNF inhibitor, % | 23.1 | 18.2 |
| Previous treatment with methotrexate, %b | 83.1 | 79.9 |
| Dosage, mean ± SD mg/weekb | 8.2 ± 2.3 | 8.1 ± 2.1 |
| Oral corticosteroid treatment, % | 59.4 | 68.6 |
| Dosage, mean ± SD mg/dayc | 4.6 ± 2.0 | 4.5 ± 2.3 |
Patients who received ≥1 dose of subcutaneous tocilizumab (TCZ‐SC) during the open‐label extension period (including 1 patient in the TCZ‐SC group who had not completed the double‐blind period of the study) are shown. IV = intravenous; DAS28‐ESR = Disease Activity Score in 28 joints using the erythrocyte sedimentation rate; anti‐CCP = anti–cyclic citrullinated peptide; anti‐TNF = anti–tumor necrosis factor.
Patients who previously received methotrexate were analyzed within 4 weeks of initial TCZ treatment.
Prednisolone or equivalent.
Three patients in the TCZ‐SC group withdrew from the study after completion of the double‐blind period without any administration of open‐label TCZ due to adverse event, protocol violation, and refusal of treatment, while 1 patient in the TCZ‐IV group withdrew due to adverse events. Two patients in the TCZ IV/SC group withdrew during the open‐label extension period, one due to an adverse event and the other due to an inadequate response. Two patients in the TCZ SC/SC group withdrew during the open‐label extension period due to an inadequate response. Of 159 patients in the TCZ SC/SC group, 158 were included in the efficacy analysis, while 1 was excluded due to noncompletion of the double‐blind period.
Efficacy results
Mean ± SD DAS28‐ESR scores in the TCZ IV/SC group were 2.5 ± 1.1 at week 24 and 2.6 ± 1.2 at week 36 (12 weeks after switching), while in the TCZ SC/SC group the scores were 2.7 ± 1.3 and 2.6 ± 1.4, respectively (Figure 1A). In addition, mean ± SD CDAI scores in the TCZ IV/SC group were 8.0 ± 7.4 at week 24 and 8.7 ± 8.3 at week 36, while in the TCZ SC/SC group they were 9.8 ± 8.5 and 9.6 ± 10.0, respectively (Figure 1B). These results suggest that overall disease control was maintained after switching from TCZ‐IV to TCZ‐SC in the open‐label extension period. ACR20, ACR50, and ACR70 responses in the TCZ IV/SC groups were 88.8%, 68.1%, and 41.3% at week 24 and 85.0%, 66.9%, and 36.9% at week 36, respectively, while in the TCZ SC/SC groups they were 81.6%, 65.8%, and 38.6% and 86.1%, 65.8%, and 39.9%, respectively (Figure 1C).
Figure 1.
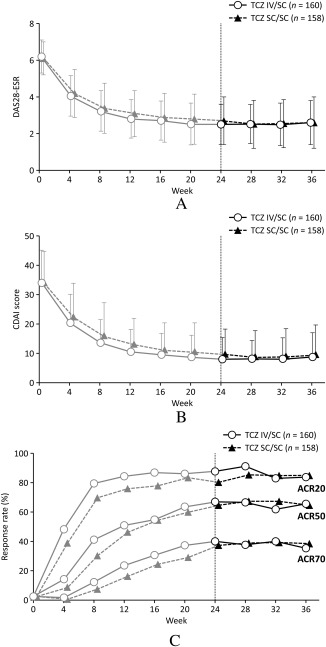
Time course of disease activity scores. A, Disease Activity Score in 28 joints using the erythrocyte sedimentation rate (DAS28‐ESR), B, Clinical Disease Activity Index (CDAI) score, and C, American College of Rheumatology 20%/50%/70% improvement criteria (ACR20/50/70) response. TCZ = tocilizumab; IV = intravenous; SC = subcutaneous.
The proportions of patients who achieved DAS28 remission (defined as DAS28‐ESR <2.6) in the TCZ IV/SC group and TCZ SC/SC group were maintained over time from 62.5% (100 of 160) and 50.0% (79 of 158) at week 24 to 62.5% (100 of 160) and 57.0% (90 of 158) at week 36, respectively (Figure 2A). The proportion of patients who achieved CDAI clinical remission (CDAI ≤2.8) or low disease activity (CDAI >2.8 to ≤10) was also maintained (Figure 2B).
Figure 2.
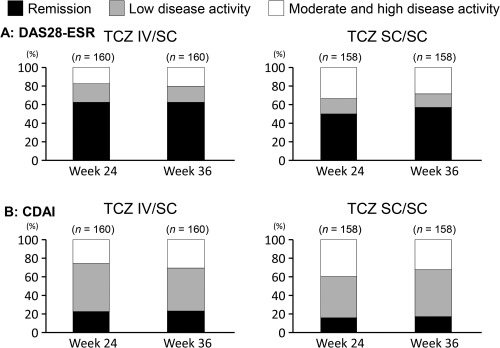
Efficacy evaluation by disease activity category in patients who switched from intravenous to subcutaneous tocilizumab (TCZ IV/SC) and who continued TCZ‐SC. A, Disease Activity Score in 28 joints using the erythrocyte sedimentation rate (DAS28‐ESR), and B, Clinical Disease Activity Index (CDAI) score.
The influence of switching from TCZ‐IV to TCZ‐SC on efficacy was analyzed based on individual patients’ disease activity at week 24, which was classified into 3 subcategories. Figure 3 demonstrates the change in DAS‐28‐ESR response for each category at week 36. In the TCZ IV/SC group, the percentages of patients at week 24 were 62.5% (100 of 160) for clinical remission (DAS28‐ESR <2.6), 20.0% (32 of 160) for low disease activity (DAS28‐ESR >2.6 to ≤3.2), and 17.5% (28 of 160) for moderate or high disease activity (DAS28‐ESR >3.2). In patients who had not achieved remission at week 24, 56.3% (18 of 32) in the low disease activity category and 14.3% (4 of 28) in the moderate or high disease activity category achieved clinical remission by week 36. In contrast, in patients who had achieved clinical remission at week 24, 78% (78 of 100) had maintained clinical remission at week 36, but 13% (13 of 100) were classified in the low disease activity category and 9% (9 of 100) in the moderate or high disease activity category. The median body weight in the latter 9 patients was 67.0 kg, numerically higher than the median body weight of 53.5 kg for the other 91 patients. In contrast, the median body weight in 4 patients who had achieved clinical remission at week 24 but were classified in the moderate or high disease activity categories at week 36 was 57.1 kg in the TCZ SC/SC group.
Figure 3.
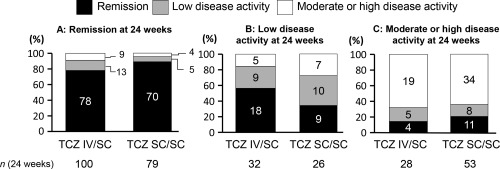
Changes in disease activity category by Disease Activity Score in 28 joints using the erythrocyte sedimentation rate in patients who switched from intravenous to subcutaneous tocilizumab (TCZ IV/SC) and who continued TCZ‐SC. A, Disease activity at week 36 in patients who achieved remission at week 24, B, Disease activity at week 36 in patients who achieved low disease activity at week 24, and C, Disease activity at week 36 in patients who achieved medium or high disease activity at week 24. The values in the columns indicate the number of patients.
In post hoc analysis, we analyzed the correlation between the efficacy and trough TCZ concentration over 4 body weight categories and 4 body mass index (BMI) categories. Clinical remission rates in the TCZ IV/SC group at week 24 and week 36 in patients stratified by 10‐kg increments in body weight are shown in Figure 4A. In the body weight categories <70 kg, the clinical remission rate was maintained after switching, while the rate in the heaviest body weight category (≥70 kg) decreased from 72.7% (8 of 11) to 27.3% (3 of 11), despite only a few Japanese patients being classified in this highest weight category. In the BMI category of ≥25 kg/m2, the remission rate decreased from 66.7% (14 of 21) to 42.9% (9 of 21) after switching (see Supplementary Figure 2, available in the online version of this article at http://onlinelibrary.wiley.com/doi/10.1002/acr.22598/abstract).
Figure 4.
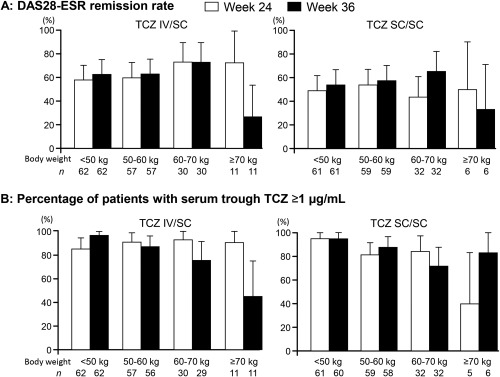
A, Changes in Disease Activity Score in 28 joints using the erythrocyte sedimentation rate (DAS28‐ESR) remission rate were stratified by 10 kg of body weight in patients who switched from intravenous to subcutaneous tocilizumab (TCZ IV/SC) and who continued TCZ‐SC. B, Percentage of patients with serum trough TCZ concentration of ≥1 μg/ml was stratified by 10 kg of body weight. The values below each column indicate the number of total patients in each weight category. The bars show the 95% confidence interval.
Changes in the C‐reactive protein (CRP) level from week 24 to week 36 were also analyzed for clinical estimation of sufficient serum level of TCZ after switching. The percentages of patients with a CRP level ≤0.05 mg/dl in the TCZ IV/SC group and the TCZ SC/SC group were also maintained from 90.0% (144 of 160) and 87.3% (138 of 158) at week 24 to 83.1% (133 of 160) and 86.7% (137 of 158) at week 36, respectively (see Supplementary Figure 3A, available in the online version of this article at http://onlinelibrary.wiley.com/doi/10.1002/acr.22598/abstract). In the TCZ IV/SC group, overall, 13.2% (19 of 144) increased to a higher level of CRP (>0.05 mg/dl) at week 36 from a lower level (≤0.05 mg/dl) at week 24, while in the heaviest body weight category (≥70 kg), 54.5% (6 of 11) increased (see Supplementary Figure 3B, available in the online version of this article at http://onlinelibrary.wiley.com/doi/10.1002/acr.22598/abstract). In contrast, in the TCZ SC/SC group, overall, 5.8% (8 of 138) increased, while in the heaviest category, 0% (0 of 3) increased. These data indicate that the proportion with an increased CRP level among a patient with heavy body weight is greater when switching from TCZ‐IV to TCZ‐SC than for continuing TCZ‐SC.
Pharmacokinetics results
Overall mean ± SD serum trough TCZ concentrations in patients in the TCZ IV/SC group and TCZ SC/SC group were maintained from 11.4 ± 8.3 μg/ml and 9.4 ± 8.0 μg/ml at week 24 to 9.2 ± 7.5 μg/ml and 9.3 ± 7.9 μg/ml at week 36, respectively. The percentages of patients with serum TCZ concentrations ≥1 μg/ml in the TCZ IV/SC group and the TCZ SC/SC group were also maintained from 89.4% (143 of 160) and 86.0% (135 of 157) at week 24 to 86.1% (136 of 158) and 87.2% (136 of 156) at week 36, respectively. The percentage of patients in the TCZ IV/SC group who maintained serum TCZ concentrations ≥1 μg/ml did not differ among any of the weight categories at week 24, while the percentage among the heaviest patients (≥70 kg) decreased from 90.9% (10 of 11) at week 24 to 45.5% (5 of 11) at week 36 (Figure 4B). In contrast, in the TCZ SC/SC group, the percentage in the heaviest patient category (≥70 kg) increased from 40.0% (2 of 5) at week 24 to 83.3% (5 of 6) at week 36.
Safety
The incidences of adverse events in the TCZ IV/SC group and the TCZ SC/SC group during the 12‐week period after switching (from week 24 to week 36) were 59.4% (95 of 160) and 57.9% (92 of 159), respectively (Table 2). The most common adverse event was infection, occurring in 27.5% (44 of 160) and 23.9% of patients (38 of 159) in the TCZ IV/SC and TCZ SC/SC groups, respectively. Serious adverse events occurred in 4.4% of patients (7 of 160) in the TCZ IV/SC group and in 2.5% (4 of 159) in the TCZ SC/SC group. These adverse events consisted of pneumonia in 2 patients and mycoplasma pneumonia, joint destruction, arthropathy, deep vein thrombosis, and urinary calculus in 1 patient each from the TCZ IV/SC group. Gastroenteritis, foot deformity, varicose veins, and peripheral embolism occurred in 1 patient each from the TCZ SC/SC group. No anaphylactoid symptoms were seen in either group.
Table 2.
Safety summary and anti‐TCZ antibody statusa
| TCZ IV/SC group | TCZ SC/SC group | |||||
|---|---|---|---|---|---|---|
| 0–12 weeks | 12–24 weeks | 24–36 weeks | 0–12 weeks | 12–24 weeks | 24–36 weeks | |
| Treatment | IV | IV | SC | SC | SC | SC |
| Total no. of patients | 173 | 167 | 160 | 173 | 167 | 159 |
| Adverse events | 137 (79.2) | 102 (61.1) | 95 (59.4) | 132 (76.3) | 105 (62.9) | 92 (57.9) |
| Infections | 40 (23.1) | 48 (28.7) | 44 (27.5) | 36 (20.8) | 49 (29.3) | 38 (23.9) |
| Adverse events leading to withdrawal | 9 (5.2) | 1 (0.6) | 1 (0.6) | 4 (2.3) | 1 (0.6) | 1 (0.6) |
| Injection site reactions | 8 (4.6)b | 4 (2.4)b | 11 (6.9) | 14 (8.1) | 9 (5.4) | 7 (4.4) |
| Systemic infusion or injection reactions | 12 (6.9) | 2 (1.2) | 3 (1.9) | 6 (3.5) | 3 (1.8) | 4 (2.5) |
| Serious adverse events | 8 (4.6) | 4 (2.4) | 7 (4.4)c | 9 (5.2) | 3 (1.8) | 4 (2.5)d |
| Death | – | – | – | – | – | – |
| Malignancy | – | – | – | – | – | – |
| Serious infections | 2 (1.2) | 3 (1.8) | 3 (1.9) | 2 (1.2) | – | 1 (0.6) |
| Serious hypersensitivity reactions | 1 (0.6) | – | – | – | – | – |
| Anti‐TCZ antibodye | – | – | 1 (0.6) | 5 (2.9) | 1 (0.6) | – |
Values are the number (%) of patients unless otherwise indicated. TCZ = tocilizumab; IV = intravenous; SC = subcutaneous.
Incidence of injection site reaction occurred due to placebo injection.
Pneumonia (2 patients) and mycoplasma pneumonia, joint destruction, arthropathy, deep vein thrombosis, and urinary calculus (1 patient each).
Gastroenteritis, foot deformity, varicose veins, and peripheral embolism (1 patient each).
Positive for both screening and confirmation assay.
The overall incidence of adverse events in both the TCZ IV/SC group and the TCZ SC/SC group from week 24 to week 36 tended to be lower than in the period from week 0 to week 24, but the incidence of injection site reactions was increased after switching (from week 24 to week 36) in the TCZ IV/SC group (Table 2). The severity of injection site reactions was mild in all instances and no patients withdrew for this reason. The incidence of systemic injection reactions was higher after switching (from week 24 to week 36) in both groups than the incidence from week 12 to week 24, but was lower than the incidence from week 0 to week 12 (Table 2). The overall safety profiles did not differ between the TCZ IV/SC and TCZ SC/SC groups. No new serious safety concerns were seen as a result of switching the route of administration.
Immunogenicity
Anti‐TCZ antibody was newly detected after switching in 1 patient in the TCZ IV/SC group, but was detected in none of the patients in the TCZ SC/SC group (Table 2). However, the efficacy in this patient was maintained after switching. No injection site reactions or anaphylactic reactions were observed in this patient.
DISCUSSION
In patients who participated in the previously reported MUSASHI study 9, we investigated the efficacy and safety of switching from TCZ‐IV 8 mg/kg/4 weeks to TCZ‐SC 162 mg/2 weeks. We found that overall favorable efficacy of TCZ‐IV monotherapy was maintained after switching to TCZ‐SC monotherapy, while no new serious safety signals were observed. The safety profile of TCZ‐SC after switching from TCZ‐IV was consistent with that of TCZ‐SC of TCZ‐naive patients in the previous study 9. These results suggest that most patients can be switched from TCZ‐IV to TCZ‐SC monotherapy with neither efficacy reduction nor safety concerns.
TCZ serum trough concentrations were not sufficiently maintained in patients with a high BMI as previously reported in the MUSASHI study 9, suggesting that the efficacy of a fixed‐dose regimen of TCZ‐SC may be reduced in RA patients with high body weights. In the present study, some patients with high body weights (≥70 kg) who had achieved remission with TCZ‐IV monotherapy could not maintain it after switching. Conversely, the percentage of patients who maintained sufficient trough concentrations of TCZ was decreased after switching, suggesting that the serum trough concentration of TCZ could not be maintained in certain patients with high body weights. It is conceivable that disease activity was increased because IL‐6 signal transduction could not be completely inhibited 14. In contrast, the trough concentration of TCZ and efficacy slightly increased after switching in patients with low body weights (<50 kg). It is noteworthy that only a few Japanese patients were classified into the high body weight group (≥70 kg; 11 of 160 in the TCZ IV/SC group). Earlier reports are comparable to the present study. In the phase III BREVACTA study, the efficacy of biweekly administration of TCZ‐SC in combination with DMARDs in patients weighing ≥100 kg was lower than the efficacy in lighter‐weight patients 11. Another phase III SUMMACTA study was designed to prevent the serum concentration of TCZ from decreasing in higher‐weight patients, and demonstrated that adequate efficacy of weekly administration of TCZ‐SC with DMARDs could be achieved in patients weighing ≥100 kg 10. Since TCZ‐SC is a fixed‐dose regimen, in order to maintain the efficacy, weekly administration of TCZ‐SC may be beneficial for restoring a sufficient serum concentration of TCZ in patients with high body weights who have inadequate responses to biweekly TCZ‐SC. Weekly administration of TCZ would be expected to be tolerable, because the safety profile of administration of TCZ‐SC 162 mg/2 weeks was similar to that of TCZ‐IV 8 mg/kg/4 weeks, except for injection site reactions, in the SUMMACTA study 10 and the open‐label extension of the MUSASHI study 15. Further studies are needed to investigate the potential benefits and adverse events of TCZ administered weekly.
The overall incidences of adverse events and serious adverse events during treatment with the TCZ‐SC regimen of every 2 weeks were similar to or lower than those of TCZ‐IV in the double‐blind period. No new safety signals were observed after switching from TCZ‐IV to TCZ‐SC. As predicted, the incidence of injection site reactions after switching to TCZ‐SC in the TCZ IV/SC group was slightly increased as compared to that in the TCZ SC/SC group, whereas the number of serious adverse events was low and comparable to that of the TCZ SC/SC group. All injection site reactions were mild, and no events led to discontinuation of treatment. The incidence of injection site reactions after switching from TCZ‐IV to TCZ‐SC was similar to that of the initial 12 weeks of the blind period in the TCZ SC/SC group. There was no unequivocal diminution of response nor were there any anaphylactic reactions after switching. The slight increase in systemic injection reactions, by only one patient in each group, was considered not clinically significant. These data demonstrate that safety after switching to TCZ‐SC is comparable to that of TCZ‐IV, and suggest that patients can switch from TCZ‐IV to TCZ‐SC with no serious safety concerns.
The expressions of antibodies to TCZ may reduce efficacy and systemic reactions to injections of anti‐TNF inhibitors would occasionally cause lack of efficacy due to the expression of antidrug antibodies 16, 17, 18, 19. In a previous study, we found the anti‐TCZ antibody frequency to be increased in TCZ‐SC patients as compared to TCZ‐IV patients in the double‐blind setting 9. In the present study, one patient newly developed anti‐TCZ antibody after switching in the TCZ IV/SC group, although this patient experienced neither a reduction of efficacy nor a systemic injection reaction. An earlier report demonstrated the incidence of anti‐TCZ antibody to be low, with no difference between TCZ‐SC and TCZ‐IV, and no correlation was observed between antibody development and either adverse events or clinical responsiveness 10. Therefore, the immunogenicity potential following switching from TCZ‐IV to TCZ‐SC monotherapy was considered to be low based on the available data.
High baseline CRP level was identified as one of the predictors of a better response to TCZ therapy in RA 20, 21. It has been reported that CRP level is normalized when the serum TCZ concentration is ≥1 μg/ml, because TCZ can be bound to ≥95% of soluble IL‐6 receptors 14. Serum TCZ concentrations can be indirectly monitored through CRP concentrations, although the serum TCZ concentration is substantially maintained with individual differences. If CRP levels were not normalized after treatment with TCZ, the serum concentration of TCZ would denote therapeutic insufficiency. Therefore, the dose of TCZ should be increased in patients without normalized CRP in the case of an inadequate response in patients with high body weights.
A limitation of the present study is that we did not explore the long‐term efficacy and safety of switching from TCZ‐IV to TCZ‐SC or those of the reverse switching from TCZ‐SC to TCZ‐IV. Other studies of bidirectional switching between TCZ‐SC and TCZ‐IV in the real‐world setting will provide further information related to sufficient maintenance of the clinical effectiveness of TCZ monotherapy.
In conclusion, the present results indicate that efficacy is adequately maintained in the majority of Japanese RA patients who switch from TCZ‐IV 8 mg/kg/4 weeks to TCZ‐SC 162 mg/2 weeks, whereas in some patients weighing ≥70 kg, efficacy might be slightly reduced. Safety was also demonstrated to be consistent with previously published 24‐week results of a TCZ‐SC monotherapy study. These results suggest that our report provides supportive evidence for TCZ‐SC as well as TCZ‐IV as being useful options for treating RA.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be submitted for publication. Dr. Ogata had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Ogata, Nomura.
Acquisition of data. Ogata, Atsumi, Fukuda, Hirabayashi, Inaba, Ishiguro, Kai, Kawabata, Kida, Kohsaka, Matsumura, Minota, Mukai, Sumida, Takasugi, Tamaki, Takeuchi, Ueda, Yamamoto, Yamanaka, Yoshifuji.
Analysis and interpretation of data. Ogata, Nomura.
ROLE OF THE STUDY SPONSOR
Chugai Pharmaceutical was involved in the study design and analysis and interpretation of the data. This manuscript was reviewed by Chugai Pharmaceutical, but the decision to submit and publish this manuscript was contingent only on the approval of the lead author and coauthors, including those employed by Chugai Pharmaceutical.
Supporting information
Supplementary Figures
ACKNOWLEDGMENTS
The authors would like to thank the investigators in/contributors to (and institutions in) the MUSASHI study: H. Takahashi (Sapporo Medical University), K. Tanimura (Hokkaido Medical Center for Rheumatic Diseases), Y. Urata (Seihoku Central Hospital, United Municipalities of Tsugaru), T. Ishii (Tohoku University), Y. Munakata (Taihaku Sakura Hospital), S. Ohta (Taga General Hospital), H. Inoue (Inoue Hospital), K. Amano (Saitama Medical Center), T. Kasama (Showa University), S. Kawai (Toho University), T. Sugimoto (National Hospital Organization, Shimoshizu Hospital), S. Tohma (National Hospital Organization, Sagamihara Hospital), N. Ogawa (Hamamatsu University), T. Miyamoto (Seirei Hamamatsu General Hospital), A. Murasawa (Niigata Rheumatic Center), K. Sugimoto, T. Ojima (Fukui General Hospital), T. Tanaka, J. Hashimoto, K. Shi (Osaka University), N. Nishimoto (CRENT Clinic), T. Koike (Osaka City University), Y. Saeki (National Hospital Organization, Osaka Minami Medical Center), T. Matsubara (Matsubara Mayflower Hospital), H. Sano (Hyogo College of Medicine), H. Dobashi (Kagawa University), M. Inoo (Utazu Hama Clinic), M. Iwahashi (Higashihiroshima Memorial Hospital), H. Tsukamoto (Kyushu University), Y. Tanaka (University of Occupational and Environmental Health), E. Suematsu, H. Miyahara (National Hospital Organization, Kyushu Medical Center), M. Kondo (Kondo Clinic of Rheumatology and Orthopedic Surgery), E. Shono (Shono Rheumatic Clinic), Y. Ueki (Sasebo Chuo Hospital), and A. Kawakami (Nagasaki University).
JAPICCTI: 101117.
REFERENCES
- 1. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh‐Hora M, Kodama T, et al. Pathogenic conversion of Foxp3 T cells into T17 cells in autoimmune arthritis. Nat Med 2014;20:62–8. [DOI] [PubMed] [Google Scholar]
- 2. Smolen JS, Beaulieu A, Rubbert‐Roth A, Ramos‐Remus C, Rovensky J, Alecock E, et al. Effect of interleukin‐6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double‐blind, placebo‐controlled, randomised trial. Lancet 2008;371:987–97. [DOI] [PubMed] [Google Scholar]
- 3. Genovese MC, McKay JD, Nasonov EL, Mysler EF, da Silva NA, Alecock E, et al. Interleukin‐6 receptor inhibition with tocilizumab reduces disease activity in rheumatoid arthritis with inadequate response to disease‐modifying antirheumatic drugs: the Tocilizumab in Combination With Traditional Disease‐Modifying Antirheumatic Drug Therapy study. Arthritis Rheum 2008;58:2968–80. [DOI] [PubMed] [Google Scholar]
- 4. Emery P, Keystone E, Tony HP, Cantagrel A, van Vollenhoven R, Sanchez A, et al. IL‐6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti‐tumour necrosis factor biologicals: results from a 24‐week multicentre randomised placebo‐controlled trial. Ann Rheum Dis 2008;67:1516–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kremer JM, Blanco R, Brzosko M, Burgos‐Vargas R, Halland AM, Vernon E, et al. Tocilizumab inhibits structural joint damage in rheumatoid arthritis patients with inadequate responses to methotrexate: results from the double‐blind treatment phase of a randomized placebo‐controlled trial of tocilizumab safety and prevention of structural joint damage at one year. Arthritis Rheum 2011;63:609–21. [DOI] [PubMed] [Google Scholar]
- 6. Jones G, Sebba A, Gu J, Lowenstein MB, Calvo A, Gomez‐Reino JJ, et al. Comparison of tocilizumab monotherapy versus methotrexate monotherapy in patients with moderate to severe rheumatoid arthritis: the AMBITION study. Ann Rheum Dis 2010;69:88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nishimoto N, Hashimoto J, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T, et al. Study of active controlled monotherapy used for rheumatoid arthritis, an IL‐6 inhibitor (SAMURAI): evidence of clinical and radiographic benefit from an x ray reader‐blinded randomised controlled trial of tocilizumab. Ann Rheum Dis 2007;66:1162–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nishimoto N, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T, Azuma J, et al. Study of active controlled tocilizumab monotherapy for rheumatoid arthritis patients with an inadequate response to methotrexate (SATORI): significant reduction in disease activity and serum vascular endothelial growth factor by IL‐6 receptor inhibition therapy. Mod Rheumatol 2009;19:12–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ogata A, Tanimura K, Sugimoto T, Inoue H, Urata Y, Matsubara T, et al. Phase 3 study of the efficacy and safety of subcutaneous versus intravenous tocilizumab monotherapy in patients with rheumatoid arthritis. Arthritis Care Res (Hoboken) 2014;66:344–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Burmester GR, Rubbert‐Roth A, Cantagrel A, Hall S, Leszczynski P, Feldman D, et al. A randomised, double‐blind, parallel‐group study of the safety and efficacy of subcutaneous tocilizumab versus intravenous tocilizumab in combination with traditional disease‐modifying antirheumatic drugs in patients with moderate to severe rheumatoid arthritis (SUMMACTA study). Ann Rheum Dis 2014;73:69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kivitz A, Olech E, Borofsky M, Zazueta BM, Navarro‐Sarabia F, Radominski SC, et al. Subcutaneous tocilizumab versus placebo in combination with disease‐modifying antirheumatic drugs in patients with rheumatoid arthritis. Arthritis Care Res (Hoboken) 2014;66:1653–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Felson DT, Anderson JJ, Boers M, Bombardier C, Chernoff M, Fried B, et al. The American College of Rheumatology preliminary core set of disease activity measures for rheumatoid arthritis clinical trials. Arthritis Rheum 1993;36:729–40. [DOI] [PubMed] [Google Scholar]
- 13. Stubenrauch K, Wessels U, Birnboeck H, Ramirez F, Jahreis A, Schleypen J. Subset analysis of patients experiencing clinical events of a potentially immunogenic nature in the pivotal clinical trials of tocilizumab for rheumatoid arthritis: evaluation of an antidrug antibody ELISA using clinical adverse event‐driven immunogenicity testing. Clin Ther 2010;32:1597–609. [DOI] [PubMed] [Google Scholar]
- 14. Nishimoto N, Terao K, Mima T, Nakahara H, Takagi N, Kakehi T. Mechanisms and pathologic significances in increase in serum interleukin‐6 (IL‐6) and soluble IL‐6 receptor after administration of an anti‐IL‐6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease. Blood 2008;112:3959–64. [DOI] [PubMed] [Google Scholar]
- 15. Ogata A, Amano K, Dobashi H, Inoo M, Ishii T, Kasama T, et al. Longterm safety and efficacy of subcutaneous tocilizumab monotherapy: results from the 2‐year open‐label extension of the MUSASHI Study. J Rheumatol 2015;42:799–809. [DOI] [PubMed] [Google Scholar]
- 16. Bartelds GM, Wijbrandts CA, Nurmohamed MT, Stapel S, Lems WF, Aarden L, et al. Clinical response to adalimumab: relationship to anti‐adalimumab antibodies and serum adalimumab concentrations in rheumatoid arthritis. Ann Rheum Dis 2007;66:921–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bartelds GM, Wijbrandts CA, Nurmohamed MT, Stapel S, Lems WF, Aarden L, et al. Anti‐infliximab and anti‐adalimumab antibodies in relation to response to adalimumab in infliximab switchers and anti‐tumour necrosis factor naive patients: a cohort study. Ann Rheum Dis 2010;69:817–21. [DOI] [PubMed] [Google Scholar]
- 18. Dore RK, Mathews S, Schechtman J, Surbeck W, Mandel D, Patel A, et al. The immunogenicity, safety, and efficacy of etanercept liquid administered once weekly in patients with rheumatoid arthritis. Clin Exp Rheumatol 2007;25:40–6. [PubMed] [Google Scholar]
- 19. Kaine J, Gladstein G, Strusberg I, Robles M, Louw I, Gujrathi S, et al. Evaluation of abatacept administered subcutaneously in adults with active rheumatoid arthritis: impact of withdrawal and reintroduction on immunogenicity, efficacy and safety (phase IIIb ALLOW study). Ann Rheum Dis 2012;71:38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pers YM, Fortunet C, Constant E, Lambert J, Godfrin‐Valnet M, De Jong A, et al. Predictors of response and remission in a large cohort of rheumatoid arthritis patients treated with tocilizumab in clinical practice. Rheumatology (Oxford) 2014;53:76–84. [DOI] [PubMed] [Google Scholar]
- 21. Kaneko A, Kida D, Saito K, Tsukamoto M, Sato T. Clinical results for tocilizumab over one year in the clinical setting as assessed by CDAI (Clinical Disease Activity Index): CRP at week 12 and MMP‐3 at week 24 are predictive factors for CDAI. Rheumatol Int 2012;32:3631–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures