

Significance
Microelectron diffraction (MicroED) is a cryo-EM method capable of determining atomic resolution macromolecular structures from nanocrystals of dose-sensitive biological macromolecules. To date, MicroED requires a reasonably accurate guess of the positions of atoms in a structure to solve it, a technique known as molecular replacement. We now show that this need for prior knowledge about a structure is eliminated in cases when crystals diffract to atomic resolution, relying only on the accuracy of the MicroED intensity measurements. Our findings provide a basis for solving structures from tiny crystals that diffract to high resolution, but are presently disregarded because of their small size.
Keywords: phasing, prion, electron diffraction, MicroED, nanocrystal
Abstract
Electrons, because of their strong interaction with matter, produce high-resolution diffraction patterns from tiny 3D crystals only a few hundred nanometers thick in a frozen-hydrated state. This discovery offers the prospect of facile structure determination of complex biological macromolecules, which cannot be coaxed to form crystals large enough for conventional crystallography or cannot easily be produced in sufficient quantities. Two potential obstacles stand in the way. The first is a phenomenon known as dynamical scattering, in which multiple scattering events scramble the recorded electron diffraction intensities so that they are no longer informative of the crystallized molecule. The second obstacle is the lack of a proven means of de novo phase determination, as is required if the molecule crystallized is insufficiently similar to one that has been previously determined. We show with four structures of the amyloid core of the Sup35 prion protein that, if the diffraction resolution is high enough, sufficiently accurate phases can be obtained by direct methods with the cryo-EM method microelectron diffraction (MicroED), just as in X-ray diffraction. The success of these four experiments dispels the concern that dynamical scattering is an obstacle to ab initio phasing by MicroED and suggests that structures of novel macromolecules can also be determined by direct methods.
Macromolecular crystallography must overcome the lack of a measurable phase for each diffraction intensity (1). In X-ray crystallography, when a sufficiently similar structure has been determined, phases from the known structure can be applied to the measured structure factors and refined to provide a new solution (2). Alternatively, the phase problem can be experimentally overcome by use of isomorphous differences, anomalous scattering, or direct methods (3, 4). Methods for ab initio structure determination from measured intensities have been applied to X-ray crystallography data since the mid-1930s (5). The direct method is an ab initio approach to phasing that relies on the relationships between measured structure factors, which inform about their phases, as well as constraints on the positions of atoms in real space (1, 6, 7). The direct-methods approach becomes feasible when a crystal diffracts strongly enough that the intensities can be measured at a resolution better than 1.2 Å (8). This limit follows from the need to resolve atoms as separate features (as required by this method). Data must be recorded from reflecting planes spaced more closely than the interatomic distances. The first structures to be solved by this approach were small atomic assemblies containing few atoms. Since then, a number of small-molecule and macromolecular structures have been determined by direct methods (6, 7, 9–11), including the structure of cytochrome C3, with 2,208 atoms (12).
Cryo-EM methods that are based on imaging, such as single-particle and tomographic techniques, benefit from the direct measurement of phases (13, 14). For these techniques, phases are encoded in the images, which are the convolution of the projected Coulomb potential of the sample with the contrast transfer function of the microscope. Such an advantage can also exist when crystals are imaged (15, 16). In contrast, phases are lost when only diffraction data are collected, either from 2D crystals by electron crystallography or from 3D crystals by microelectron diffraction (MicroED), just as in X-ray crystallography (15, 17). That is because electron-diffraction micrographs are measurements of the back focal plane of the electromagnetic objective lens in an electron microscope (18). They represent the squared magnitudes of a Fourier transform of the crystal being imaged.
When high-resolution intensities collected using MicroED are combined with phases calculated from preexisting models by the method of molecular replacement, they result in atomic resolution structures (19–22). However, to date, no other X-ray crystallographic method for experimental phase determination has been adapted to MicroED. Because MicroED is capable of producing atomic resolution data, direct methods approaches used in X-ray crystallography should be applicable to MicroED. The influence of dynamical scattering on the measured intensities was thought to limit accurate determination of structure factors (23–26). The hypothesis of whether these effects limit ab initio determination of macromolecular structures from protein nanocrystals has been difficult to test because a crystal formed from macromolecules has yet to diffract to better than 1.2 Å by electron-based techniques. Using ultra-low-dose cryo-EM data collection, we now demonstrate experimental phasing of peptide nanocrystals for MicroED at 1.0-Å resolution—an important step in the determination of novel macromolecular structures and a milestone for atomic resolution cryo-EM of dose-sensitive samples.
Results
We investigated nanocrystals of peptide segments from the amyloid core of the Sup35 prion protein (27–30), Zn2+-NNQQNY, Cd2+-NNQQNY, and GNNQQNY, using MicroED (Fig. 1). Some of these had been previously determined by X-ray crystallography (27–31). We chose these nanocrystals to assess the feasibility of experimental phasing by isomorphous replacement using MicroED. Because these nanocrystals diffract to atomic resolution (Fig. 1), we also chose to explore structure determination by direct methods (27). By using MicroED, resolution was significantly improved for all structures over previous X-ray studies, from 1.3 to 1.0 Å for Zn2+-NNQQNY (PDB ID code 1YJO), from 1.8 to 1.1 Å for GNNQQNY form 1 (PDB ID code 1YJP), and from 2.0 to 1.05 Å for GNNQQNY form 2 (PDB ID code 2OMM).
Fig. 1.

Atomic resolution electron diffraction from amyloid nanocrystals. Diffraction images of Zn2+-NNQQNY (A), Cd2+-NNQQNY (B), GNNQQNY-P21 (C), and GNNQQNY-P212121 (D). A cross indicates the beam position, rings indicate resolution ranges, and arrows point to high-resolution reflections. Insets show images of crystals (indicated by black arrows) from which the high-resolution diffraction patterns were obtained. (Scale bars, 2 μm.)
To limit the radiation damage experienced by the crystals, we measured continuous rotation MicroED data at a dose rate <0.01e− per square angstrom per second. From the illuminated area and size of our crystals, we estimate that, on average, ∼170 × 4,100 × 170 unit cells are illuminated in our experiments. Each frame of the diffraction movie represents a 0.6° wedge of reciprocal space collected during a 2-s exposure. Full datasets were acquired in minutes without exceeding a total accumulated dose of 5e− per square angstrom and processed by X-ray crystallography software to 1.0-Å resolution (Table 1). Multiple datasets were merged to achieve adequate completeness for each of the segments, at high resolution. In the case of GNNQQNY, a single crystal belonging to a different space group, P212121 instead of P21 (27), produced high-resolution data with 77.6% completeness at 1.0 Å (Table 1).
Table 1.
Crystallographic structure determination
| Crystal | Zn2+-NNQQNY | Cd2+-NNQQNY | GNNQQNY-P21 | GNNQQNY-P212121 |
| Data collection | ||||
| Radiation source | Electron FEG | Electron FEG | Electron FEG | Electron FEG |
| Dose upper limit, e−/ Å2 | 5 | 5 | 5 | 5 |
| Space group | P21 | P21 | P21 | P212121 |
| Cell dimensions | ||||
| a,b,c, Å | 21.5, 4.9, 23.9 | 22.1, 4.9, 23.5 | 22.9, 4.9, 24.2 | 23.2, 4.9, 40.5 |
| α,β,γ, ° | 90, 104.0, 90 | 90, 104.3, 90 | 90, 107.8, 90 | 90, 90, 90 |
| Resolution limit, Å | 1.00 (1.06–1.00) | 1.00 (1.05–1.00) | 1.10 (1.16–1.10) | 1.05 (1.11–1.05) |
| Wavelength, Å | 0.0251 | 0.0251 | 0.0251 | 0.0251 |
| No. of crystals merged | 6 | 4 | 4 | 1 |
| Rmerge | 0.151 (0.245) | 0.180 (0.282) | 0.265 (0.450) | 0.199 (0.586) |
| Rr.i.m. | 0.163 (0.280) | 0.203 (0.363) | 0.286 (0.482) | 0.230 (0.669) |
| Rp.i.m. | 0.057 (0.111) | 0.087 (0.181) | 0.139 (0.223) | 0.109 (0.305) |
| I/σI | 10.2 (6.6) | 7.3 (3.7) | 4.6 (3.0) | 4.2 (1.8) |
| CC1/2, % | 98.5 (90.6) | 96.6 (53.9) | 98.4 (88.4) | 98.5 (69.3) |
| Completeness, % | 82.6 (55.3) | 84.5 (66.0) | 94.8 (96.0) | 78.6 (79.3) |
| Multiplicity | 7.1 (5.1) | 4.6 (3.3) | 6.6 (7.2) | 3.7 (3.7) |
| Unique reflections | 2,360 (202) | 2,542 (257) | 2,219 (326) | 1,837 (257) |
| Refinement | ||||
| Resolution range, Å | 22–1.0 | 22–1.0 | 23–1.1 | 20–1.05 |
| No. of reflections (work) | 2,124 | 2,308 | 1,997 | 1,653 |
| Rwork | 0.156 | 0.220 | 0.187 | 0.177 |
| Rfree | 0.194 | 0.242 | 0.224 | 0.186 |
| CCwork | 0.955 | 0.922 | 0.954 | 0.966 |
| CCfree | 0.912 | 0.934 | 0.947 | 0.979 |
| No. of atoms (incl. hydrogen) | 111 | 113 | 114 | 113 |
| Protein | 97 | 98 | 107 | 107 |
| Water | 6 | 7 | 7 | 6 |
| Cation | 1 | 1 | 0 | 0 |
| Acetate | 7 | 7 | 0 | 0 |
| B factors, Å2 | ||||
| Protein | 4.0 | 4.9 | 4.5 | 4.2 |
| Water | 12.9 | 16.0 | 14.5 | 14.7 |
| Cation | 3.7 | 11.3 | n/a | n/a |
| Acetate | 4.1 | 17.2 | n/a | n/a |
| Wilson B, Å2 | 3.2 | 3.4 | 6.1 | 5.7 |
| Rms deviations | ||||
| Bond lengths, Å | 0.020 | 0.021 | 0.019 | 0.017 |
| Bond angles, ° | 1.5 | 1.7 | 1.3 | 1.6 |
Numbers reported in parentheses correspond to values in the highest-resolution shell. incl., including; n/a, not applicable.
Strong anomalous signals are not expected for electron diffraction because the energy of the electron beam is far from any known resonance phenomenon in the elements composing the crystal. Indeed, no significant anomalous signal was detected in our datasets, as evidenced by the poor correlation in Friedel differences between randomly chosen half datasets, at <30% in all cases (Table S1). In contrast, anomalous signals are routinely used in X-ray diffraction experiments to assist in phase determination (3, 4) and are enhanced by tuning the X-ray energy to an absorbance maximum specific to a particular element in the crystal. As a case in point, an anomalous signal sufficient for phasing was reported from Zn2+-NNQQNY crystals using a microfocus synchrotron X-ray source (27). As expected, the anomalous correlations from this experiment are significantly higher than for MicroED (Table S1). A detectable anomalous signal has been reported from electron diffraction experiments in the gas phase and attributed to the modification of the potential field by the presence of the incident electron (32). Evidently, the magnitude of this effect (second Born approximation) proved too small to measure in our experiments.
Table S1.
Anomalous correlation coefficients (CCano) comparing anomalous differences between randomly selected half-datasets
| Resolution | CCano, % | ||||
| X-ray | Electron | ||||
| Zn2+-NNQQNY | Zn2+-NNQQNY | Cd2+-NNQQNY | GNNQQNY-P21 | GNNQQNY-P212121 | |
| 20.0–2.60 | 55 | 13 | 23 | −34 | −5 |
| 2.60–2.00 | 27 | 10 | 19 | −10 | −2 |
| 2.00–1.70 | 32 | 9 | 15 | −36 | −10 |
| 1.70–1.50 | 32 | 26 | 11 | −20 | 1 |
| 1.50–1.35 | 20 | −1 | 5 | −29 | 4 |
| 1.35–1.25 | — | 15 | 2 | −36 | −4 |
| 1.25–1.15 | — | 7 | 1 | −15 | −7 |
| 1.15–1.10 | — | 6 | −12 | −14 | −11 |
The low correlation values indicate that there is no significant anomalous signal from any of the crystals. X-ray data for Zn2+-NNQQNY originates from Nelson et al. (27). Data not measured are indicated by a dash.
The data collected by continuous rotation MicroED and processed by X-ray crystallography software show nearly kinematic scattering devoid of dynamical scattering artifacts. Reflections forbidden by symmetry are overall absent or present at <8% of the measured intensities for their allowed counterparts on average (Table S2). Furthermore, comparisons of reflections related by Friedel symmetry (RFriedel) and non-Friedel symmetry (Rnon-Friedel) gave no strong indication of dynamical scattering (Table S3). We found the differences between RFriedel and Rnon-Friedel are small: at <1% in many datasets (Table S3).
Table S2.
Measured intensities for [0, n*K, 0] reflections in space group P21
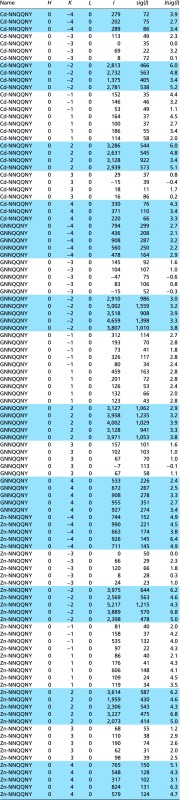 |
Allowed reflections are colored in blue.
Table S3.
Comparisons of RFriedel and Rnon-Friedel offer insight into the magnitude of dynamical scattering
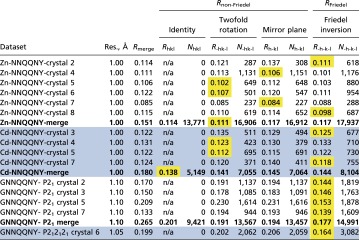 |
Statistics for composite datasets (merges of multiple crystals) are shown in bold. Component datasets (entirely from a single crystal) are shown in regular font. Alternate white and blue shading is used to group together crystals belonging to the same project, and distinguish the four different projects. A conventional Rmerge was calculated by using all reciprocal space symmetry operators appropriate for the space group and Rhkl, R-hk-l, Rh-kl, and R-h-k-l were calculated by using individual symmetry operators. The contribution of dynamical effects to violations of Friedel symmetry (RFriedel or R-h-k-l) has been reported as negligible in electron diffraction experiments with bacteriorhodopsin (24, 43), presumably because electron beam paths are equivalent for Friedel mates (i.e., Ihkl and I–h-k-l originate from opposite sides of the same Bragg plane). In contrast, dynamical effects can potentially contribute to violations of non-Friedel symmetry (Rnon-Friedel) because electron beam paths for non-Friedel symmetry-related reflections (e.g., Ihkl and I–hk-l) do not share the same path. Comparison of RFriedel and Rnon-Friedel therefore offer an estimate of the magnitude of dynamical scattering. If dynamical scattering is strong, then we would expect RFriedel << Rnon-Friedel. Instead, we find RFriedel ∼ Rnon-Friedel. The symmetry operator that produces the lowest R factor is highlighted with a yellow background. In the cases where RFriedel < Rnon-Friedel, the difference is small in comparison with the increase in Rmerge incurred by merging together crystals. Hence, dynamical scattering effects on Rmerge appear to be similar or less than typical levels of crystal nonisomorphism. Reflections containing an index of zero were not included in Rsym calculations because they are related by multiple symmetry operators. Nsym reports the number of pairs of reflections compared. The merging R factors were calculated by using the following equation: Σhkl Σj |<Ihkl> − Ihkl,j|/ Σhkl Σj Ihkl,j. For Rhkl, R-hk-l, Rh-kl, and R-h-k-l, j ranges from 1 to 2 only.
We evaluated our hypothesis that intensities measured at atomic resolution by MicroED from all four peptide nanocrystals are sufficient for structure determination using ab initio structure determination methods. We executed the direct-methods protocol with standard parameters using the SHELXD software package (SI Methods) (12). Direct methods solutions were obtained for all four datasets: Zn2+-NNQQNY, Cd2+-NNQQNY, GNNQQNY-P21 and GNNQQNY-P212121 (Fig. S1). In each case, the ensembles of atoms generated by SHELXD were correctly assigned and interpretable as peptide segments in the amyloid state (Fig. 2 and Table S4). Their structures were refined (Fig. 2) to fit the diffraction measurements (Methods). Each refined structure included hydrogen atoms that fit well into high-resolution density maps (Fig. S2 and Table S5). Refined structures also showed strong densities for the coordinated Zn2+ and Cd2+ ions, which scaled in proportion to their atomic numbers.
Fig. S1.

Histograms of combined figure of merit (CFOM) scores indicate the frequency of obtaining correct solutions by direct methods using ShelxD. Each histogram indicates the distribution of 5,000 independent CFOM scores obtained for each of the four sup35 structures. Correct solutions have CFOM scores > 80. A clear separation between incorrect and correct structures is indicated by the bimodal distributions of CFOM scores. The smaller of the two peaks in the distribution correspond to correct solutions (higher CFOMs). The two metal-containing structures have a higher frequency of obtaining a correct structure compared with the two non-metal-containing structures. The presence of the metal may be a factor in facilitating a correct direct methods solution. We also note that the two structures that contain metals correspond to higher-resolution (higher-quality) datasets, which also facilitate a correct direct-methods solution.
Fig. 2.

Density maps from ab initio and final refined model. Structures of Zn2+-NNQQNY (A), Cd2+-NNQQNY (B), GNNQQNY-P21 (C), and GNNQQNY-P212121 (D) are shown. Note that a majority of the peptide atoms were correctly placed by direct methods. The σA-weighted 2mFo − DFc maps (gray) are contoured at 1.8 σ. The σA-weighted mFo − DFc maps are contoured at +3.0 σ (green) and −3.0 (red) σ. Modeled water molecules are depicted as cyan-colored spheres.
Table S4.
Ab initio structure determination statistics
| Crystal | Zn-NNQQNY | Cd-NNQQNY | GNNQQNY-P21 | GNNQQNY-P212121 |
| Best CFOM | 92.8 | 84.3 | 90.8 | 89.4 |
| Best FCC | 63.3 | 61.0 | 62.1 | 64.8 |
| Peptide atoms (excluding hydrogen) | 55 | 55 | 59 | 59 |
| Peptide atoms correctly placed (excluding hydrogen) | 54 | 54 | 58 | 54 |
Table shows CFOM, final correlation coefficient (FCC), number of atoms identified in the ab initio solution by SHELXD, number of atoms placed correctly, and number of atoms in the final map.
Fig. S2.

Hydrogen atom positions are revealed by a σA weighted Fo − Fc map. (A) The residual map is contoured at +3.0 σ (green) and −3.0 σ (red). The map was calculated by using phases from a model of GNNQQNY (P212121), which had no hydrogens included, nor had any hydrogens been introduced at any point leading to this model. (B) GNNQQNY with hydrogen atoms (black) modeled in riding positions. All of these peaks (20) are within 1.0 Å of the geometrically positioned hydrogen atoms and are labeled by the hydrogen atom. These account for 42% of the total hydrogen atoms present. No hydrogen atoms were detected on water molecules (cyan spheres).
Table S5.
Number of hydrogen atoms verified by residual peaks in a simulated omit hydrogen map
| Structure | Peptide hydrogens | Water hydrogens | ||
| Located | Possible | Located | Possible | |
| Zn2+-NNQQNY | 16 | 45 | 0 | 12 |
| Cd2+-NNQQNY | 9 | 45 | 0 | 14 |
| GNNQQNY- P21 | 16 | 48 | 0 | 14 |
| GNNQQNY- P212121 | 19 | 48 | 0 | 12 |
Peaks must be 3σ or greater in height and <1.0 Å distant from refined hydrogen position to be counted as “located.” Simulated annealing temperature was 5,000 K (44).
SI Methods
Sample Input Parameters for Ab Initio Structure Determination Using SHELXD.
TITL Zn-NNQQNY in P2(1)
CELL 0.0251 21.4800 4.9500 23.8700 90.000 103.952 90.000
ZERR 0.001 0.0083 0.0008 0.0082 0.000 0.100 0.000
LATT -1
SYMM -X, 0.5+Y, -Z
SFAC C H N O Zn
UNIT 31 45 11 13 1
NTRY 5000
FIND 75
PLOP 50 80 120 160 160
HKLF 4
END
Search Parameters for Identification of X-Ray Structures in PDB with Resolution Better than or Equal to 1.1 Å.
The pdbU server at the University of California, Los Angeles (services.mbi.ucla.edu/pdbU) provides a method of searching the Research Collaboratory for Structural Bioinformatics (118,763 structures; last updated April 13, 2016) for structures matching specific criteria. We used this server to filter all PDB entries by resolution cutoff and X-ray technique. The server provides the number of current entries that satisfy our search criteria and lists the details of each entry.
Discussion
The structures of Zn2+-NNQQNY, Cd2+-NNQQNY, GNNQQNY-P21, and GNNQQNY-P212121 demonstrate that MicroED can satisfy the stringent requirements for ab initio phasing of macromolecules—specifically, the measurement of accurate high-resolution intensities. Several experimental challenges were overcome to achieve this. We minimized the total electron dose delivered to a crystal to maintain its high-resolution diffraction. Continuous rotation MicroED data collection was used to minimize dynamical scattering. We collected diffraction data from multiple crystals in varied orientations to fill the reciprocal space. We selected crystals for merging based on isomorphism; nonisomorphism can limit the accuracy of the final intensities. In one case, we determined an ab initio structure from a single nanocrystal in space group P212121.
Our structures are of small macromolecules, but the principles that govern our direct methods approach are applicable to larger proteins. Currently, 1,540 structures present in the PDB diffract to better than 1.1 Å (SI Methods). An unknown number of proteins like these could be future targets for ab initio phasing by MicroED. Phasing by direct methods is computationally more challenging for larger macromolecules, but the technique has already been widely demonstrated on structures that range in complexity from a few atoms to several thousand (7, 10). Ultimately, crystal size, number of unit cells, and crystalline order can all limit high-resolution diffraction to a range below that required by direct methods. We show here that the resolution limit for successful direct methods phasing is the same for MicroED and X-ray diffraction––better than 1.2-Å resolution (8). Indeed, success of direct methods using various resolution cutoffs on our GNNQQNY-P21 data showed that, of 5,000 trials, we found 126 successes at 1.1 Å, 2 successes at 1.15 Å, and 0 successes at 1.2 Å. Thus, a more general approach for phasing of MicroED data may be required for routine de novo phasing of macromolecular structures from submicrometer crystals.
Electrons, because of their strong interaction with matter, can potentially produce high-resolution diffraction patterns from tiny 3D crystals. Therefore, MicroED is well suited for answering questions that require high-resolution diffraction from dose-sensitive biological macromolecular samples studied under cryogenic conditions. The atomic arrangements solved by direct methods present a unique opportunity for further improvement of our current estimates of electron scattering factors. Our atomic structures include hydrogen atoms, which play a central role in many biochemical processes; knowing their positions has provided important insights into catalytic mechanisms (33, 34) and greatly aids rational drug design (35). Lastly, by using direct methods, MicroED can also provide ab initio solutions for small-molecule organic compounds, such as rare pharmaceutical polymorphs (36) and designer peptoids (37) that challenge other means of investigation.
Conclusion
We present four ab initio structures determined by cryo-EM. At 1-Å resolution, these are the highest-resolution cryo-EM structures to date, determined by either imaging or diffraction. Our proof-of-principle determination of these structures speaks to the broader potential of applying ab initio phasing to MicroED. Future targets include new structures yet to be solved by traditional crystallographic means from well-ordered nanocrystals that may diffract to high resolution, but are presently overlooked because of their small size.
Methods
Preparation of Peptide Nanocrystals.
Lyophilized, synthetic GNNQQNY and NNQQNY peptides were purchased from Genscript and dissolved in ultrapure water. GNNQQNY was prepared at a concentration of 10 mg/mL, and NNQQNY was prepared at a concentration of 30 mg/mL The dissolved peptides were then filtered by using a 0.22-μm cutoff filter.
Crystals of Zn2+- and Cd2+-NNQQNY were grown by hanging-drop vapor diffusion at ∼20 °C. The reservoir solution contained 100 mM Hepes (pH 7.0) and 1 M sodium acetate (pH not adjusted). Drops were prepared by pipetting 5 μL of peptide solution, 4 μL of reservoir solution, and 1 μL of 0.1 M ZnSO4 or CdSO4 onto a glass coverslip. Crystals of Zn2+-NNQQNY grew within a day.
Crystals of Zn2+-NNQQNY were used to seed growth of GNNQQNY crystals to ensure isomorphism (27). Crystals of GNNQQNY were grown in batch at ∼20 °C in ultrapure water.
Collection of MicroED Data by Continuous Rotation.
MicroED data from Zn2+-, Cd2+-NNQQNY, and GNNQQNY nanocrystals were collected in an identical way, and similar to what has been described for other nanocrystals (18, 21, 38). Briefly, for each of these samples, nanocrystals were harvested, and a single crystal suspension was generated by mixing of crystal-containing solutions. A small amount (∼2 μL) of the crystal suspension was placed on quantifoil EM grids (R2/2, 300#; EMS). Grids were blotted and plunge-frozen into liquid ethane by using a Vitrobot Mark IV (FEI). Frozen grids were then transferred into liquid nitrogen for storage and later imaging. Frozen hydrated grids were placed in a cryo-TEM (transmission electron microscope) Gatan 626 cryo-holder for imaging and diffraction.
Diffraction patterns and crystal images were collected by using a FEI Tecnai F20 transmission cryo-electron microscope equipped with a field emission electron gun operating at 200 keV and used in diffraction mode. All data were collected by using a bottom-mount TVIPS TemCam-F416 CMOS camera with a sensor size of 4,096 × 4,096 pixels, each 15.6 × 15.6 μm. Diffraction patterns were recorded by operating the detector in rolling shutter mode with 2 × 2-pixel binning, producing a final image 2,048 × 2,048 pixels in size. Individual image frames were taken with exposure times of 2 s per image, by using a selected area aperture with an illuminating spot size of ∼1 μm. This geometry equates to an electron dose rate of <0.01 e− per square angstrom per second. During each exposure, crystals were continuously rotated within the beam at a rate of 0.3° per second, corresponding to a 0.6° wedge of data per frame. Diffraction data were collected from several crystals, each oriented differently with respect to the rotation axis. These datasets each spanned wedges of reciprocal space ranging from 50° to 80°.
We estimate that there are ∼1.0 × 108 unit cells illuminated on average per crystal, per dataset. This value was obtained by approximating the crystal shape as a cylindrical rod with a 200-nm radius and length limited by the 2-μm aperture diameter. Thus, the illuminated volume of the crystal is the volume of a cylinder, πr2h = π × (200 nm)2 × (2,000 nm) = 2.5 × 108 nm3. The unit cell volume for the GNNQQNY crystal is 2.29 nm × 0.49 nm × 2.42 nm × sin(104°) = 2.4 nm3. The number of unit cells illuminated by the beam is simply the ratio of the illuminated volume of the crystal to the unit cell volume; this ratio is ∼1.0 × 108 unit cells. This value corresponds approximately to 170 × 4,100 × 170 unit cells. By comparison, the illuminated volume in the original synchrotron X-ray dataset reported in 2005 was ∼16 times greater (27), but did not yield as high resolution (1.8 vs. 1.1 Å).
We did not find it beneficial to illuminate a larger area because any gain in diffracting intensity from a greater volume of diffracting unit cells is offset by the inclusion of background regions and potentially other crystal fragments that would pose a challenge for indexing and integration of measured reflections.
MicroED Data Processing.
Diffraction images were collected as TVIPS movies and converted to SMV crystallographic format as described (18). These images were processed with both XDS/XSCALE (39) and Denzo/Scalepack (40). These programs were primarily developed for X-ray crystallography, but can be adapted for indexing and integration of MicroED data. The two programs yielded comparable statistics in each of the four crystal projects. We chose the Denzo/Scalepack statistics for Zn2+- and Cd2+-NNQQNY datasets, and XDS/XSCALE for the GNNQQNY in space group P21 and P212121. Indexing and merging statistics are presented in Table 1.
Ab Initio Structure Determination and Refinement.
The SHELX macromolecular structure determination suite was used for phasing the measured intensities (12). The input parameters for the phasing trials by SHELXD are provided in the sample input script (SI Methods). Results for ab initio structure determination using direct methods protocols by SHELXD are presented in Table S1. Refinement was carried out with electron scattering form factors using the program Refmac (41) starting from the atomic coordinates determined by the direct-methods protocol. We used atomic form factors calculated using the electron’s rest mass. These values reported in the International Tables of Crystallography (volume C, table 4.3.1.1) originate from Doyle and Turner (42). The relativistic correction for 200-keV electrons is reported in table 4.3.2.1 as a scaling factor of 1.39138. Application of this correction to the refinement of GNNQQNY-P21 produced only insignificant differences. Comparing coordinates refined with and without the correction revealed an rmsd of 0.002 Å over 107 atom pairs. Average B factors differed by only 0.001 Å2. Refinement and structure validation statistics are presented in Table 1.
Acknowledgments
We thank Tom Holton for assistance with the pdbU server; Dr. Todd Yeates for helpful discussions; and Dr. Melinda Balbirnie for her preliminary work on these structures. This work was supported by National Science Foundation Award MCB-0445429; National Institutes of Health Award 1R01-AG029430; Alzheimer’s Disease Research Center at University of California, Los Angeles (UCLA) Award NIH-016570; the Howard Hughes Medical Institute and their Collaborative Innovation Award; and the Janelia Research Campus visitor program. M.R.S, M.J.C., and D.C are part of the UCLA-Department of Energy (DOE) X-Ray Crystallization and Crystallography Core Facilities, which are supported by DOE Grant DE-FC02-02ER63421. J.R. was supported by the Giannini Foundation.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 5K2E, 5K2F, 5K2G, 5K2H) and the Electron Microscopy Data Bank, https://www.ebi.ac.uk/pdbe/emdb (EMDB codes EMD-8196, EMD-8197, EMD-8198, EMD-8199).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1606287113/-/DCSupplemental.
References
- 1.Hauptman HA. 2001. The Phase Problem of X-Ray Crystallography. Twentieth Century Harmonic Analysis—A Celebration, NATO Science Series, ed Byrnes JS (Springer, Dordrecht, The Netherlands), pp 163–171.
- 2.Rossmann MG, Blow DM. The detection of sub-units within the crystallographic asymmetric unit. Acta Crystallogr. 1962;15(1):24–31. [Google Scholar]
- 3.Perutz MF. Isomorphous replacement and phase determination in non-centrosymmetric space groups. Acta Crystallogr. 1956;9(11):867–873. [Google Scholar]
- 4.Hendrickson WA. Determination of macromolecular structures from anomalous diffraction of synchrotron radiation. Science. 1991;254(5028):51–58. doi: 10.1126/science.1925561. [DOI] [PubMed] [Google Scholar]
- 5.Patterson AL. A Fourier series method for the determination of the components of interatomic distances in crystals. Phys Rev. 1934;46(5):372–376. [Google Scholar]
- 6.Hauptman H. The direct methods of X-ray crystallography. Science. 1986;233(4760):178–183. doi: 10.1126/science.233.4760.178. [DOI] [PubMed] [Google Scholar]
- 7.Usón I, Sheldrick GM. Advances in direct methods for protein crystallography. Curr Opin Struct Biol. 1999;9(5):643–648. doi: 10.1016/s0959-440x(99)00020-2. [DOI] [PubMed] [Google Scholar]
- 8.Sheldrick GM. Phase annealing in SHELX-90: Direct methods for larger structures. Acta Crystallogr A. 1990;46(6):467–473. [Google Scholar]
- 9.Dorset DL, Hauptman HA. Direct phase determination for quasi-kinematical electron diffraction intensity data from organic microcrystals. Ultramicroscopy. 1976;1(3):195–201. doi: 10.1016/0304-3991(76)90034-6. [DOI] [PubMed] [Google Scholar]
- 10.Hauptman H. Phasing methods for protein crystallography. Curr Opin Struct Biol. 1997;7(5):672–680. doi: 10.1016/s0959-440x(97)80077-2. [DOI] [PubMed] [Google Scholar]
- 11.van Genderen E, et al. Ab initio structure determination of nanocrystals of organic pharmaceutical compounds by electron diffraction at room temperature using a Timepix quantum area direct electron detector. Acta Crystallogr A Found Adv. 2016;72(Pt 2):236–242. doi: 10.1107/S2053273315022500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64(Pt 1):112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 13.Lučič V, Rigort A, Baumeister W. Cryo-electron tomography: The challenge of doing structural biology in situ. J Cell Biol. 2013;202(3):407–419. doi: 10.1083/jcb.201304193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng Y, Grigorieff N, Penczek PA, Walz T. A primer to single-particle cryo-electron microscopy. Cell. 2015;161(3):438–449. doi: 10.1016/j.cell.2015.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henderson R, Unwin PNT. Three-dimensional model of purple membrane obtained by electron microscopy. Nature. 1975;257(5521):28–32. doi: 10.1038/257028a0. [DOI] [PubMed] [Google Scholar]
- 16.Nederlof I, Li YW, van Heel M, Abrahams JP. Imaging protein three-dimensional nanocrystals with cryo-EM. Acta Crystallogr D Biol Crystallogr. 2013;69(Pt 5):852–859. doi: 10.1107/S0907444913002734. [DOI] [PubMed] [Google Scholar]
- 17.Nannenga BL, Gonen T. Protein structure determination by MicroED. Curr Opin Struct Biol. 2014;27:24–31. doi: 10.1016/j.sbi.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hattne J, et al. MicroED data collection and processing. Acta Crystallogr A Found Adv. 2015;71(Pt 4):353–360. doi: 10.1107/S2053273315010669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi D, Nannenga BL, Iadanza MG, Gonen T. Three-dimensional electron crystallography of protein microcrystals. eLife. 2013;2:e01345. doi: 10.7554/eLife.01345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nannenga BL, Shi D, Hattne J, Reyes FE, Gonen T. Structure of catalase determined by MicroED. eLife. 2014;3:e03600. doi: 10.7554/eLife.03600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodriguez JA, et al. Structure of the toxic core of α-synuclein from invisible crystals. Nature. 2015;525(7570):486–490. doi: 10.1038/nature15368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hattne J, Shi D, de la Cruz MJ, Reyes FE, Gonen T. Modeling truncated pixel values of faint reflections in MicroED images. J Appl Cryst. 2016;49(Pt 3):1029–1034. doi: 10.1107/S1600576716007196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cowley JM, Fields PM. Dynamical theory for electron scattering from crystal defects and disorder. Acta Crystallogr A. 1979;35(1):28–37. [Google Scholar]
- 24.Glaeser RM, Ceska TA. High-voltage electron diffraction from bacteriorhodopsin (purple membrane) is measurably dynamical. Acta Crystallogr A. 1989;45(Pt 9):620–628. doi: 10.1107/s0108767389004599. [DOI] [PubMed] [Google Scholar]
- 25.Grigorieff N, Henderson R. Comparison of calculated and observed dynamical diffraction from purple membrane: Implications. Ultramicroscopy. 1996;65(1–2):101–107. [Google Scholar]
- 26.Subramanian G, Basu S, Liu H, Zuo J-M, Spence JCH. Solving protein nanocrystals by cryo-EM diffraction: Multiple scattering artifacts. Ultramicroscopy. 2015;148:87–93. doi: 10.1016/j.ultramic.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 27.Nelson R, et al. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435(7043):773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balbirnie M, Grothe R, Eisenberg DS. An amyloid-forming peptide from the yeast prion Sup35 reveals a dehydrated β-sheet structure for amyloid. Proc Natl Acad Sci USA. 2001;98(5):2375–2380. doi: 10.1073/pnas.041617698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brewster AS, et al. Indexing amyloid peptide diffraction from serial femtosecond crystallography: New algorithms for sparse patterns. Acta Crystallogr D Biol Crystallogr. 2015;71(Pt 2):357–366. doi: 10.1107/S1399004714026145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diaz-Avalos R, et al. Cross-beta order and diversity in nanocrystals of an amyloid-forming peptide. J Mol Biol. 2003;330(5):1165–1175. doi: 10.1016/s0022-2836(03)00659-4. [DOI] [PubMed] [Google Scholar]
- 31.Sawaya MR, et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447(7143):453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 32.Schomaker V, Glauber R. The Born approximation in electron diffraction. Nature. 1952;170(4320):290–291. [Google Scholar]
- 33.Wan Q, et al. Toward resolving the catalytic mechanism of dihydrofolate reductase using neutron and ultrahigh-resolution X-ray crystallography. Proc Natl Acad Sci USA. 2014;111(51):18225–18230. doi: 10.1073/pnas.1415856111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klinman JP, Kohen A. Hydrogen tunneling links protein dynamics to enzyme catalysis. Annu Rev Biochem. 2013;82:471–496. doi: 10.1146/annurev-biochem-051710-133623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaufmann KW, Meiler J. Using RosettaLigand for small molecule docking into comparative models. PLoS One. 2012;7(12):e50769. doi: 10.1371/journal.pone.0050769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neumann MA, van de Streek J, Fabbiani FPA, Hidber P, Grassmann O. Combined crystal structure prediction and high-pressure crystallization in rational pharmaceutical polymorph screening. Nat Commun. 2015;6:7793. doi: 10.1038/ncomms8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mannige RV, et al. Peptoid nanosheets exhibit a new secondary-structure motif. Nature. 2015;526(7573):415–420. doi: 10.1038/nature15363. [DOI] [PubMed] [Google Scholar]
- 38.Shi D, et al. The collection of MicroED data for macromolecular crystallography. Nat Protoc. 2016;11(5):895–904. doi: 10.1038/nprot.2016.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol Macromol Crystallogr A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 41.Murshudov GN, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Doyle PA, Turner PS. Relativistic Hartree–Fock X-ray and electron scattering factors. Acta Crystallogr A. 1968;24(3):390–397. [Google Scholar]
- 43.Glaeser RM, Downing KH. High-resolution electron crystallography of protein molecules. Ultramicroscopy. 1993;52(3-4):478–486. doi: 10.1016/0304-3991(93)90064-5. [DOI] [PubMed] [Google Scholar]
- 44.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]