

Abstract
Introduction
Charcot–Marie–Tooth disease (CMT) is a heterogeneous inherited neuropathy. The number of known CMT genes is rapidly increasing mainly due to next‐generation sequencing technology, at present more than 70 CMT‐associated genes are known. We investigated whether variants in the DCTN2 could cause CMT.
Material and methods
Fifty‐nine Norwegian CMT families from the general population with unknown genotype were tested by targeted next‐generation sequencing (NGS) for variants in DCTN2 along with 32 CMT genes and 19 other genes causing other inherited neuropathies or neuronopathies, due to phenotypic overlap. In the family with the DCTN2 variant, exome sequencing was then carried out on all available eight family members to rule out the presence of more potential variants.
Results
Targeted NGS identified in one family a variant of DCTN2, c.337C>T, segregating with the phenotype in five affected members, while it was not present in the three unaffected members. The DCTN2 variant c.337C>T; p.(His113Tyr) was neither found in in‐house controls nor in SNP databases. Exome sequencing revealed a singular heterozygous shared haplotype containing four genes, DCTN2,DNAH10,LRIG3, and MYO1A, with novel sequence variants. The haplotype was shared by all the affected members, while the unaffected members did not have it.
Conclusions
This is the first time a haplotype on chromosome 12 containing sequence variants in the genes DCTN2,DNAH10,LRIG3, and MYO1A has been linked to an inherited neuropathy in humans.
Keywords: Charcot–Marie–Tooth disease, inherited neuropathy, DCTN2, targeted next‐generation sequencing, exome sequencing
Introduction
Charcot–Marie–Tooth disease (CMT) is the most common inherited neuropathy with a prevalence ratio of 15–82/100,000 in different European settings 1, 2, 3, 4. CMT is genetically heterogeneous (The Mutation Database of Inherited Peripheral Neuropathies (IPNMDB) (http://www.molgen.ua.ac.be/cmtmutations/) and Neuromuscular Disease Center (http://neuromuscular.wustl.edu/)). At present, variants in more than 70 different genes can cause CMT, and the number is rapidly increasing mainly due to next‐generation sequencing technology 5, 6.
The overexpression of DCTN2 in transgenic mice disrupts the dynein (DYNC1H1)–dynactin (DCTN1) complex and results in a late‐onset slowly progressive motor neuron degenerative disease 7. Although not yet implicated in CMT, it was hypothesized that the gene DCTN2 might cause CMT.
Despite the high number of currently known CMT genes, there are indications that many genes are yet to be identified; for example, in a Norwegian study investigating 51 neuropathy genes, only 46% of the families received a molecular diagnosis 1, 8.
We tested 59 Norwegian CMT families without identified gene mutations for the presence of DCTN2 mutations. Then, we did exome sequencing of the identified DCTN2 family looking for other shared sequence variants that may explain the phenotype.
Material and methods
Study population
We analyzed 59 Norwegian CMT families from eastern Akershus County, irrespective of their neurophysiological phenotype with targeted next‐generation sequencing (NGS). These families had previously tested negative for PMP22 duplication by real‐time quantitative PCR followed by sequential point mutation analysis of the genes PMP22, GJB1, MPZ, LITAF, MFN2, and EGR2 by conventional Sanger sequencing 1. Detailed description of the study population, clinical interview, genetic and neurologic examination, neurophysiology, and genetic laboratory tests has been published elsewhere 1, 8. In this study, the 59 families (22 CMT1 families, 29 CMT2 families, one intermediate CMT family, and seven families with unknown neurophysiological phenotype) were investigated for mutations in DCTN2. A control group of 180 healthy individuals were included to help differentiate between normal and pathogenic variation 9.
Neuropathy impairment score
Cranial nerves, muscle weakness, reflexes, and sensation were scored according to the Neuropathy Impairment Score (NIS) 10. Muscle strength is scored linearly from 0 to 4; normal strength is scored 0; paralysis is scored 4; and 25% muscle weakness, 50% muscle weakness, and 75% muscle weakness are scored 1, 2, and 3. Movement against gravity, movement with gravity eliminated, and muscle flicker without movement are scored 3.25, 3.5, and 3.75, respectively.
Targeted capture and DNA sequencing
Mutations in DCTN2 were investigated by adding DCTN2 to a gene panel for NGS sequencing along with 51 other known inherited neuropathy or neuronopathy genes. The results from the known neuropathy genes have been published elsewhere, such as gene names, GenBank accession and version number, average coverage, % bases ≥2× coverage, and % bases ≥30× coverage 11. Exome sequencing was performed in the eight family members from the DCTN2 family according to methods published elsewhere 12.
Sequence analysis
Bioinformatic analysis consisted of a standard protocol including image analysis and base calling by Illumina RTA 1.12.4.2, demultiplexing by CASAVA 1.8 (Illumina Inc, San Diego, CA, USA), and alignment of sequence reads to the reference genome GRCh37/hg19 by BWA 13. Picard was used for removing PCR duplicates (http://picard.sourceforge.net/). The genome analysis toolkit (GATK) was applied for base quality score recalibration, insertion and deletion (INDEL) realignment, and single‐nucleotide polymorphism (SNP) and INDEL discovery 14, 15. Annotation of sequence variants was performed by Annovar 16. Variants present in exons ± 10‐bp intron sequences, 3′untranslated regions (UTR), and 5′UTR were included in further analysis. Variants were filtered based on frequency data from dbSNP 135, 1000 genomes, 180 in‐house normal controls, pathogenicity predictions through the Alamut interface v2.2‐0 (Interactive Biosoftware, Rouen, France), and reports in the Human Genome Mutation Database (HGMD), the Mutation Database of Inherited Peripheral Neuropathies (IPNMDB), and published reports.
Verification by Sanger sequencing
The variants identified in the DCTN2 family were verified by Sanger sequencing in all available family members to establish genotype–phenotype correlation. Primer design and sequence analysis were performed using CLC Main Workbench (CLC bio, Aarhus, Denmark), and the sequencing was carried out using standard procedures and sequenced on the ABI3130XL (Life Technologies Ltd, Paisley, UK) 1.
The variants identified in this family have been submitted to the ClinVar database (http://www.ncbi.nlm.nih.gov/clinvar/).
Variant interpretation
Several tools were utilized for variant effect prediction. Multiple sequence alignment (MSA) was visualized using Alamut to see conservation across several species (http://www.interactive-biosoftware.com/). The mutation effect predictors Align GVGD, MutationTaster, PolyPhen, SIFT, and SNAP were utilized 17, 18, 19, 20, 21. Nucleotide conservation was assessed using phylop 22, 23. Evaluation of biochemical differences was carried out by Grantham distance 24.
Ethics
The study was approved by the Norwegian regional committees for medical and health research ethics. Participation was based on informed consent.
Results
Clinical features
Fig. 1 shows the pedigree. The mode of inheritance is compatible with dominant inheritance. The parents (I‐1 and I‐2) were second cousins, while II‐7 and II‐8 were unrelated. I‐1 and I‐2 had gait problems of unknown cause. I‐1 had gait problems and markedly reduced balance, while I‐2 had steppage gait. II‐1 and II‐9 had intellectual deficiency due to birth complications.
Figure 1.
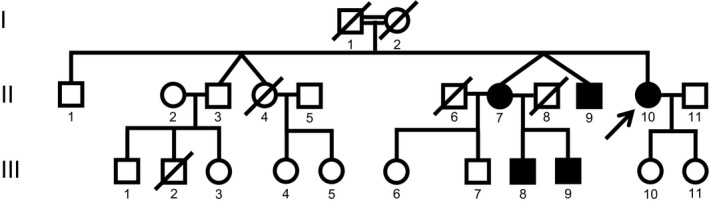
Pedigree of the CMT family with DCTN2 variant c.337C>T.
The available family members II‐3, II‐7, II‐9, II‐10, III‐7, III‐8, III‐9, and III‐10 were neurologically examined. II‐3, III‐7, and III‐10 were unaffected. Table 1 shows clinical characteristics of those affected members, that is, II‐7, II‐9, II‐10, III‐8, and III‐9. Age at onset was late in 4th and 5th decade. The motor signs and severely reduced balance were present in three affected members (II‐7, II‐10, and III‐8), while one affected member (III‐9) had minor signs 2 years after the onset, and one (II‐9) had an inconclusive neurological examination due to lack of cooperation. One affected member (II‐10) also had multiple sclerosis affecting eye movement, and asymmetrical facial and soft palate weakness. Table 2 shows the neurophysiological investigation. The results are compatible with intermediate CMT.
Table 1.
Clinical characteristics of affected members carrying the DCTN2variant
| Family members | II‐7 | II‐9 | II‐10 | III‐8 | III‐9 |
|---|---|---|---|---|---|
| Gender | ♀ | ♂ | ♀ | ♂ | ♂ |
| Age at onset | 40 | – | 35 | 30 | 42 |
| Age at investigation | 71 | 63 | 64 | 45 | 44 |
| Clinical characteristics | |||||
| Muscle wasting 1 | |||||
| Underarm | 0 | 1 | 0 | 0 | 0 |
| Hand | 2 | 2 | 0 | 0 | 0 |
| Thigh | 1 | 1 | 0 | 0 | 0 |
| Leg | 1 | 1 | 1 | 0 | 0 |
| Feet | 1 | 1 | 1 | 1 | 0 |
| Muscle weaknessNIS | |||||
| Facial muscles | 0 | – | 1/0 2 | 0 | 0 |
| Soft palate | 0 | – | 1/0 2 | 0 | 0 |
| Finger spread | 2 | – | 0 | 0 | 0 |
| Thumb abduction | 2 | – | 0 | 0 | 0 |
| Knee flexion | 1 | – | 0 | 0 | 0 |
| Knee extension | 1 | – | 0 | 0 | 0 |
| Ankle dorsiflexors | 3 | – | 1 | 0 | 0 |
| Ankle plantarflexors | 3 | – | 0 | 0 | 0 |
| Toe extensors | 3/3.25 2 | – | 1 | 2/3 2 | 0 |
| Toe flexors | 3 | – | 1 | 2 | 0 |
| Sensory loss | |||||
| Touch 1 | |||||
| Feet, leg | 1 | – | 1 | 0 | 0 |
| Pain 1 | |||||
| Overarm | 1 | – | 0 | 0 | 0 |
| Hand, underarm | 1 | – | 1 | 0 | 1 |
| Thigh | 1 | – | 0 | 0 | 0 |
| Feet, leg | 2 | – | 1 | 1 | 1 |
| Vibration 1 | |||||
| Hand | 2 | – | 1 | 2 | 1 |
| 2. finger | 2 | 1 | 0 | 1 | |
| Knee | 2 | – | 2 | 2 | 1 |
| Ankle | 2 | – | 2 | 2 | 1 |
| 1. metatarsal | 2 | – | 2 | 2 | 2 |
| 1.toe | 2 | – | 2 | 2 | 2 |
| Proprioceptive 1 | |||||
| Toe | 1 | – | 1 | 1 | 0 |
| Reflexes 1 | |||||
| Biceps | 2 | 2 | 1 | 2 | 0 |
| Triceps | 0 | 2 | 1 | 1 | 0 |
| Brachioradialis | 0 | 1 | 0 | 1 | 0 |
| Patellar | 2 | 2 | 2 | 2 | 1 |
| Achilles | 2 | 2 | 2 | 2 | 1 |
| Deformities 1 | |||||
| Pescavus | 2 | 1 | 0/1 2 | 2 | 0 |
| Hammertoes | 1 | – | 1 | 0 | 0 |
| Pesplanus | 0 | – | 1/0 2 | 0 | 0 |
| Slow eye movement 1 | 0 | – | 1 | 0 | 0 |
| Romberg 1 | 2 | – | 2 | 2 | 0 |
| NIS | 58.25 | 16 3 | 32 | 37 | 14 |
–, not informative; 10, normal; 1, mildly/moderately affected modality; 2, severely affected modality. 2Asymmetrical signs right/left. Neuropathy Impairment Score (NIS): For details, see Methods section. 3Muscle weakness and sensory signs unknown due to mental retardation.
Table 2.
Neurophysiology in affected members carrying the DCTN2 variant. Abnormal values are in bold
| Sex | Age at | R/L | Motor nerves | Sensory nerves | EMG chronic dener‐vation | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Onset (yrs) | Examination (yrs) | Median | Ulnar | Peroneal | Tibial | Median | Ulnar | Sural | |||||||||||
| CMAP | CV | CMAP | CV | CMAP | CV | CMAP | CV | SNAP | CV | SNAP | CV | SNAP | CV | ||||||
| Normal values→ | 4.0 | 49.0 | 4.0 | 49.0 | 3.0 | 41.0 | 3.0 | 41.0 | 12.0 | 46.0 | 17.0 | 47.0 | 17.0 | 44.0 | |||||
| II‐7 | ♀ | 40 | 67 | R | 3.6 | 40.0 | 4.7 | 40.0 | A | A | A | A | ↓ | 29.0 | 0.5 | 29.0 | A | A | Present |
| L | – | – | – | – | A | A | A | A | – | – | – | – | A | A | |||||
| II‐9 | ♂ | – | 64 | R | 4.2 | 42.0 | 2.9 | 37.3 | A | A | A | A | 2.4 | 29.6 | A | A | A | A | Present |
| L | 1.5 | 35.5 | – | – | A | A | A | A | – | – | – | – | A | A | Present | ||||
| II‐10 | ♀ | 35 | 46 | R | 4.0 | 50.0 | – | – | 2.0 | 30.0 | 0.4 | 31.0 | 10.0 | 56.0 | – | – | A | A | Present |
| III‐8 | ♂ | 30 | 40 | R | – | – | – | – | 0.4 | 35.2 | 1.3 | 35.5 | – | – | – | – | A | A | Present |
| L | 6.8 | 45.9 | 5.1 | 44.9 | 0.7 | 29.8 | 0.7 | 32.2 | A | A | A | A | 1.6 | 29.7 | |||||
CMAP, compound motor action potential (mV); SNAP, sensory nerve action potential (μV); CV, conduction velocity (m/s); A, absent evoked response; –, not measured; R/L, right/left.
Genetic analyses
We identified a DCTN2 variant in one of 59 families. In this family, targeted NGS identified two heterozygous variants DCTN2, c.337C>T, and GARS, c.95T>C, but only the DCTN2 variant segregated with the phenotype. DNA was available in five affected (II‐7, II‐9, II‐10, III‐8, and III‐9) and three unaffected (II‐3, III‐7, and III‐10) family members. The DCTN2 variant c.337C>T, p.(His113Tyr) replaces histidine with tyrosine.
The GARS variant c.95T>C, p.(Leu32Pro) replaces leucine with proline and was carried by one unaffected member (III‐7) and four affected members (II‐7, II‐9, II‐10, and III‐9). III‐7 was unaffected at the age of 50, while III‐8 was affected and did not carry the GARS variant. Thus, the GARS variant did not segregate with the CMT phenotype.
To rule out the presence of more potentially pathogenic variants, exome sequencing was performed and analyzed using both dominant and recessive models. There were no homozygous or compound heterozygous variants shared among all of the affected or some of the affected members. The only shared haplotype among the affected is situated on chromosome 12 containing four genes with sequence variants. It is heterozygous, consistent with a dominant model. The haplotype was shared by all the affected members, while the unaffected members did not have it.
Exome sequencing revealed four novel heterozygous sequence variants in different genes on chromosome 12 that were shared among the affected members, while the unaffected members did not have these sequence variants. The genes and transcripts were NM_006400.3 DCTN2:c.337C>T (p.His113Tyr), NM_207437.3 DNAH10:c.9510C>G (p.Ile3170Met), NM_153377.3 LRIG3:c.65G>A (p.Gly22Glu), and NM_005379.3 MYO1A:c.3128A>T (p.Gln1043Leu). The chromosomal gDNA position for the four heterozygous sequence variants on chromosome 12 ranged from 57422543 to 124393856 with MYO1A variant at 57422543, DCTN2 variant at 57928880, LRIG3 variant at 59313952, and DNAH10 variant at 124393856. The affected members did not share homozygous sequence variants nor compound heterozygous variants.
Variant interpretation
The DCTN2 His113 is a moderately conserved amino acid (Fig. 2), situated in dynamitin subunit 2 and also moderately conserved on the nucleotide level, as shown by a phylop score of 2.95. The DCTN2 variant, c.337C>T, infers an amino acid change from the basic histidine to a polar uncharged tyrosine, which is a biochemically moderate change with a Grantham distance of 83 (0–215). The variant is predicted neutral by Align GVGD (score: C0 (GV: 160.13 – GD: 0.00)), disease causing by MutationTaster (P‐value: 0.999), possibly damaging by PolyPhen (score: 0.802), tolerated by SIFT (score: 1), and neutral by SNAP (Table 3). DNAH10 3170Ile is a moderately conserved amino acid. The DNAH10 c.9510C>G infers an amino acid change from the hydrophobic isoleucine to the hydrophobic methionine, which is a biochemically weak change with a Grantham distance of 10. The variant is predicted neutral by Align GVGD (score: C0), disease causing by MutationTaster (P‐value: 0.998), probably damaging by PolyPhen (score: 0.972), and deleterious by SIFT (score: 0.01).
Figure 2.
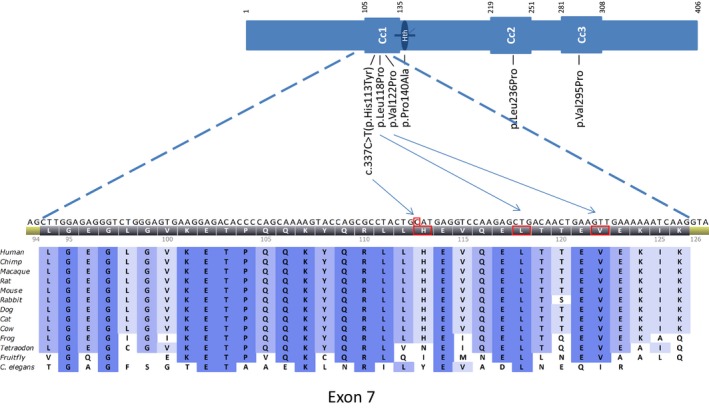
Multiple sequence alignment for DCTN2, exon 7, DCTN2 (dynamitin) protein overview with coiled‐coil motifs (Cc1, Cc2, Cc3), helix‐to‐helix motif (Hth), and position of the in silico tested variants; it was adapted from references 24 and 30.
Table 3.
In silico tests of our variant and dynamitin mutants used in targeted mutagenesis to evaluate how dynamitin's different structural domains contribute to its ability to self‐associate, interact with dynactin, and assemble into a complex with its close binding partner, p24
| Motif | Side chain alteration | Phylop score | Grantham distance | Align GVGD (Class) | Mutation Taster | PolyPhen (score) | SIFT (score) | SNAP | |
|---|---|---|---|---|---|---|---|---|---|
| Our variant | |||||||||
| His113Tyr | Coiled‐coil (Cc1) | Polar→hydrophobic | 2.95 | 83 | C0 (GV:160.13 – GD:0.00) | Disease causing | Possibly damaging (0.802) | Tolerated (1.00) | Neutral |
| Dynamitin mutants | |||||||||
| Leu11 8Pro (C1) | Coiled‐coil (Cc1) | Hydrophobic→ hydrophobic | 4.08 | 98 | C65 (GV: 0.00 – GD: 97.78) | Disease causing | Probably damaging (0.999) | Deleterious (0.00) | Non‐neutral |
| Val122Pro (C1) | Coiled‐coil (Cc1) | Hydrophobic→ hydrophobic | 2.55 | 68 | C25 (GV: 28.68‐ GD: 68.57) | Disease causing a | Probably damaging (0.999) | Deleterious (0.00) | Neutral |
| Pro140Ala | Helix‐turn‐helix | Hydrophobic→ hydrophobic | 3.84 | 27 | C0 (GV: 173.88 – GD: 13.77) | Disease causing | Possibly damaging (0.802) | Tolerated(0.45) | Neutral |
| Leu236Pro (C2) | Coiled‐coil (Cc2) | Hydrophobic→ hydrophobic | 4.40 | 98 | C65 (GV: 4.86 – GD: 95.38) | Disease causing | Probably damaging(0.994) | Deleterious(0.00) | Non‐neutral |
| Val295Pro (C3) | Coiled‐coil (Cc3) | Hydrophobic→ hydrophobic | 4.48 | 68 | C0 (GV: 235.27 – GD: 33.99) | Disease causing a | Possibly damaging(0.574) | Deleterious (0.02) | Non‐neutral |
Side chain charges carried at physiological pH 7.4. Phylop score: values between −14 and +6, with positive scores indicating conserved sites. Grantham distance ranges from 0 to 215 (possible scores 5‐215). Scores of <50 small biochemical difference, 51 to 151 moderate biochemical difference, and values above 151 have a large biochemical difference. Align GVGD: classes (C0, C15, C25, C35, C45, C55, C65) are computed based on both biochemical difference and multiple sequence alignments. The higher the class, the more likely the change is deleterious. MutationTaster outputs prediction of the effect of the change, along with a probability of 0–1, where a value close to 1 indicates a high probability of the prediction being correct, all with P‐value ≥0.999. PolyPhen: values >0.5 is likely to affect the protein. SIFT ranges from 0 to 1; scores <0.05 are predicted deleterious, while scores ≥ are tolerated. SNAP outputs an interpretation of the effect of the mutation along with a reliability of this interpretation, all with accuracy >50%. Values affecting protein function in bold.
Two bases were substituted to achieve this amino acid change.
LRIG3 22Gly is a moderately conserved amino acid. The LRIG3 c.65G>A infers an amino acid change from the non‐polar glycine to a polar glutamic acid, which is a biochemically moderate change with a Grantham distance of 98. The variant is predicted neutral by Align GVGD (score: C0), polymorphism by MutationTaster (P‐value: 0.969), benign by PolyPhen (score: 0.272), and deleterious by SIFT (score: 0).
MYO1A 1043Gln is a moderately conserved amino acid. The MYO1A c.3128A>T infers an amino acid change from the polar glutamine to the hydrophobic leucine, which is a biochemically moderate change with a Grantham distance of 113. The variant is predicted neutral by Align GVGD (score: C0), polymorphism by MutationTaster (P‐value: 1), benign by PolyPhen (score: 0.012), and tolerated by SIFT (score: 0.19).
Discussion
Methodological considerations
DCTN2 was included in the panel as the overexpression of DCTN2 in transgenic mice disrupts the dynein (DYNC1H1)–dynactin (DCTN1) complex and results in a late‐onset slowly progressive motor neuron degenerative disease 7. Although not yet implicated in CMT, it was hypothesized that the gene DCTN2 might cause CMT.
Despite the high number of currently known CMT genes, there are indications that many genes are yet to be identified, for example, in a Norwegian study investigating 51 neuropathy genes, only 46% of the families received a molecular diagnosis 1, 8.
A novel heterozygous variant in DCTN2 c.337C>T was detected in one of the 59 families investigated. The variant was confirmed by Sanger sequencing in all five affected members, while three unaffected members did not have it. The variant was neither seen in the 180 neurologically assessed in‐house Norwegian controls nor in the Single Nucleotide Polymorphism Database (dbSNP) 135 (http://www.ncbi.nlm.nih.gov/snp/), Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org), GEnomes Management Application (GEM.app) (https://genomics.med.miami.edu/gem-app/), and 1000 genomes (http://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/).
Then, we did exome sequencing of the identified DCTN2 family looking for other shared sequence variants that may explain the phenotype. Exome sequencing revealed three other heterozygous sequence variants on chromosome 12. Probably, the affected members share an ancestral haplotype containing the four heterozygous sequence variants in the genes MYO1A, DCTN2, LRIG3, and DNAH10. A shared haplotype is compatible with dominant inheritance seen in the pedigree (Fig. 1), and the affected members did not share homozygous sequence variants nor compound heterozygous variants.
Genotype–phenotype correlation
We observed perfect cosegregation of the DCTN2 variant with the disease in the eight subjects for whom DNA was available A segregation pattern observed in our pedigree could occur by chance in (½)7 = 1/128. The variant was situated in a conserved domain among several species (Fig. 2). Five tools were used to investigate the in silico predicted pathogenicity of the detected variant in DCTN2 (Table 3). Of these, three predicted a neutral effect, while two predicted a disease causing or possible damaging effect. Similar findings were seen for the dynamitin mutagenesis protein variants that were used to investigate the protein–protein interactions important for dynactin structure (Table 3) 25.
The identified GARS variant, c.95T>C, did not segregate with the phenotype. The GARS variant, c.95T>C, was neither seen in in‐house controls nor in dbSNP and ExAC databases. The variant was weakly conserved among species, and it was predicted benign in all variant prediction tools.
Exome sequencing revealed three other heterozygous variants on chromosome 12 in all five affected members probably sharing an ancestral haplotype.
The DNAH10 gene is the force‐generating protein of respiratory cilia and is a paralog to DYNC1H1, another CMT gene (ExAC and GeneCards (http://www.genecards.org/) databases).
DNAH10 is also expressed in brain, and it is not clear whether the ortholog in rat (Dlp10) is only involved in cilia function 26, 27. In addition, DNAH10 might be expressed in peripheral nerves (http://www.proteinatlas.org/ENSG00000197653-DNAH10/tissue/soft+tissue).
Four tools were used to investigate the in silico predicted pathogenicity of the detected variant in DNAH10. Of these, one predicted a neutral effect, while three predicted a disease causing or probably damaging effect.
The LRIG3 gene may play a role in craniofacial and inner ear morphogenesis during embryonic development (ExAC and GeneCards (http://www.genecards.org/) databases). Four tools were used to investigate the in silico predicted pathogenicity of the detected variant in LRIG3. Of these, three predicted a neutral effect, while one predicted a damaging effect.
The MYO1A gene is associated with non‐syndromic hearing loss and deafness (ExAC and GeneCards (http://www.genecards.org/) databases). Four tools were used to investigate the in silico predicted pathogenicity of the detected variant in MYO1A. Of these, all predicted a neutral effect and the inferred change was in the last amino acid before stop.
Variant interpretation considerations
These variant prediction tools tend to disagree and are generally cautious in labeling a variant as having physical impact on the function of the gene (Table 3) 28. This further underlines the fact that there is to date no perfect in silico tools for mutation effect prediction and that genotype–phenotype correlation and functional assays are still the most important way to evaluate novel variants. The DCTN2 gene is moderately conserved on the amino acid level (data not shown), and Fig. 2 shows the protein and the conservation on the exon 7 surrounding the c.337C>T variant are high for all mammals in the multiple sequence alignment. In addition, there is little normal variation in the gene (dbSNP and 1000 genomes). Our CMT family and C. elegans do share the Tyr residue at position 113 in DCTN2. The three other genes identified with heterozygous variants on chromosome 12 seem to have functions related to other organ systems.
Dynein–dynactin complex
DCTN2 overexpression in transgenic mice disrupts the dynein (DYNC1H1)–dynactin 0(DCTN1) complex 7. DYNC1H1 variants cause CMT2 and spinal muscular atrophy, while DCTN1 variants cause distal hereditary motor neuronopathy (dHMN) 29, 30, 31, 32. Thus, heterozygous variants in both DYNC1H1 and DCTN1 cause inherited neuropathies and neuronopathies. Neurophysiology in our family with the DCTN2 variant is compatible with intermediate CMT 8.
DCTN2 encodes the protein dynamitin (p50), which is an element of dynactin, a highly conserved multiprotein complex 25, 33. Dynactin works in conjugation with the molecular motor dynein (encoded by DYNC1H1) and is responsible for retrograde transport in neurons, moving cargoes such as mitochondria, organelles, and proteins along the microtubules 34, 35, 36, 37. Dynactin also has other functions independent of dynein such as attachment of microtubules to several subcellular structures 25, 33. Dynamitin is the core element of dynactin, four dynamitin subunits sit at the junction between dynactins two functional domains, the Arp1 filament binding cargo and the p150Glued (encoded by DCTN1), interacting with dynein and microtubules 25, 33. The expression of the dynactin molecule is highly regulated where dynamitin plays a key role probably due to its strong propensity for self‐association. The overexpression of dynamitin monomers leads to disassembly of the dynactin‐bound dynamitin, and association with the free monomers, this releases the p150Glued side chain which renders the remaining dynactin molecule non‐functional 25, 33. Also, the depletion of dynamitin leaves the other subunits of dynactin unstable, leading to downregulation of the complex 33. The overexpression of DCTN2 in transgenic mice results in a late‐onset slowly progressive motor neuron degenerative disease 7.
The dynamitin molecule consists of three coiled‐coil motifs, of which the two‐first are predicted to mediate in self‐association and stabilization of the whole dynactin complex. Sequence analysis predicts that particularity the first coiled‐coil motif is especially important as this is the most conserved among species (Fig. 2) 25, 33.
The variant at position 113 (isoform 2) found in this family is situated in the first coiled‐coil motif (Fig. 2).
The DYNC1H1 paralog DNAH10 is expressed in the nervous system and may influence the dynein–dynactin complex or similar multiprotein complexes.
Conclusions
This is the first time a variant in the genes DCTN2, DNAH10, LRIG3, and MYO1A has been linked to an inherited neuropathy in humans.
However, our CMT family and C. elegans do share the Tyr residue at position 113 in DCTN2 and DNAH10 may play a role in the nervous system. Hence, functional studies will be needed to confirm that these variants could be deleterious in humans. At the moment, no conclusion can be drawn concerning the causal role of the identified variants in the family phenotype. The haplotype with singular variants in DCTN2, DNAH10, LRIG3, and MYO1A is merely associated with CMT.
Conflicts of interest
The authors declare that they have no conflict of interests.
Authors' contribution
GJB acquired the material, conceived the study, participated in the design of the study, and drafted the manuscript. HH conceived the study, participated in the design of the study, and carried out the molecular genetic studies, sequence alignment, and bioinformatic analyses. ØLB and KT carried out the molecular genetic studies and bioinformatic analyses. CFS participated in the design of the study. MBR participated in the design of the study and drafted the manuscript. All authors critically read and approved the final manuscript.
Acknowledgement
Øystein L. Holla and Linda Strand contributed in NGS, Hilde H. Hilmarsen contributed in Sanger sequencing and analysis, and Marit V. Svendsen designed the database. The study was financially supported by South‐Eastern Norway Regional Health Authority, Akershus University Hospital HF, Telemark Hospital HF, and Nansen Foundation.
Braathen GJ, Høyer H, Busk ØL, Tveten K, Skjelbred CF, Russell MB. Variants in the genes DCTN2, DNAH10, LRIG3, and MYO1A are associated with intermediate Charcot–Marie–Tooth disease in a Norwegian family. Acta Neurol Scand 2016: 134: 67–75. © 2015 The Authors. Acta Neurologica Scandinavica Published by John Wiley & Sons Ltd.
Correction added on 29 October 2015 after first online publication: The 3rd affiliation was previously omitted for authors G.J Braathen and H. Høyer, and this is now corrected in the current version.
References
- 1. Braathen GJ, Sand JC, Lobato A, Hoyer H, Russell MB. Genetic epidemiology of Charcot‐Marie‐Tooth in the general population. Eur J Neurol 2011;18:39–48. [DOI] [PubMed] [Google Scholar]
- 2. Skre H. Genetic and clinical aspects of Charcot‐Marie‐Tooth's disease. Clin Genet 1974;6:98–118. [DOI] [PubMed] [Google Scholar]
- 3. Combarros O, Calleja J, Polo JM, Berciano J. Prevalence of hereditary motor and sensory neuropathy in Cantabria. Acta Neurol Scand 1987;75:9–12. [DOI] [PubMed] [Google Scholar]
- 4. Foley C, Schofield I, Eglon G, Bailey G, Chinnery PF, Horvath R. Charcot‐Marie‐Tooth disease in Northern England. J Neurol Neurosurg Psychiatry 2012;83:572–3. [DOI] [PubMed] [Google Scholar]
- 5. Saporta AS, Sottile SL, Miller LJ, Feely SM, Siskind CE, Shy ME. Charcot‐Marie‐Tooth disease subtypes and genetic testing strategies. Ann Neurol 2011;69:22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Murphy SM, Laura M, Fawcett K et al. Charcot‐Marie‐Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. J Neurol Neurosurg Psychiatry 2012;83:706–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. LaMonte BH, Wallace KE, Holloway BA et al. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late‐onset progressive degeneration. Neuron 2002;34:715–27. [DOI] [PubMed] [Google Scholar]
- 8. Braathen GJ. Genetic epidemiology of Charcot‐Marie‐Tooth disease. Acta Neurol Scand Suppl 2012;193:iv‐22. [DOI] [PubMed] [Google Scholar]
- 9. Collins JS, Schwartz CE. Detecting polymorphisms and mutations in candidate genes. Am J Hum Genet 2002;71:1251–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dyck PJ, Turner DW, Davies JL, O'Brien PC, Dyck PJ, Rask CA. Electronic case‐report forms of symptoms and impairments of peripheral neuropathy. Can J Neurol Sci 2002;29:258–66. [DOI] [PubMed] [Google Scholar]
- 11. Høyer H, Braathen GJ, Busk ØL et al. Genetic diagnosis of Charcot‐Marie‐Tooth disease in a population by next‐generation sequencing. BioMed Research International 2014;2014:210401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Holla OL, Bock G, Busk OL, Isfoss BL. Familial visceral myopathy diagnosed by exome sequencing of a patient with chronic intestinal pseudo‐obstruction. Endoscopy 2014;46:533–7. [DOI] [PubMed] [Google Scholar]
- 13. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. DePristo MA, Banks E, Poplin R et al. A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat Genet 2011;43:491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McKenna A, Hanna M, Banks E et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res 2010;20:1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Adzhubei IA, Schmidt S, Peshkin L et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hicks S, Wheeler DA, Plon SE, Kimmel M. Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed. Hum Mutat 2011;32:661–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res 2001;11:863–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease‐causing potential of sequence alterations. Nat Methods 2010;7:575–6. [DOI] [PubMed] [Google Scholar]
- 21. Johnson AD, Handsaker RE, Pulit SL, Nizzari MM, O'Donnell CJ, de Bakker PI. SNAP: a web‐based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics 2008;24:2938–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 2010;20:110–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Siepel A, Bejerano G, Pedersen JS et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 2005;15:1034–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grantham R. Amino acid difference formula to help explain protein evolution. Science 1974;185:862–4. [DOI] [PubMed] [Google Scholar]
- 25. Maier KC, Godfrey JE, Echeverri CJ, Cheong FK, Schroer TA. Dynamitin mutagenesis reveals protein‐protein interactions important for dynactin structure. Traffic 2008;9:481–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maiti AK, Mattéi MG, Jorissen M, Volz A, Zeigler A, Bouvagnet P. Identification, tissue specific expression, and chromosomal localisation of several human dynein heavy chain genes. Eur J Hum Genet 2000;8:923–32. [DOI] [PubMed] [Google Scholar]
- 27. Tanaka Y, Zhang Z, Hirokawa N. Identification and molecular evolution of new dynein‐like protein sequences in rat brain. J Cell Sci 1995;108(Pt 5):1883–93. [DOI] [PubMed] [Google Scholar]
- 28. Castellana S, Mazza T. Congruency in the prediction of pathogenic missense mutations: state‐of‐the‐art web‐based tools. Briefings in Bioinformatics 2013;14:448–59. [DOI] [PubMed] [Google Scholar]
- 29. Weedon MN, Hastings R, Caswell R et al. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot‐Marie‐Tooth disease. Am J Hum Genet 2011;89:308–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harms MB, Ori‐McKenney KM, Scoto M et al. Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology 2012;78:1714–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Puls I, Jonnakuty C, LaMonte BH et al. Mutant dynactin in motor neuron disease. Nat Genet 2003;33:455–6. [DOI] [PubMed] [Google Scholar]
- 32. Munch C, Sedlmeier R, Meyer T et al. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 2004;63:724–6. [DOI] [PubMed] [Google Scholar]
- 33. Jacquot G, Maidou‐Peindara P, Benichou S. Molecular and functional basis for the scaffolding role of the p50/dynamitin subunit of the microtubule‐associated dynactin complex. J Biol Chem 2010;285:23019–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schnapp BJ, Reese TS. Dynein is the motor for retrograde axonal transport of organelles. Proceedings of the National Academy of Sciences of the USA 1989;86:1548–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Eschbach J, Dupuis L. Cytoplasmic dynein in neurodegeneration. Pharmacol Ther 2011;130:348–63. [DOI] [PubMed] [Google Scholar]
- 36. Hafezparast M, Klocke R, Ruhrberg C et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science 2003;300:808–12. [DOI] [PubMed] [Google Scholar]
- 37. Schroer TA, Steuer ER, Sheetz MP. Cytoplasmic dynein is a minus end‐directed motor for membranous organelles. Cell 1989;56:937–46. [DOI] [PubMed] [Google Scholar]