

Abstract
Neuroendocrine tumors (NETs) comprise a heterogeneous group of malignancies, with differences in prognosis and effective therapies. Traditionally, NETs have been characterized by tumor grade, site of primary tumor, functional status, and presence of underlying familial syndrome. However, increased feasibility and utilization of next-generation sequencing and other methodologies have revealed new genomic and epigenetic aberrations. In the last decade, treatment options available for metastatic well-differentiated gastroenteropancreatic (GEP) NETs have expanded, with approval of antiangiogenic and mTOR-directed targeted therapies, and our armamentarium of active therapies is likely to further increase. Cytotoxic therapies also are an important option for pancreatic NETs, and MGMT promoter methylation and protein expression may be an important biomarker for efficacy of alkylating agents. Peptide receptor radioligand therapy is an emerging treatment that uses functional imaging to personalize dosimetry to the tumor and avoid nephrotoxicity. Nevertheless, there is a critical need for further biomarkers, particularly multianalyte biomarkers, to aid in prognostication and predict efficacy of therapies.
Keywords: Carcinoid tumor, neuroendocrine tumors (NETs), islet cell carcinoma, precision medicine
Introduction
The FDA approval of the targeted therapies sunitinib and everolimus for advanced pancreatic neuroendocrine tumors (NETs) in 2011 was a reflection of our growing understanding of the biology and heterogeneity of NETs and their responses to various therapies (Table 1). Improvements in technology have continued to increase the feasibility and utilization of next-generation sequencing and other methodologies in NETs, revealing new genetic and biologic aberrations. These findings lead to testable hypotheses to determine relevant prognostic and ideally predictive biomarkers that may guide personalization of existing therapy and shed light on novel potential targets.
Table 1. United States Food and Drug Administration approval of agents for NETs within the last 10 years.
| Date of FDA approval | Drug | Indication |
|---|---|---|
| 05/06/2011 | Everolimus | Unresectable, locally advanced or metastatic, progressive pancreatic NET |
| 05/20/2011 | Sunitinib | Unresectable, locally advanced or metastatic, progressive pancreatic NET |
| 12/16/2014 | Lanreotide | Unresectable, well or moderately differentiated, locally advanced or metastatic GEP-NETs to improve progression-free survival |
| 02/26/2016 | Everolimus | Unresectable, well differentiated, locally advanced or metastatic, non-functional, progressive gastrointestinal or lung NET |
FDA, Food and Drug Administration; GEP, gastroenteropancreatic; NETs, neuroendocrine tumors.
NETs have increased in incidence from the mid-1970s through the 2000s, with an incidence of 5.25/100,000 in 2004 (1). However, given prolonged overall survival for the majority of patients with NETs, the estimated prevalence of disease as of 2004 was 35/100,000 in the United States, outstripping the prevalence of esophageal cancer, gastric cancer, and pancreatic cancer (1). Encouragingly, survival of patients with metastatic NETs has improved in more modern treatment eras, with significant improvement in survival after 1988, likely attributable to the development and use of octreotide (1). Though unresectable metastatic NETs are invariably incurable malignancies, there is marked heterogeneity in the speed of progression and the responses to therapy. Classification of gastroenteropancreatic (GEP) NETs by histologic grade, primary site, and functional hormone secretion status provides some insight into this heterogeneity and is important to consider in determining an individual’s sequence of therapies. We expect that molecular classification may in the future provide a more refined treatment algorithm.
Clinical classification of NETs
WHO grade
Pathologic assessment of WHO grade is critical in determining prognosis and thus serving as the most basic requirement for individualizing therapy of GEP NETs (Table 2) (2). Prognosis is dismal for patients with poorly differentiated disease, with median survival of 14 months with regional disease and only 5 months with distant metastatic disease in SEER data (1). Conversely, prognosis is markedly better for patients with well or moderately differentiated NETs, with median survival of 111 months with regional disease and 33 months even with distant metastatic disease in SEER data prior to 2004 (1). These survival rates have only improved with more modern treatment paradigms, as median survival in the PROMID study of octreotide was 84.7 months (3). However, even within well-differentiated NETs, there is significant heterogeneity of disease aggressiveness and prognosis. Stage-for-stage, grade 1 NETs had superior outcomes than grade 2 NETs in SEER analysis (1). Retrospective analysis of pathologic specimens from patients with metastatic GEP NETs also showed low-grade NETs had superior survival than intermediate-grade NETs, with 5-year survival rate of 87% vs. 38%, and strong correlation between both Ki-67 index and mitotic rate with overall survival (4). Additional studies also demonstrated correlation between Ki-67 and overall survival for metastatic pancreatic and midgut NETs (5).
Table 2. WHO grading system of GEP NETs (2).
| Differentiation | Grade | Proliferative rate |
|---|---|---|
| Well differentiated | G1 (low) | <2 mitoses/10 hpf and Ki-67 index <3% |
| G2 (intermediate) | 2–20 mitoses/10 hpf or Ki-67 index 3–20% | |
| Poorly differentiated | G3 (high) | >20 mitoses/10 hpf or Ki-67 index >20% |
GEP, gastroenteropancreatic; NETs, neuroendocrine tumors; hpf, high-power field.
Primary tumor site
The primary tumor site bears prognostic importance and may determine therapy options, particularly when determining pancreatic versus extrapancreatic primary. Among metastatic well-differentiated GEP NETs, those with duodenal and small bowel primaries have the best median survival (over 4.5 years), while pancreatic primaries confer median survival of 2 years and colorectal primaries have the worst survival at 5 months (Table 3) (1).
Table 3. Median overall survival in months, by disease stage, of G1/G2 NETs based on SEER data from 1973 to 2004 (1).
| Embryologic | Primary site | Localized | Regional | Distant |
|---|---|---|---|---|
| Foregut | Lung | 227 | 154 | 16 |
| Thymus | 110 | 68 | 40 | |
| Gastric | 154 | 71 | 13 | |
| Pancreas | 136 | 77 | 24 | |
| Duodenum | 107 | 101 | 57 | |
| Midgut | Small bowel | 111 | 105 | 56 |
| Appendix | >360 | >360 | 27 | |
| Cecum | 135 | 107 | 41 | |
| Hindgut | Colon | 261 | 36 | 5 |
| Rectum | 290 | 90 | 22 |
NETs, neuroendocrine tumors.
Traditionally, NETs were classified by the embryologic origin of their primary site as either foregut, midgut, or hindgut. Indeed, these classifications formerly defined inclusion criteria for specific clinical trials; for example, the PROMID study only enrolled patients with midgut NETs (6). While this classification was based on presumed differences in histologic structure, argentaffin staining patterns, association with carcinoid syndrome, and metastatic patterns (7), SEER data demonstrates marked heterogeneity in prognosis even within each embryologic grouping (Table 3). At present, the anatomic distinction that is most clinically important for metastatic NETs is pancreatic versus non-pancreatic primary. Pancreatic NETs are usually non-functional (90.8%), though non-functional tumors have inferior prognosis to functional tumors as they are usually discovered at a more advanced stage (8). Recent phase III clinical trials specifically enrolled either pancreatic or non-pancreatic NETs given the differential clinical and biologic characteristics of these distinct groups of tumors (9-11).
Functional syndromes
The presence of functional syndromes due to secretion of active hormones is an important consideration in treatment. Carcinoid syndrome, comprised classically of flushing, secretory diarrhea, bronchospasm, and right-sided cardiac valvular fibrosis, is attributed to excessive secretion of serotonin by neoplastic cells and is most frequently found in patients with well-differentiated midgut NETs with hepatic metastases (12). Additionally, functional pancreatic NETs can secrete insulin, glucagon, vasoactive intestinal peptide (VIP), gastrin, or somatostatin. For patients with functional syndromes, symptomatic control of the functional syndrome is an important goal of management. Somatostatin analogs (SSAs) are used to inhibit secretion of bioactive peptides and are a mainstay of symptomatic control for carcinoid syndrome (13) and are also effective for VIPomas (14), glucagonomas (15), and somatostatinomas (16). The use of SSAs in insulinomas can paradoxically transiently aggravate hypoglycemia by suppressing counterregulatory glucagon and growth hormone secretion during fasting, causing symptomatic hypoglycemia, and thus should be used only with caution (17). Instead, diazoxide, which suppresses insulin release from pancreatic beta cells, is commonly used to control hypoglycemia in unresectable symptomatic insulinoma (18), and the use of SSAs is often reserved for cases refractory to diazoxide. Telotristat etiprate, an oral inhibitor of serotonin synthesis, significantly improved symptoms of refractory carcinoid syndrome and decreased urinary 5-HIAA levels in early phase clinical trials (19,20), and the randomized, double-blind, placebo-controlled phase III TELESTAR trial in patients with refractory carcinoid syndrome demonstrated that adding telotristat etiprate to SSA significantly improved symptoms of carcinoid syndrome (21).
Octreotide scintigraphy
An important additional imaging biomarker is positive uptake on octreotide-labeled nuclear imaging. Indium-111-pentetreotide (octreoscan) scintigraphy uses a radiolabeled SSA that can serve as a functional imaging biomarker for expression of somatostatin receptors overexpressed in the majority of GEP NETs (22), with 78% sensitivity to detect abdominal carcinoids and 67% sensitivity to detect pancreatic NETs (23), and frequently is able to detect metastatic tumors overlooked on conventional cross-sectional imaging (24,25).
Gallium-68-DOTA-peptide PET/CT has more recently been developed, using SSAs labelled with the positron-emitting 68Ga isotope. A recent prospective study of imaging in patients with metastatic GEP NETs found 68Ga-DOTATATE PET/CT imaging successfully identified 95.1% of lesions, compared to only 30.9% identified by 111In-pentetreotide imaging (26). These results may result in more widespread use of 68Ga-DOTA-peptide PET imaging in the U.S. Octreotide-targeted therapies such as peptide receptor radiotherapy (PRRT) routinely require positive functional somatostatin receptor imaging such as 111In-pentetreotide or 68Ga-DOTA-peptide imaging to serve as a surrogate biomarker indicating sufficiently high somatostatin receptor expression (27). Indeed, in an early study of PRRT, higher rates of tumor remission were observed in patients with greater uptake on baseline 111In-pentetreotide imaging (28). Thus, radiolabeled somatostatin imaging serves as an example of noninvasive imaging serving as a predictive biomarker.
Inherited syndromes
Finally, NETs arising in the context of a familial inherited syndrome have distinct underlying pathobiology and differential risk for development of second primary NETs than sporadic NETs. In multiple endocrine neoplasia-1 (MEN-1) syndrome, germline inactivating mutations of the MEN1 tumor suppressor promote development of parathyroid hyperplasia, pituitary adenomas, and pancreatic NETs. MEN1 encodes menin, a nuclear scaffolding protein that interacts with many partners, including transcription factors, cytoskeletal proteins, and DNA repair proteins, to effect cell growth and alter transcriptional programs (29). Generally, treatment of metastatic MEN-1-associated pancreatic NETs is not different than for sporadic pancreatic NETs, though MEN1-associated pancreatic NETs may be slower growing than sporadic pancreatic NETs (30,31).
NET biology and genetics
NETs are believed to derive from enterochromaffin cells that originate embryologically within the neural crest and disperse throughout the respiratory and gastrointestinal mucosal tracts (32,33). NETs overexpress somatostatin receptors, a family of G protein coupled receptors that bind the somatotropin-release inhibiting factors with nanomolar affinity. There are five subtypes of somatostatin receptor, sst1-5, which are typically all expressed in normal pancreatic islets. Ligand binding triggers intracellular signal transduction changes, inhibiting activity of adenylyl cyclase. Ultimately, somatostatin receptor activation inhibits hormone secretion and can inhibit proliferation and induce apoptosis (34). NETs broadly express the somatostatin receptors, particularly sst2 (35). The SSAs octreotide and lanreotide both are agonists that bind sst2 with high affinity, and also bind sst3 and sst5 with intermediate affinity (34).
Pancreatic NETs
Several lines of evidence point toward the AKT-mTOR pathway as a key driver of pancreatic NETs. Gene expression microarrays of pancreatic NETs demonstrated activation of the AKT-mTOR pathway in both insulinomas and non-functional pancreatic NETs, along with downregulation of TSC2, an inhibitor of the AKT-mTOR pathway (Figure 1). Altered protein levels of TSC2, PTEN, or both have been found in 85% of primary tumors, and none of 20 patients whose primary tumors had normal levels of both TSC2 and PTEN proteins developed metastatic disease, while 8/25 of those whose tumors had low levels of TSC2 and PTEN did develop metastases (36). Thus, activation of the AKT-mTOR pathway was thought to be a potentially prognostic, targetable aberration in pancreatic NETs.
Figure 1.
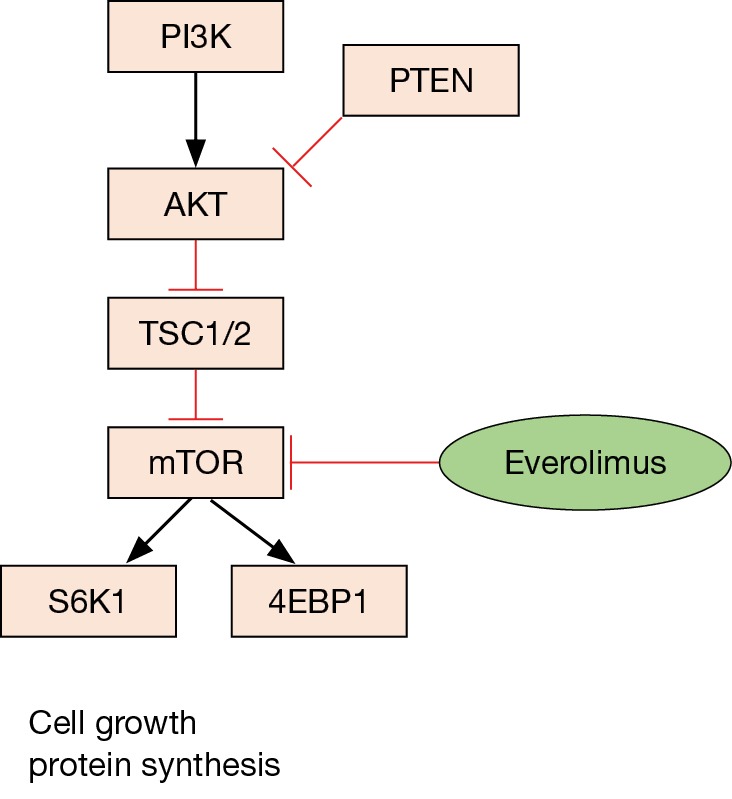
The PI3K/AKT/mTOR pathway is a critical pro-survival cell signaling pathway within NETs that mediates cell growth and peptide translation. Targeted mTOR inhibiting therapies such as everolimus have demonstrated efficacy in pancreatic and extrapancreatic well-differentiated NETs. NETs, neuroendocrine tumors.
Epigenetic dysregulation is increasingly appreciated as a major regulator of pancreatic NET biology. Indeed, hypermethylation, which is commonly associated with gene silencing, has been described in 87% of pancreatic NETs, with the most common hypermethylated genes including RASSF1A (75%), CDKN2A (40%), MGMT (40%), RARB (25%), and MLH1 (23%) (37). These are potentially clinically significant, as CDKN2A encodes the p16INK4A cyclin dependent kinase inhibitor, MGMT impacts efficacy of alkylating chemotherapy agents, and hMLH1 loss causes microsatellite instability.
Genomic sequencing studies of pancreatic NETs also demonstrate aberrations within the AKT-mTOR pathway along with additional novel mutations. A seminal study of 68 sporadic pancreatic NETs revealed recurrent mutations in MEN1 (44%), DAXX or ATRX (43%), and genes in the mTOR pathway (15%) (38). The high frequency of mutations of MEN1 even in sporadic pancreatic NETs underscores the importance of MEN1 as an important common driver mutation in pancreatic NETs. Inactivating DAXX and ATRX mutations appear to be mutually exclusive. DAXX and ATRX encode interacting proteins that are involved in the alternative lengthening of telomeres phenotype (39), and DAXX or ATRX protein loss also are associated with chromosomal instability (40). Studies conflict on whether the presence of DAXX/ATRX mutations confers a favorable (38) or unfavorable prognosis (40). Mutations within the mTOR pathway included PTEN (5/68 cases), TSC2 (6/68), and PIK3CA (1/68) (38). The prognostic or predictive effect of these mutations on therapies used for pancreatic NETs remains unclear, and future work will almost certainly focus on associating genotype with prognosis or outcome with specific therapies.
Small bowel NETs
Consistent with the clinical differences in these entities, small intestinal NETs appear to have a distinct biology and mutational landscape compared with pancreatic NETs. Whole exome sequencing on 48 small bowel NET samples showed there were few recurrent mutations identified, though copy number variation (CNV) aberrations were detected. Like pancreatic tumors, 29% of small bowel NETs had genetic alterations in the PI3K-AKT-mTOR pathway, including amplification of AKT1 or AKT2 in 13 cases (27%) and amplification of MTOR in 4 cases. Unlike the pancreatic NETs, SRC was amplified in 11/48 cases (23%), and mutation or deletion in SMAD2 or SMAD4 was found in 22/48 cases (46%) (41). Additionally, recurrent somatic mutations and deletions in CDKN1B were found in 14/180 (8%) small intestinal NETs. CDKN1B encodes p27, a cyclin-dependent kinase inhibitor that binds to and inhibits CDK2 and CDK4 (42). As such, inhibition of AKT-mTOR pathway components, SRC, or cell cycle pathway components is a rational consideration.
Rather than harboring significant mutational burdens, small intestinal NETs were recently found to have marked epigenetic dysregulation. A comprehensive genomic, epigenetic, and gene expression profiling study of 97 small intestinal NETs revealed three subtypes of disease with markedly different prognoses. The largest group (55%) was characterized by loss of heterozygosity of chromosome 18, CpG island methylator phenotype (CIMP) negative status, higher proportion of CDKN1B mutations, and superior progression-free survival (PFS) after resection of localized tumors. Another subgroup (19%) had no CNV, high rate of CIMP positivity, and intermediate PFS after resection of localized tumors. A third subgroup (26%) had multiple CNV aberrations including several chromosomal amplifications, and worst PFS after resection of localized tumors. These findings may motivate personalization of therapeutic strategies based on risk stratification after resection and may provide rationale for clinical trials that modulate epigenetics, such as hypomethylating agents (43).
Additionally, a study of IHC expression of several PI3K pathway components in archived NETs, the majority of which had small intestine primary, showed correlation between expression of PIK3CA, MTOR, and phospho-EIF4EBP1 and high Ki-67. Notably, high expression of MTOR or its downstream activated targets phospho-RPS6KB1, phospho-RPS6, or phospho-EIF4EBP1 was associated with inferior outcomes (44). Whether treatment of these tumors with mTOR inhibitors will provide greater benefit compared to those without abnormalities of the PI3K-mTOR axis remains to be determined.
Systemic therapies to slow progression of GEP NETs
SSAs
While SSAs are used to control carcinoid syndrome and other syndromes due to release of bioactive amines from functional NETs, their ability to slow the growth of advanced NETs is also now established. In the randomized, double-blind, placebo-controlled PROMID clinical trial, octreotide LAR prolonged PFS compared to placebo in 85 patients with metastatic well-differentiated midgut NETs (PFS 14.3 vs. 6 mo; HR 0.34, 95% CI: 0.20–0.59). In subgroup analysis, there was no significant difference in benefit based on presence of carcinoid syndrome, chromogranin A (CgA) elevation, or age (6). There was no evidence of benefit in OS, though the majority of patients on the placebo arm crossed over, confounding results (3). Notably, essentially all patients enrolled on PROMID had Ki-67 <2%, and thus results from the study could only reliably be generalized to G1 midgut NETs.
The subsequent CLARINET study was a larger randomized, double-blind, placebo-controlled phase III study of 204 patients with well or moderately differentiated, nonfunctional pancreatic, midgut, or hindgut NETs who were randomized to receive either lanreotide or placebo, and confirmed the findings of PROMID. Treatment with lanreotide significantly improved PFS (HR 0.47, 95% CI: 0.30–0.73), and subgroup analysis showed nearly all subgroups demonstrated significant or strong trend toward improved PFS with lanreotide, including midgut and pancreas primaries, grade 1 and 2 (all had Ki-67 <10%), and hepatic tumor volume ≤25% or >25% (45).
Everolimus in pancreatic NETs
Given the evidence for mutations in and activation of the AKT-mTOR pathway in sporadic NETs, a series of clinical trials evaluated the efficacy of everolimus (RAD001) in well-differentiated NETs. An early single-arm study at MD Anderson Cancer Center of everolimus combined with octreotide LAR in 30 pancreatic and 30 non-pancreatic well-differentiated NET patients, found median PFS of 60 weeks and overall response rate of 20%, with decrease in Ki-67 index detected in 2-week on-treatment biopsies in 5/7 patients (46). The subsequent multinational single-arm phase II RADIANT-1 study of everolimus among patients with pancreatic NETs who failed prior cytotoxic chemotherapy found median PFS with everolimus ranging from 9.7 to 16.7 months among subgroups (47). The confirmatory randomized phase III RADIANT-3 trial in 410 patients with well-differentiated pancreatic NETs that had progressed within the prior 12 months verified there was indeed improvement in the PFS primary endpoint with everolimus compared to placebo (11.0 mo with everolimus vs. 4.6 mo with placebo; HR 0.35, 95% CI: 0.27–0.45). Though there was no significant difference in response rates (5% vs 2%), 64% of patients receiving everolimus had at least some tumor shrinkage compared to 21% of patients receiving placebo. Notably, patients randomized to the placebo arm were allowed to crossover, and 73% did in fact crossover, confounding overall survival (9). The positive results of the RADIANT-3 study motivated FDA approval of everolimus for well-differentiated pancreatic NETs.
Notably, the PFS benefit on treatment with everolimus was present for all clinically defined subgroups in RADIANT-3, including both well and moderately differentiated NETs, prior SSA therapy, and prior chemotherapy (9,48). In the RADIANT-1 and MD Anderson studies, elevated levels of CgA and neuron-specific enolase (NSE), markers likely associated with higher disease burden, were each associated with inferior median PFS. Moreover, patients who had a response in their CgA or NSE levels, defined as 30% decrease compared to baseline at week 4, were more likely to experience some degree of tumor shrinkage (49). Thus, the baseline level of CgA or NSE appears to be a prognostic biomarker in pancreatic NETs, and early CgA or NSE response is associated with radiographic tumor shrinkage and may be a predictor of treatment outcome. Pharmacodynamic studies from the MD Anderson trial using 17 paired pretreatment and 2-weeks on-treatment tumor biopsies found significant decrease in phosphorylated-S6 on-treatment, demonstrating on-target effect of everolimus. Additionally, samples with greater feedback activation of AKT, demonstrated by greater phospho-AKT S473 levels, actually had greater degree of tumor response to everolimus (50). Thus, feedback activation of AKT, at least after two weeks of treatment, actually appears to be a biomarker of greater efficacy of everolimus rather than a mechanism of resistance as has been described in other settings (51). Finally, treatment with everolimus was associated with decrease in detectable VEGF pathway components, such as soluble VEGF receptor 2 and placental growth factor, though it remains unclear if these changes have prognostic or predictive implications (52).
Everolimus in non-pancreatic NETs
Everolimus also appears effective in non-pancreatic NETs. In the randomized, double-blind, placebo-controlled RADIANT-2 phase III clinical trial, 429 patients with well-differentiated NETs with carcinoid syndrome that progressed within the past 12 months were randomized to receive either everolimus or placebo in combination with octreotide LAR. Median PFS was 16.4 months in the everolimus arm and 11.3 months in the placebo arm (HR 0.77, 95% CI: 0.59–1.00) (53). Subsequently, in the randomized, double-blind, placebo-controlled RADIANT-4 phase III trial, 302 patients with well-differentiated, non-functional, progressive lung or GI NETs were randomized to receive either everolimus or placebo. Median PFS was 11.0 months in the everolimus arm and 3.9 months in the placebo arm (HR 0.48, 95% CI: 0.35–0.67) (10). This data recently culminated in FDA approval of everolimus for gastrointestinal and lung NETs.
Subgroup analysis of RADIANT-2 showed benefit with everolimus in all prespecified subgroups, independent of tumor grade, primary tumor site (53), prior SSA use (54), colorectal primaries (55), and lung primaries (56). Notably, preclinical data had suggested that the presence of the FGFR4-G388R single-nucleotide polymorphism may be associated with more aggressive NET biology and decreased susceptibility to everolimus (57). FGFR4 interacts with the adhesion molecule N-cadherin, which is related to tumor invasion and metastasis and activates downstream signaling cascades including the PI3K-AKT-mTOR pathway (57). However, in retrospective clinical data, the presence of the FGFR4 polymorphism was not associated with PFS or OS (58).
Sunitinib and anti-angiogenics
NETs have been well recognized to be particularly vascular tumors, and IHC shows increased expression of vascular endothelial growth factor (VEGF) and VEGF receptor (VEGFR) subtypes in both pancreatic and non-pancreatic NETs (59). Sunitinib malate is a multitarget tyrosine kinase inhibitor that can inhibit several proangiogenic kinases including VEGFR1, VEGFR2, PDGFRα, and PDGFRβ. The efficacy of sunitinib in extending PFS in treatment of pancreatic NETs was confirmed in a placebo-controlled, randomized, double-blind phase III clinical trial comparing sunitinib to placebo. In the study, 171 patients with well-differentiated pancreatic NETs were randomized to receive sunitinib or placebo, and the study was halted after interim analysis after 154 patients’ data was analyzed due to increased deaths and serious adverse events in the placebo arm. The primary endpoint of PFS was superior in patients who received sunitinib (11.4 vs. 5.5 months with placebo, HR 0.42, 95% CI: 0.26–0.66). ORR with sunitinib was 9.3%, including two complete responses (11). This study resulted in FDA approval for sunitinib in pancreatic NETs.
Pazopanib has been investigated in several single-arm phase II studies in pancreatic and extrapancreatic NETs, with PFS ranging from 9.1 to 14.4 months (60-62). A placebo-controlled randomized phase II clinical trial, A021202, is ongoing to evaluate the efficacy of pazopanib in progressive well-differentiated non-pancreatic NETs (NCT01841736).
As is the case in nearly all tumor types, correlative studies to identify prognostic and potentially predictive biomarkers of outcome with treatment with VEGFR TKIs are needed. Plasma cytokine analysis from the phase II trial of sunitinib in pancreatic and non-pancreatic NETs (63) revealed that after 28 days of sunitinib treatment, there was a significant increase in VEGF, IL-8, and stromal cell-derived factor-1α (SDF-1α) levels, and a significant decrease in sVEGFR-2 and sVEGFR-3 levels. High baseline SDF-1α level was associated with inferior PFS and OS in pancreatic and non-pancreatic NETs, and high baseline IL-8 and sVEGFR3 were also associated with inferior PFS and OS in non-pancreatic NETs (64). Notably, SDF-1α, or CXCL12, is a chemokine that binds its receptor CXCR4 to promote vascular endothelial cell migration during neovascularization and also promotes leukocyte chemotaxis (65). In another phase II study of pazopanib in patients with pancreatic or non-pancreatic well-differentiated NETs, there were non-significant trends toward improved PFS in patients with no baseline circulating tumor cells, lower soluble-VEGFR2 levels, absence of the VEGFR3 rs307821 (R1324L) polymorphism, and absence of the VEGFR3 rs307826 (T494A) polymorphism (61). The results were not significant, though could be underpowered. Nevertheless, these results would be at best prognostic rather than predictive, and further study in well-powered prospective studies would be required.
Selection of targeted therapy
At present, in pancreatic NET patients, generally selection of everolimus or sunitinib for initial systemic therapy is guided by the toxicity profiles of the drugs rather than by biologic susceptibility of an individual’s tumor to the drug, other than metastatic insulinomas, in which case everolimus is recommended to better control hypoglycemia (66-68). A prospective open-label phase II clinical trial is planned for well-differentiated pancreatic or non-pancreatic NETs by the National Cancer Institute in which patients will have tumor genotyping performed and be assigned to receive either sunitinib or everolimus based on the germline or somatic mutations present. Patients with mutations in MEN1, PDGFR, KIT, or FLT3 would be assigned to receive sunitinib, while patients with mutations in NF1, PTEN, PI3K, AKT, MTOR, VHL, or TP53 will be assigned to receive everolimus. Those with multiple of the above mutations or those with no mutations will be assigned to receive sunitinib. Patients will crossover to the other drug upon first progression (69).
Cytotoxic therapies for pancreatic NETs
While everolimus and sunitinib improve PFS, they have low response rates, and several consensus guidelines recommend or suggest the use of cytotoxic chemotherapy for patients with bulky or rapidly progressive metastatic pancreatic NETs (30,70,71). It should be noted, however, that cytotoxic chemotherapies have never been shown definitively to improve survival.
Streptozocin is an alkylating agent approved by the U.S. FDA in 1982 for treatment of metastatic pancreatic NETs. Streptozocin is an analog of glucose and undergoes cell transport via the glucose transporter GLUT2 (72), which is expressed in essentially all pancreatic NETs (73). The efficacy of streptozocin-based combination chemotherapy regimens in treatment of pancreatic NETs was established in early clinical trials (74,75), and further retrospective studies using modern response criteria demonstrated response rate of 35–40% (76-78).
Temozolomide-based regimens also demonstrate efficacy on retrospective trials, with capecitabine/temozolomide having 70% response rate among pancreatic NET patients in a retrospective study of 30 patients (79). Prognostic or predictive biomarkers or characteristics for efficacy of alkylating agents remain under investigation. A retrospective study of patients who received streptozocin/5-fluorouracil/doxorubicin found that patients who had positive octreoscan and who experienced biochemical response were more likely to have an objective response (80).
Promoter methylation and gene silencing of the O6-methylguanine DNA MGMT gene is a likely biomarker of susceptibility to temozolomide or other alkylating agents (81), and MGMT was one of the most commonly hypermethylated genes noted in a study of pancreatic NETs (37). Alkylating agents induce DNA damage by covalently attaching alkyl or methyl groups to guanine residues, causing interstrand crosslinks and ultimately inducing cell death. MGMT repairs this DNA damage by removing the aberrant methyl groups, impairing the efficacy of alkylator chemotherapy (82). The presence of MGMT promoter methylation and gene silencing is a predictive biomarker for improved outcomes on treatment with temozolomide in glioblastoma (83,84). Indeed, a retrospective study of MGMT expression by IHC in archival pancreatic and non-pancreatic NET samples showed that 51% of pancreatic NETs were deficient in MGMT, while 0% of the non-pancreatic NETs were deficient in MGMT. An independent cohort of patients treated with temozolomide-based chemotherapy indeed demonstrated superior response rate in pancreatic NET patients (34%) compared to non-pancreatic NET patients (2%). Twenty-one of these 101 temozolomide-treated patients had their tumors tested for MGMT expression, and 4/5 pancreatic NET patients with deficient MGMT had response, while 0/3 pancreatic NET patients with intact MGMT had response. Conversely, 13 patients with non-pancreatic NETs all had intact MGMT, and none had a response (85). Similarly, 12/99 pancreatic and non-pancreatic NET samples had MGMT promoter methylation when detected by methyl-specific PCR and 24/99 samples had promoter methylation when detected by pyrosequencing, while 29/89 (33%) of samples had MGMT loss by IHC. PFS since the start of alkylator chemotherapy, including temozolomide, dacarbazine, or streptozocin-based regimens, was superior in patients with MGMT protein loss or methylation by pyrosequencing, though was not associated with OS (86). Thus, the superior response rate of alkylating agents in pancreatic NETs may be accentuated by MGMT promoter methylation and protein deficiency.
Cytotoxic chemotherapy has been thought to have low response rates in non-pancreatic NETs. Streptozocin/5-FU and doxorubicin/5-FU regimens both had response rates of 16%, though streptozocin/5-FU had superior PFS (87). A retrospective study of capecitabine/temozolomide in non-pancreatic NETs showed 2/4 patients had response (88), and similarly a phase II prospective study showed 5/12 non-pancreatic NET patients had a response (89). Generally, however, cytotoxic chemotherapy is only considered for non-pancreatic NETs for patients without other therapeutic options, and its efficacy remains unproven (30).
PRRT
The use of 90Y- or 177Lu-radiolabeled octreotide analogs, though considered investigational in the United States, is standard in Europe and incorporated into European Society of Medical Oncology (ESMO) clinical practice guidelines (30,90). Several single-arm trials have been completed. A single-center single-arm study of 90Y-DOTATOC in 1,109 patients with positive octreoscan and progressive metastatic NET, showed 29.7% clinical response rate, including 23–38% for well-differentiated non-pancreatic NETs depending on primary site and 29% for nonfunctioning pancreatic NETs. Greater tumor radiopeptide uptake was significantly associated with superior overall survival. Kidney injury was dose-limiting, with 9.2% rate of grade 4–5 permanent nephrotoxicity (91). An independent single-arm study of 177Lu-DOTATATE was performed in 458 patients with metastatic well-differentiated GEP NETs and positive octreoscan, 310 of whom were evaluable. A total of 29% of patients had response, including 22% of patients with well-differentiated non-pancreatic GEP NETs, 42% of patients with non-functional pancreatic NETs, 5/12 patients with gastrinoma, and 3/5 patients with insulinoma. Median PFS was 33 months and median OS was 46 months (92). In a previously published subset of these patients, the degree of tumor uptake on octreoscan was significantly associated with the proportion of patients who had tumor shrinkage (28).
While the intensity of uptake on baseline octreoscan is associated with response rate, it remains unclear if uptake on 68Ga-DOTA-peptide PET imaging is similarly associated. About 27/32 patients with no uptake and 14/16 patients with only faint uptake seen on octreoscan had positive 68Ga-DOTATATE PET/CT, allowing 20 of the patients to proceed to PRRT (93). However, there are no well-validated cutoff criteria for PRRT using baseline 68Ga-DOTA-peptide PET scanning (94). The issue is further complicated because of the use of multiple SSA peptides for imaging, including 68Ga-DOTATATE, 68Ga-DOTATOC, and 68Ga-DOTANOC. Each of these agents has a different specificity among the 5 subtypes of somatostatin receptors: 68Ga-DOTANOC binds sst2,3,5, while 68Ga-DOTATATE and 68Ga-DOTATOC only bind sst2 (95,96).
To personalize therapy with PRRT, individualizing dosimetry to maximize effect while minimizing toxicity, particularly renal toxicity, is important. There is a significant association between severe nephrotoxicity and renal uptake on baseline somatostatin scintigraphy, especially with 90Y-DOTATOC (91). Personalizing dosing based on calculation of renal biological expected dose (BED) using 111In-pentetreotide with concurrent amino acid infusion could be performed, and if the renal BED after four cycles exceeded 37 Gy, subsequent cycles were dose-reduced or cancelled, thus preventing the development of long-term renal failure at 15 months after treatment with 90Y-DOTATOC (97). Indeed, dosimetry to various organs may dynamically evolve over the course of subsequent cycles of PRRT, with a case report of a patient having a marked increase in plasma half-life of 111In-octreotide after her first cycle of treatment with 90Y-DOTATOC resulting in an increase in BED to kidneys and bone marrow, indicating the importance of serial reassessment of dosimetry (98).
The multicenter, randomized phase III NETTER-1 trial compared treatment with 177Lu-DOTATATE combined with SSAs compared to octreotide LAR 60 mg in patients with unresectable, advanced, progressive well-differentiated midgut NETs with positive 111In-octreotide scintigraphy. The study enrolled 229 patients, with marked improvement in PFS [median estimated 40 months with 177Lu-DOTATATE vs. 8.4 months with high-dose octreotide, HR 0.21 (0.129–0.338) and overall response rate (18% vs. 3%, P=0.0008) (99). Subgroup analyses are pending, but this comprises the first randomized, controlled trial demonstrating the efficacy of PRRT. Notably, NETTER-1 did not include pancreatic or other foregut NETs, but as described above, previous single-arm studies showed efficacy of PRRT in both functional and nonfunctional pancreatic NETs, with even higher response rates than in small intestinal NETs (92). Hopefully, the results of NETTER-1 will make PRRT more accessible for North American patients with metastatic NETs.
Immunotherapy
Historically, interferon-α has been used in therapy of carcinoid tumors, since an early study of treatment with leukocyte interferon in 9 patients with small intestinal carcinoid tumors showed improvement in the symptoms of carcinoid syndrome and decreased in urinary 5-HIAA levels (100). However, the data is unclear on the true efficacy of interferon-α, as randomized studies have been small and likely underpowered. One study showed that combination octreotide and interferon-α decreased risk of progression compared to octreotide alone (HR 0.28, 95% CI: 0.16–0.45) (101), but other studies found that adding interferon-α to SSA did not improve 12-month disease stability rate (102) or time to treatment failure (103). Based on these studies, some authors argue that there is at least a trend toward improved survival with combination interferon-α and SSA compared to SSA alone (104). At present, consensus guidelines state interferon alfa-2b may be considered for unresectable or metastatic GI tract, lung, or thymus NETs (30).
With the discovery of the efficacy of immune checkpoint inhibitors, such as CTLA-4 inhibitors, PD-1 inhibitors, and PDL-1 inhibitors in other malignancies, there has been consideration of investigating these therapies in NETs. Retrospective analysis of a series of archival well-differentiated pancreatic NETs found that 68% of NETs had T-cell infiltration, while 34% of NET samples had infiltrating FoxP3+ regulatory T cells (Tregs). Among the subset of intermediate grade NET samples, the extent of T cell infiltrate was significantly associated with recurrence. Within the subset of NET samples obtained from hepatic metastasectomy, the degree of FoxP3+ T cell infiltration was inversely associated with overall survival (105). Additionally, studies have found varying rates of microsatellite instability (MSI-high) in well-differentiated NETs, with 0–10% of pancreatic NETs (106,107) and 33% of insulinomas MSI-high, usually driven by MLH1 promoter methylation and loss of heterozygosity (108). Immune checkpoint inhibitors appear effective in MSI-high tumors, as the PD-1 inhibitor pembrolizumab yielded 50% response rate in MSI-high metastatic non-colorectal cancers (109,110). Given that MLH1 hypermethylation was one of the most commonly hypermethylated genes in pancreatic NETs (37), further study of immune checkpoint inhibitors in metastatic NETs, especially pancreatic NETs, is rational. Notably, essentially 0% of small bowel NETs were MSI-high (106,111). Nevertheless, the phase I study of pembrolizumab included a patient with carcinoid tumor with 19 weeks of stable disease and a patient with pancreatic NET who had 11 weeks of stable disease (112). Studies using immune checkpoint inhibitors in pancreatic and non-pancreatic NET patients are planned, including pembrolizumab in well and moderately-differentiated pancreatic, small intestinal, appendiceal, and colorectal NETs (NCT02628067).
Conclusions
Our understanding of the genomic and epigenetic underpinnings of pancreatic and extrapancreatic NETs has grown significantly in the last decade. The positive phase III clinical trials for everolimus and sunitinib in metastatic pancreatic NETs confirmed the biologically driven hypothesis that targeting mTOR signaling and angiogenic pathways results in clinically meaningful tumor growth suppression. With the positive RADIANT-4 and NETTER-1 trials, our therapy options against extrapancreatic NETs also continue to grow. While we can personalize therapy of metastatic GEP NETs by clinical characteristics, by biochemical response, and by somatostatin based imaging, there is a critical need for improved biomarkers. A recent National Cancer Institute summit concluded that monoanalyte biomarkers have generally been lacking in sufficient prognostic or predictive value, and instead multianalyte biomarkers, particularly incorporating genomics, should be further developed (113). We need to prospectively assess the predictive role that underlying genomic mutations, especially in pancreatic NETs, has in guiding response to targeted therapies. The potential of achieving durable disease control and response with immunotherapy is also exciting, and development of clinical trials to assess the efficacy of immune checkpoint therapy is ongoing.
Acknowledgements
This work was supported by the Conquer Cancer Foundation Young Investigator Award to MS Lee.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Yao JC, Hassan M, Phan A, et al. One hundred years after "carcinoid": epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol 2008;26:3063-72. 10.1200/JCO.2007.15.4377 [DOI] [PubMed] [Google Scholar]
- 2.Bosman FT, World Health Organization. International Agency for Research on Cancer. WHO classification of tumours of the digestive system. 4th ed. Lyon: International Agency for Research on Cancer, 2010:417. [Google Scholar]
- 3.Rinke A, Wittenberg M, Schade-Brittinger C, et al. Placebo Controlled, Double Blind, Prospective, Randomized Study on the Effect of Octreotide LAR in the Control of Tumor Growth in Patients with Metastatic Neuroendocrine Midgut Tumors (PROMID): Results on Long Term Survival. Neuroendocrinology 2016. [Epub ahead of print]. 10.1159/000443612 [DOI] [PubMed] [Google Scholar]
- 4.Strosberg J, Nasir A, Coppola D, et al. Correlation between grade and prognosis in metastatic gastroenteropancreatic neuroendocrine tumors. Hum Pathol 2009;40:1262-8. 10.1016/j.humpath.2009.01.010 [DOI] [PubMed] [Google Scholar]
- 5.Khan MS, Luong TV, Watkins J, et al. A comparison of Ki-67 and mitotic count as prognostic markers for metastatic pancreatic and midgut neuroendocrine neoplasms. Br J Cancer 2013;108:1838-45. 10.1038/bjc.2013.156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rinke A, Muller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol 2009;27:4656-63. 10.1200/JCO.2009.22.8510 [DOI] [PubMed] [Google Scholar]
- 7.Williams ED, Sandler M. The classification of carcinoid tum ours. Lancet 1963;1:238-9. 10.1016/S0140-6736(63)90951-6 [DOI] [PubMed] [Google Scholar]
- 8.Halfdanarson TR, Rabe KG, Rubin J, et al. Pancreatic neuroendocrine tumors (PNETs): incidence, prognosis and recent trend toward improved survival. Ann Oncol 2008;19:1727-33. 10.1093/annonc/mdn351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med 2011;364:514-23. 10.1056/NEJMoa1009290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yao JC, Fazio N, Singh S, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet 2016;387:968-77. 10.1016/S0140-6736(15)00817-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raymond E, Dahan L, Raoul JL, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med 2011;364:501-13. 10.1056/NEJMoa1003825 [DOI] [PubMed] [Google Scholar]
- 12.Feldman JM. Carcinoid tumors and the carcinoid syndrome. Curr Probl Surg 1989;26:835-85. 10.1016/0011-3840(89)90010-5 [DOI] [PubMed] [Google Scholar]
- 13.Kvols LK, Moertel CG, O'Connell MJ, et al. Treatment of the malignant carcinoid syndrome. Evaluation of a long-acting somatostatin analogue. N Engl J Med 1986;315:663-6. 10.1056/NEJM198609113151102 [DOI] [PubMed] [Google Scholar]
- 14.Nikou GC, Toubanakis C, Nikolaou P, et al. VIPomas: an update in diagnosis and management in a series of 11 patients. Hepatogastroenterology 2005;52:1259-65. [PubMed] [Google Scholar]
- 15.Frankton S, Bloom SR. Gastrointestinal endocrine tumours. Glucagonomas. Baillieres Clin Gastroenterol 1996;10:697-705. 10.1016/S0950-3528(96)90019-6 [DOI] [PubMed] [Google Scholar]
- 16.Angeletti S, Corleto VD, Schillaci O, et al. Use of the somatostatin analogue octreotide to localise and manage somatostatin-producing tumours. Gut 1998;42:792-4. 10.1136/gut.42.6.792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stehouwer CD, Lems WF, Fischer HR, et al. Aggravation of hypoglycemia in insulinoma patients by the long-acting somatostatin analogue octreotide (Sandostatin). Acta Endocrinol (Copenh) 1989;121:34-40. [DOI] [PubMed] [Google Scholar]
- 18.Gill GV, Rauf O, MacFarlane IA. Diazoxide treatment for insulinoma: a national UK survey. Postgrad Med J 1997;73:640-1. 10.1136/pgmj.73.864.640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kulke MH, O'Dorisio T, Phan A, et al. Telotristat etiprate, a novel serotonin synthesis inhibitor, in patients with carcinoid syndrome and diarrhea not adequately controlled by octreotide. Endocr Relat Cancer 2014;21:705-14. 10.1530/ERC-14-0173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pavel M, Horsch D, Caplin M, et al. Telotristat etiprate for carcinoid syndrome: a single-arm, multicenter trial. J Clin Endocrinol Metab 2015;100:1511-9. 10.1210/jc.2014-2247 [DOI] [PubMed] [Google Scholar]
- 21.Kulke MH, Hörsch D, Caplin M, et al. Telotristat Etiprate Shows Benefit in Treating Patients With Carcinoid Syndrome That is Inadequately Controlled by Somatostatin Analog Therapy in the Phase 3 TELESTAR Clinical Trial. 2015 European Cancer Congress. Abstract 37LBA. Presented September 29, 2015. [Google Scholar]
- 22.Jamar F, Fiasse R, Leners N, et al. Somatostatin receptor imaging with indium-111-pentetreotide in gastroenteropancreatic neuroendocrine tumors: safety, efficacy and impact on patient management. J Nucl Med 1995;36:542-9. [PubMed] [Google Scholar]
- 23.Koopmans KP, Neels ON, Kema IP, et al. Molecular imaging in neuroendocrine tumors: molecular uptake mechanisms and clinical results. Crit Rev Oncol Hematol 2009;71:199-213. 10.1016/j.critrevonc.2009.02.009 [DOI] [PubMed] [Google Scholar]
- 24.Scherübl H, Bäder M, Fett U, et al. Somatostatin-receptor imaging of neuroendocrine gastroenteropancreatic tumors. Gastroenterology 1993;105:1705-9. 10.1016/0016-5085(93)91066-Q [DOI] [PubMed] [Google Scholar]
- 25.Olsen JO, Pozderac RV, Hinkle G, et al. Somatostatin receptor imaging of neuroendocrine tumors with indium-111 pentetreotide (Octreoscan). Semin Nucl Med 1995;25:251-61. 10.1016/S0001-2998(95)80014-X [DOI] [PubMed] [Google Scholar]
- 26.Sadowski SM, Neychev V, Millo C, et al. Prospective Study of 68Ga-DOTATATE Positron Emission Tomography/Computed Tomography for Detecting Gastro-Entero-Pancreatic Neuroendocrine Tumors and Unknown Primary Sites. J Clin Oncol 2016;34:588-96. 10.1200/JCO.2015.64.0987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bodei L, Mueller-Brand J, Baum RP, et al. The joint IAEA, EANM, and SNMMI practical guidance on peptide receptor radionuclide therapy (PRRNT) in neuroendocrine tumours. Eur J Nucl Med Mol Imaging 2013;40:800-16. 10.1007/s00259-012-2330-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kwekkeboom DJ, Teunissen JJ, Bakker WH, et al. Radiolabeled somatostatin analog [177Lu-DOTA0,Tyr3]octreotate in patients with endocrine gastroenteropancreatic tumors. J Clin Oncol 2005;23:2754-62. 10.1200/JCO.2005.08.066 [DOI] [PubMed] [Google Scholar]
- 29.Matkar S, Thiel A, Hua X. Menin: a scaffold protein that controls gene expression and cell signaling. Trends Biochem Sci 2013;38:394-402. 10.1016/j.tibs.2013.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kulke MH, Shah MH, Benson AB, 3rd, et al. Neuroendocrine tumors, version 1.2015. J Natl Compr Canc Netw 2015;13:78-108. [DOI] [PubMed] [Google Scholar]
- 31.Thakker RV, Newey PJ, Walls GV, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab 2012;97:2990-3011. 10.1210/jc.2012-1230 [DOI] [PubMed] [Google Scholar]
- 32.Strosberg JR, Nasir A, Hodul P, et al. Biology and treatment of metastatic gastrointestinal neuroendocrine tumors. Gastrointest Cancer Res 2008;2:113-25. [PMC free article] [PubMed] [Google Scholar]
- 33.Rosai J. The origin of neuroendocrine tumors and the neural crest saga. Mod Pathol 2011;24 Suppl 2:S53-7. 10.1038/modpathol.2010.166 [DOI] [PubMed] [Google Scholar]
- 34.Weckbecker G, Lewis I, Albert R, et al. Opportunities in somatostatin research: biological, chemical and therapeutic aspects. Nat Rev Drug Discov 2003;2:999-1017. 10.1038/nrd1255 [DOI] [PubMed] [Google Scholar]
- 35.Hofland LJ, Lamberts SW. Somatostatin receptor subtype expression in human tumors. Ann Oncol 2001;12 Suppl 2:S31-6. 10.1093/annonc/12.suppl_2.S31 [DOI] [PubMed] [Google Scholar]
- 36.Missiaglia E, Dalai I, Barbi S, et al. Pancreatic endocrine tumors: expression profiling evidences a role for AKT-mTOR pathway. J Clin Oncol 2010;28:245-55. 10.1200/JCO.2008.21.5988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.House MG, Herman JG, Guo MZ, et al. Aberrant hypermethylation of tumor suppressor genes in pancreatic endocrine neoplasms. Ann Surg 2003;238:423-31; discussion 31-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiao Y, Shi C, Edil BH, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011;331:1199-203. 10.1126/science.1200609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Wilde RF, Heaphy CM, Maitra A, et al. Loss of ATRX or DAXX expression and concomitant acquisition of the alternative lengthening of telomeres phenotype are late events in a small subset of MEN-1 syndrome pancreatic neuroendocrine tumors. Mod Pathol 2012;25:1033-9. 10.1038/modpathol.2012.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marinoni I, Kurrer AS, Vassella E, et al. Loss of DAXX and ATRX are associated with chromosome instability and reduced survival of patients with pancreatic neuroendocrine tumors. Gastroenterology 2014;146:453-60.e5. 10.1053/j.gastro.2013.10.020 [DOI] [PubMed] [Google Scholar]
- 41.Banck MS, Kanwar R, Kulkarni AA, et al. The genomic landscape of small intestine neuroendocrine tumors. J Clin Invest 2013;123:2502-8. 10.1172/JCI67963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Francis JM, Kiezun A, Ramos AH, et al. Somatic mutation of CDKN1B in small intestine neuroendocrine tumors. Nat Genet 2013;45:1483-6. 10.1038/ng.2821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karpathakis A, Dibra H, Pipinikas C, et al. Prognostic Impact of Novel Molecular Subtypes of Small Intestinal Neuroendocrine Tumor. Clin Cancer Res 2016;22:250-8. 10.1158/1078-0432.CCR-15-0373 [DOI] [PubMed] [Google Scholar]
- 44.Qian ZR, Ter-Minassian M, Chan JA, et al. Prognostic significance of MTOR pathway component expression in neuroendocrine tumors. J Clin Oncol 2013;31:3418-25. 10.1200/JCO.2012.46.6946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Caplin ME, Pavel M, Ruszniewski P. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med 2014;371:1556-7. 10.1056/NEJMoa1316158 [DOI] [PubMed] [Google Scholar]
- 46.Yao JC, Phan AT, Chang DZ, et al. Efficacy of RAD001 (everolimus) and octreotide LAR in advanced low- to intermediate-grade neuroendocrine tumors: results of a phase II study. J Clin Oncol 2008;26:4311-8. 10.1200/JCO.2008.16.7858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yao JC, Lombard-Bohas C, Baudin E, et al. Daily oral everolimus activity in patients with metastatic pancreatic neuroendocrine tumors after failure of cytotoxic chemotherapy: a phase II trial. J Clin Oncol 2010;28:69-76. 10.1200/JCO.2009.24.2669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lombard-Bohas C, Yao JC, Hobday T, et al. Impact of prior chemotherapy use on the efficacy of everolimus in patients with advanced pancreatic neuroendocrine tumors: a subgroup analysis of the phase III RADIANT-3 trial. Pancreas 2015;44:181-9. 10.1097/MPA.0000000000000262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yao JC, Pavel M, Phan AT, et al. Chromogranin A and neuron-specific enolase as prognostic markers in patients with advanced pNET treated with everolimus. J Clin Endocrinol Metab 2011;96:3741-9. 10.1210/jc.2011-0666 [DOI] [PubMed] [Google Scholar]
- 50.Meric-Bernstam F, Akcakanat A, Chen H, et al. PIK3CA/PTEN mutations and Akt activation as markers of sensitivity to allosteric mTOR inhibitors. Clin Cancer Res 2012;18:1777-89. 10.1158/1078-0432.CCR-11-2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rodrik-Outmezguine VS, Chandarlapaty S, Pagano NC, et al. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov 2011;1:248-59. 10.1158/2159-8290.CD-11-0085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yao JC, Phan AT, Jehl V, et al. Everolimus in advanced pancreatic neuroendocrine tumors: the clinical experience. Cancer Res 2013;73:1449-53. 10.1158/0008-5472.CAN-12-3923 [DOI] [PubMed] [Google Scholar]
- 53.Pavel ME, Hainsworth JD, Baudin E, et al. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): a randomised, placebo-controlled, phase 3 study. Lancet 2011;378:2005-12. 10.1016/S0140-6736(11)61742-X [DOI] [PubMed] [Google Scholar]
- 54.Anthony LB, Pavel ME, Hainsworth JD, et al. Impact of Previous Somatostatin Analogue Use on the Activity of Everolimus in Patients with Advanced Neuroendocrine Tumors: Analysis from the Phase III RADIANT-2 Trial. Neuroendocrinology 2015;102:18-25. 10.1159/000381715 [DOI] [PubMed] [Google Scholar]
- 55.Castellano D, Bajetta E, Panneerselvam A, et al. Everolimus plus octreotide long-acting repeatable in patients with colorectal neuroendocrine tumors: a subgroup analysis of the phase III RADIANT-2 study. Oncologist 2013;18:46-53. 10.1634/theoncologist.2012-0263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fazio N, Granberg D, Grossman A, et al. Everolimus plus octreotide long-acting repeatable in patients with advanced lung neuroendocrine tumors: analysis of the phase 3, randomized, placebo-controlled RADIANT-2 study. Chest 2013;143:955-62. 10.1378/chest.12-1108 [DOI] [PubMed] [Google Scholar]
- 57.Serra S, Zheng L, Hassan M, et al. The FGFR4-G388R single-nucleotide polymorphism alters pancreatic neuroendocrine tumor progression and response to mTOR inhibition therapy. Cancer Res 2012;72:5683-91. 10.1158/0008-5472.CAN-12-2102 [DOI] [PubMed] [Google Scholar]
- 58.Cros J, Moati E, Raffenne J, et al. Gly388Arg FGFR4 Polymorphism is not Predictive of Everolimus Efficacy in Well-Differentiated Digestive Neuroendocrine Tumors. Neuroendocrinology 2016;103:495-9. 10.1159/000440724 [DOI] [PubMed] [Google Scholar]
- 59.Turner HE, Harris AL, Melmed S, et al. Angiogenesis in endocrine tumors. Endocr Rev 2003;24:600-32. 10.1210/er.2002-0008 [DOI] [PubMed] [Google Scholar]
- 60.Ahn HK, Choi JY, Kim KM, et al. Phase II study of pazopanib monotherapy in metastatic gastroenteropancreatic neuroendocrine tumours. Br J Cancer 2013;109:1414-9. 10.1038/bjc.2013.470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grande E, Capdevila J, Castellano D, et al. Pazopanib in pretreated advanced neuroendocrine tumors: a phase II, open-label trial of the Spanish Task Force Group for Neuroendocrine Tumors (GETNE). Ann Oncol 2015;26:1987-93. 10.1093/annonc/mdv252 [DOI] [PubMed] [Google Scholar]
- 62.Phan AT, Halperin DM, Chan JA, et al. Pazopanib and depot octreotide in advanced, well-differentiated neuroendocrine tumours: a multicentre, single-group, phase 2 study. Lancet Oncol 2015;16:695-703. 10.1016/S1470-2045(15)70136-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kulke MH, Lenz HJ, Meropol NJ, et al. Activity of sunitinib in patients with advanced neuroendocrine tumors. J Clin Oncol 2008;26:3403-10. 10.1200/JCO.2007.15.9020 [DOI] [PubMed] [Google Scholar]
- 64.Zurita AJ, Khajavi M, Wu HK, et al. Circulating cytokines and monocyte subpopulations as biomarkers of outcome and biological activity in sunitinib-treated patients with advanced neuroendocrine tumours. Br J Cancer 2015;112:1199-205. 10.1038/bjc.2015.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kryczek I, Wei S, Keller E, et al. Stroma-derived factor (SDF-1/CXCL12) and human tumor pathogenesis. Am J Physiol Cell Physiol 2007;292:C987-95. 10.1152/ajpcell.00406.2006 [DOI] [PubMed] [Google Scholar]
- 66.Kulke MH, Bergsland EK, Yao JC. Glycemic control in patients with insulinoma treated with everolimus. N Engl J Med 2009;360:195-7. 10.1056/NEJMc0806740 [DOI] [PubMed] [Google Scholar]
- 67.Bernard V, Lombard-Bohas C, Taquet MC, et al. Efficacy of everolimus in patients with metastatic insulinoma and refractory hypoglycemia. Eur J Endocrinol 2013;168:665-74. 10.1530/EJE-12-1101 [DOI] [PubMed] [Google Scholar]
- 68.Fiebrich HB, Siemerink EJ, Brouwers AH, et al. Everolimus induces rapid plasma glucose normalization in insulinoma patients by effects on tumor as well as normal tissues. Oncologist 2011;16:783-7. 10.1634/theoncologist.2010-0222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Neychev V, Steinberg SM, Cottle-Delisle C, et al. Mutation-targeted therapy with sunitinib or everolimus in patients with advanced low-grade or intermediate-grade neuroendocrine tumours of the gastrointestinal tract and pancreas with or without cytoreductive surgery: protocol for a phase II clinical trial. BMJ Open 2015;5:e008248. 10.1136/bmjopen-2015-008248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Falconi M, Eriksson B, Kaltsas G, et al. ENETS Consensus Guidelines Update for the Management of Patients with Functional Pancreatic Neuroendocrine Tumors and Non-Functional Pancreatic Neuroendocrine Tumors. Neuroendocrinology 2016;103:153-71. 10.1159/000443171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kunz PL, Reidy-Lagunes D, Anthony LB, et al. Consensus guidelines for the management and treatment of neuroendocrine tumors. Pancreas 2013;42:557-77. 10.1097/MPA.0b013e31828e34a4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schnedl WJ, Ferber S, Johnson JH, et al. STZ transport and cytotoxicity. Specific enhancement in GLUT2-expressing cells. Diabetes 1994;43:1326-33. 10.2337/diab.43.11.1326 [DOI] [PubMed] [Google Scholar]
- 73.Tomita T. Immunocytochemical Localization of Glucose Transporter-2 (GLUT-2) in Pancreatic Islets and Islet Cell Tumors. Endocr Pathol 1999;10:213-21. 10.1007/BF02738882 [DOI] [PubMed] [Google Scholar]
- 74.Moertel CG, Lefkopoulo M, Lipsitz S, et al. Streptozocin-doxorubicin, streptozocin-fluorouracil or chlorozotocin in the treatment of advanced islet-cell carcinoma. N Engl J Med 1992;326:519-23. 10.1056/NEJM199202203260804 [DOI] [PubMed] [Google Scholar]
- 75.Moertel CG, Hanley JA, Johnson LA. Streptozocin alone compared with streptozocin plus fluorouracil in the treatment of advanced islet-cell carcinoma. N Engl J Med 1980;303:1189-94. 10.1056/NEJM198011203032101 [DOI] [PubMed] [Google Scholar]
- 76.Kouvaraki MA, Ajani JA, Hoff P, et al. Fluorouracil, doxorubicin, and streptozocin in the treatment of patients with locally advanced and metastatic pancreatic endocrine carcinomas. J Clin Oncol 2004;22:4762-71. 10.1200/JCO.2004.04.024 [DOI] [PubMed] [Google Scholar]
- 77.Dilz LM, Denecke T, Steffen IG, et al. Streptozocin/5-fluorouracil chemotherapy is associated with durable response in patients with advanced pancreatic neuroendocrine tumours. Eur J Cancer 2015;51:1253-62. 10.1016/j.ejca.2015.04.005 [DOI] [PubMed] [Google Scholar]
- 78.Delaunoit T, Ducreux M, Boige V, et al. The doxorubicin-streptozotocin combination for the treatment of advanced well-differentiated pancreatic endocrine carcinoma; a judicious option? Eur J Cancer 2004;40:515-20. 10.1016/j.ejca.2003.09.035 [DOI] [PubMed] [Google Scholar]
- 79.Strosberg JR, Fine RL, Choi J, et al. First-line chemotherapy with capecitabine and temozolomide in patients with metastatic pancreatic endocrine carcinomas. Cancer 2011;117:268-75. 10.1002/cncr.25425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Krug S, Boch M, Daniel H, et al. Streptozocin-Based Chemotherapy in Patients with Advanced Neuroendocrine Neoplasms--Predictive and Prognostic Markers for Treatment Stratification. PLoS One 2015;10:e0143822. 10.1371/journal.pone.0143822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005;352:997-1003. 10.1056/NEJMoa043331 [DOI] [PubMed] [Google Scholar]
- 82.Liu L, Gerson SL. Targeted modulation of MGMT: clinical implications. Clin Cancer Res 2006;12:328-31. 10.1158/1078-0432.CCR-05-2543 [DOI] [PubMed] [Google Scholar]
- 83.Esteller M, Garcia-Foncillas J, Andion E, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med 2000;343:1350-4. 10.1056/NEJM200011093431901 [DOI] [PubMed] [Google Scholar]
- 84.Hegi ME, Diserens AC, Godard S, et al. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin Cancer Res 2004;10:1871-4. 10.1158/1078-0432.CCR-03-0384 [DOI] [PubMed] [Google Scholar]
- 85.Kulke MH, Hornick JL, Frauenhoffer C, et al. O6-methylguanine DNA methyltransferase deficiency and response to temozolomide-based therapy in patients with neuroendocrine tumors. Clin Cancer Res 2009;15:338-45. 10.1158/1078-0432.CCR-08-1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Walter T, van Brakel B, Vercherat C, et al. O6-Methylguanine-DNA methyltransferase status in neuroendocrine tumours: prognostic relevance and association with response to alkylating agents. Br J Cancer 2015;112:523-31. 10.1038/bjc.2014.660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sun W, Lipsitz S, Catalano P, et al. Phase II/III study of doxorubicin with fluorouracil compared with streptozocin with fluorouracil or dacarbazine in the treatment of advanced carcinoid tumors: Eastern Cooperative Oncology Group Study E1281. J Clin Oncol 2005;23:4897-904. 10.1200/JCO.2005.03.616 [DOI] [PubMed] [Google Scholar]
- 88.Fine RL, Gulati AP, Krantz BA, et al. Capecitabine and temozolomide (CAPTEM) for metastatic, well-differentiated neuroendocrine cancers: The Pancreas Center at Columbia University experience. Cancer Chemother Pharmacol 2013;71:663-70. 10.1007/s00280-012-2055-z [DOI] [PubMed] [Google Scholar]
- 89.Fine RL, Gulati AP, Tsushima D, et al. Prospective phase II study of capecitabine and temozolomide (CAPTEM) for progressive, moderately, and well-differentiated metastatic neuroendocrine tumors. J Clin Oncol 2014;32:abstr 179.
- 90.Öberg K, Knigge U, Kwekkeboom D, et al. Neuroendocrine gastro-entero-pancreatic tumors: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2012;23 Suppl 7:vii124-30. [DOI] [PubMed] [Google Scholar]
- 91.Imhof A, Brunner P, Marincek N, et al. Response, survival, and long-term toxicity after therapy with the radiolabeled somatostatin analogue [90Y-DOTA]-TOC in metastasized neuroendocrine cancers. J Clin Oncol 2011;29:2416-23. 10.1200/JCO.2010.33.7873 [DOI] [PubMed] [Google Scholar]
- 92.Kwekkeboom DJ, de Herder WW, Kam BL, et al. Treatment with the radiolabeled somatostatin analog [177 Lu-DOTA 0,Tyr3]octreotate: toxicity, efficacy, and survival. J Clin Oncol 2008;26:2124-30. 10.1200/JCO.2007.15.2553 [DOI] [PubMed] [Google Scholar]
- 93.Srirajaskanthan R, Kayani I, Quigley AM, et al. The role of 68Ga-DOTATATE PET in patients with neuroendocrine tumors and negative or equivocal findings on 111In-DTPA-octreotide scintigraphy. J Nucl Med 2010;51:875-82. 10.2967/jnumed.109.066134 [DOI] [PubMed] [Google Scholar]
- 94.van Essen M, Sundin A, Krenning EP, et al. Neuroendocrine tumours: the role of imaging for diagnosis and therapy. Nat Rev Endocrinol 2014;10:102-14. 10.1038/nrendo.2013.246 [DOI] [PubMed] [Google Scholar]
- 95.Poeppel TD, Binse I, Petersenn S, et al. 68Ga-DOTATOC versus 68Ga-DOTATATE PET/CT in functional imaging of neuroendocrine tumors. J Nucl Med 2011;52:1864-70. 10.2967/jnumed.111.091165 [DOI] [PubMed] [Google Scholar]
- 96.Wild D, Bomanji JB, Benkert P, et al. Comparison of 68Ga-DOTANOC and 68Ga-DOTATATE PET/CT within patients with gastroenteropancreatic neuroendocrine tumors. J Nucl Med 2013;54:364-72. 10.2967/jnumed.112.111724 [DOI] [PubMed] [Google Scholar]
- 97.Van Binnebeek S, Baete K, Vanbilloen B, et al. Individualized dosimetry-based activity reduction of (0)Y-DOTATOC prevents severe and rapid kidney function deterioration from peptide receptor radionuclide therapy. Eur J Nucl Med Mol Imaging 2014;41:1141-57. [DOI] [PubMed] [Google Scholar]
- 98.Van Binnebeek S, Deroose CM, Baete K, et al. Altered biodistribution of somatostatin analogues after first cycle of Peptide receptor radionuclide therapy. J Clin Oncol 2011;29:e579-81. 10.1200/JCO.2010.34.3384 [DOI] [PubMed] [Google Scholar]
- 99.Strosberg JR, Wolin EM, Chasen B, et al. NETTER-1 phase III: Progression-free survival, radiographic response, and preliminary overall survival results in patients with midgut neuroendocrine tumors treated with 177-Lu-Dotatate. J Clin Oncol 2016;34:abstr 194.
- 100.Oberg K, Funa K, Alm G. Effects of leukocyte interferon on clinical symptoms and hormone levels in patients with mid-gut carcinoid tumors and carcinoid syndrome. N Engl J Med 1983;309:129-33. 10.1056/NEJM198307213090301 [DOI] [PubMed] [Google Scholar]
- 101.Kölby L, Persson G, Franzén S, et al. Randomized clinical trial of the effect of interferon alpha on survival in patients with disseminated midgut carcinoid tumours. Br J Surg 2003;90:687-93. 10.1002/bjs.4149 [DOI] [PubMed] [Google Scholar]
- 102.Faiss S, Pape UF, Bohmig M, et al. Prospective, randomized, multicenter trial on the antiproliferative effect of lanreotide, interferon alfa, and their combination for therapy of metastatic neuroendocrine gastroenteropancreatic tumors--the International Lanreotide and Interferon Alfa Study Group. J Clin Oncol 2003;21:2689-96. 10.1200/JCO.2003.12.142 [DOI] [PubMed] [Google Scholar]
- 103.Arnold R, Rinke A, Klose KJ, et al. Octreotide versus octreotide plus interferon-alpha in endocrine gastroenteropancreatic tumors: a randomized trial. Clin Gastroenterol Hepatol 2005;3:761-71. 10.1016/S1542-3565(05)00481-7 [DOI] [PubMed] [Google Scholar]
- 104.Fazio N, de Braud F, Delle Fave G, et al. Interferon-alpha and somatostatin analog in patients with gastroenteropancreatic neuroendocrine carcinoma: single agent or combination? Ann Oncol 2007;18:13-9. 10.1093/annonc/mdl144 [DOI] [PubMed] [Google Scholar]
- 105.Katz SC, Donkor C, Glasgow K, et al. T cell infiltrate and outcome following resection of intermediate-grade primary neuroendocrine tumours and liver metastases. HPB (Oxford) 2010;12:674-83. 10.1111/j.1477-2574.2010.00231.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Arnason T, Sapp HL, Rayson D, et al. Loss of expression of DNA mismatch repair proteins is rare in pancreatic and small intestinal neuroendocrine tumors. Arch Pathol Lab Med 2011;135:1539-44. 10.5858/arpa.2010-0560-OA [DOI] [PubMed] [Google Scholar]
- 107.House MG, Herman JG, Guo MZ, et al. Prognostic value of hMLH1 methylation and microsatellite instability in pancreatic endocrine neoplasms. Surgery 2003;134:902-8; discussion 9. 10.1016/S0039-6060(03)00412-4 [DOI] [PubMed] [Google Scholar]
- 108.Mei M, Deng D, Liu TH, et al. Clinical implications of microsatellite instability and MLH1 gene inactivation in sporadic insulinomas. J Clin Endocrinol Metab 2009;94:3448-57. 10.1210/jc.2009-0173 [DOI] [PubMed] [Google Scholar]
- 109.Le DT, Uram JN, Wang H, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 2015;372:2509-20. 10.1056/NEJMoa1500596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Le DT, Uram JN, Wang H, et al. PD-1 blockade in mismatch repair deficient non-colorectal gastrointestinal cancers. J Clin Oncol 2016;34:abstr 195.
- 111.Kidd M, Eick G, Shapiro MD, et al. Microsatellite instability and gene mutations in transforming growth factor-beta type II receptor are absent in small bowel carcinoid tumors. Cancer 2005;103:229-36. 10.1002/cncr.20750 [DOI] [PubMed] [Google Scholar]
- 112.Patnaik A, Kang SP, Rasco D, et al. Phase I Study of Pembrolizumab (MK-3475; Anti-PD-1 Monoclonal Antibody) in Patients with Advanced Solid Tumors. Clin Cancer Res 2015;21:4286-93. 10.1158/1078-0432.CCR-14-2607 [DOI] [PubMed] [Google Scholar]
- 113.Oberg K, Modlin IM, De Herder W, et al. Consensus on biomarkers for neuroendocrine tumour disease. Lancet Oncol 2015;16:e435-46. 10.1016/S1470-2045(15)00186-2 [DOI] [PMC free article] [PubMed] [Google Scholar]