

Abstract
Functional genomics approaches such as gain- and loss-of-function screening can efficiently reveal genes that control cancer cell growth, survival, signal transduction, and drug resistance, but distilling the results of large-scale screens into actionable therapeutic strategies is challenging given our incomplete understanding of the functions of many genes. Research over several decades, including the results of large-scale cancer sequencing projects, has made it clear that many oncogenic properties are controlled by a common set of core oncogenic signaling pathways. By directly screening this core set of pathways, rather than much larger numbers of individual genes, it may be possible to more directly and efficiently connect functional genomic screening results with therapeutic targets. Here, we describe the recent development of methods to directly screen oncogenic pathways in high-throughput. We summarize the results of studies that have used pathway-centric screening to map the pathways of resistance to targeted therapies in diverse cancer types, then conclude by expanding on potential future applications of this approach.
Keywords: functional genomics, gain of function screens, drug resistance, cancer genomics, signaling pathways
INTRODUCTION
Characterizing the genetic and functional landscape of cancer cells
The advent of high-throughput sequencing led to an explosion of data on the nature and number of genetic alterations present across a broad array of cancer types. Thanks to the efforts of the Cancer Genome Atlas, the International Cancer Genome Consortium, and other groups, researchers now have more knowledge than ever about the genetic events that trigger the transition from normal cell to cancer cell [1]. Through this body of work, we now know much more about the variety of mutation rates and patterns among different cancer types, the involvement of processes not originally thought to impact cancer, and even the existence of new cancer-relevant genes [2]. Many of these genetic discoveries have had profound impacts on the treatment of cancer patients. For example, the discovery of activating mutations in the epidermal growth factor receptor (EGFR) helped explain the success of EGFR inhibitors in a subset of lung cancer patients, in the process revolutionizing the screening and treatment of these patients [3–5]. Further, while some genetic insights have led to a decrease in reliance on older histopathology-based tumor classifications, other discoveries have served to deepen our understanding of the genetic bases of these classifications [6, 7].
However, still much remains to be discovered about cancer genomics. Beyond contemporary questions like how genetic heterogeneity impacts tumor biology, the importance of rarely mutated genes, and the function of non-coding regions of the genome, there is much that we simply do not understand about the workings of cancer-relevant mammalian genes. For instance, in a 70 gene expression signature of poor prognosis in breast tumors, the function of 20 genes is poorly understood [8]. Similar proportions of poorly understood genes can be found in lists of the most commonly altered genes in cancer, predictors of relapse on therapy, and others [9]. Additionally, apart from large scale sequencing efforts, hypothesis driven research has demonstrated the functional importance of many non-mutated genes such as those required for signaling downstream of oncogenes and tumor suppressors. The ability to identify these functionally important yet non-genetically altered genes could be crucial to the development of new therapies. Given that such genes are unlikely to be directly identified through sequencing alone, there is a clear role for complementary analysis techniques.
Toward this end, functional genomics methods enabling the specific and rapid manipulation of individual genes, first through cDNA expression, then siRNA- and shRNA-mediated RNA interference (RNAi), and more recently using CRISPR technology, have profoundly impacted the functional dissection of cellular processes, particularly those involved in cancer. As it had for whole genome sequencing, the emergence of affordable massively parallel sequencing allowed for the true rise of large-scale functional genomic screening, bypassing the throughput limitations of arrayed screens. Broadly, functional genomic screens can be categorized as either gain-of-function or loss-of-function based on their effects on target gene function. As loss-of-function screens echo the action of the majority of pharmacological agents available for cancer therapy, these screens have received the most attention from researchers. Targeted or whole genome libraries of siRNA, shRNA, or CRISPR constructs can be utilized to assess the impacts of genetic loss on biological processes of interest. For example, numerous published screens have identified genes whose loss impairs tumor progression, cell cycle control, growth factor signaling, and migration and invasion [10–17]. Inversely, gain-of-function screens can be effectively used to screen the effects of ectopic or endogenous gene expression on phenotypes of interest. Although assembly and physiological expression of gain-of-function ORF libraries remains challenging, these approaches have successfully identified, for example, kinases whose activation can maintain phosphoinositide-3-kinase (PI3K) signaling [18] and proteins whose overexpression confers sensitivity or resistance to cytotoxic and targeted cancer therapies [19–26]. More recently, new screening methods making use of CRISPR-based gene activation have made it possible to perform gain-of-function screens from endogenous promoters without ectopic cDNA expression [27–29].
Finally, within the space of functional genomic screening, a particularly important subset of work has borrowed concepts from yeast genetics to search for interactions between either genes or chemical compounds. Termed ‘synthetic lethal’ screens, these approaches involve the introduction of libraries of genetic reagents into a cell population bearing a gene alteration or treated with a compound, where the effect of each genetic perturbation is compared to a matched control population to identify genes whose alteration provides a specific outcome, such as cell death, only in the context of the studied gene alteration or chemical compound. These screens have provided important information on potential combination therapies with the potential to improve initial therapeutic responses or suppress resistance. Many groups have identified potential targets to enhance the cytotoxic effects of cancer therapeutics such as paclitaxel in lung cancer, PARP inhibitors in breast cancer, HDAC inhibitors in osteosarcoma, imatinib in CML, and BRAF inhibitors in colon cancer and melanoma among others [30–37].
APPROACH
High-throughput mapping of the signaling pathways driving resistance
With respect to the development of targeted cancer therapies, the results of the past decade of cancer genomics raise important questions. How can gene products such as RAS family members and other proteins lacking pharmacologically addressable active sites be targeted? Perhaps more broadly, given that a diverse array of genomic alterations can initiate tumorigenesis, cause drug resistance, spark metastasis, or provide escape from immune surveillance, combined with the established capacity of tumors to evolve resistance to drugs targeting individual alterations, how will it be possible to design robust therapies?
Our growing understanding of cancer cell signaling networks provides a potential answer to these thorny problems. While diverse genomic alterations can drive phenotypic outcomes such as drug resistance, the net result of each alteration is often the redundant modulation of a single signaling pathway. In just one example, the wide array of resistance mechanisms in BRAF-mutant melanoma to RAF inhibitors (RAFi) include mutations in NRAS, MEK and ERK and the amplification and alternative splicing of BRAF [20, 38–41]. However, as diverse as these resistance mechanisms may seem, they all result in the reactivation of mitogen-activated protein kinase (MAPK) signaling in the presence of the original RAF inhibitor. This seems to suggest a hardwired predilection for MAPK signaling itself, regardless of the specific modification instrumental to its stimulation. Other resistance mechanisms to RAFi have been identified in melanoma such as alterations in IGF-R, PIK3CA, PTEN and AKT [42–44]. In all of these cases, the alteration is likely to drive resistance through activation of the PI3K pathway, an alternative signaling pathway capable of rescuing growth and survival in the context of MAPK pathway inhibition. Outside the setting of melanoma, most of the identified resistance mechanisms to targeted therapy involve either bypass and reactivation of the original driver pathway or activation of a similar pathway. The ability to cut through the overabundance of specific alterations capable of activating canonical growth, survival, differentiation and apoptosis pathways, and instead focus solely on the specific pathways themselves, may provide much needed simplicity to the field of drug resistance and the broader pursuits of cancer biology research.
With this goal in mind, our group set out to devise a method of systematically interrogating signaling pathways for their impact on oncogenic properties, with a specific focus on drug resistance. We first assembled a set of 17 signaling pathways that had been previously found to be frequent players in oncogenic processes (Table 1). This list was composed of the MAPK and PI3K pathways as introduced above as well as major pathways contributing to proliferation (JAK-STAT, estrogen receptor (ER), androgen receptor (AR), TGF-β, ERK5, Ral), survival (p53, BCL-2 family members, p38, Hippo), differentiation (Wnt, Hedgehog, Notch), and inflammation (JNK, NF-κB), with many of these pathways also having impacts on multiple phenotypes [45]. For each of these pathways, we next selected 1-3 well validated methods of either activating (oncogenic pathways) or deactivating (tumor suppressive pathways) each signaling pathway. For instance, in the case of PI3K signaling, a total of three activating constructs were selected. These include myristoylated-PIK3CA and -AKT1, which localize constitutively at the cell membrane to initiate downstream signaling, and the Q64L mutant of RHEB which locks the GTPase in its active, GTP-bound state, facilitating activation of mTORC1. All of the activating and deactivating strategies are summarized in Table 1. We then barcoded and cloned these constructs into lentiviral vectors in which transgene expression is driven by the human phosphoglycerate kinase 1 (PGK) promoter and selection can be achieved using the puromycin resistance gene.
Table 1. cDNAs activating defined oncogenic signaling pathways.
| Signaling pathway | Protein | Activating strategy | Validation method |
| Ras-MAPK | KRAS | G12V mutation | Western (P-ERK) |
| HRAS | G12V mutation | Western (P-ERK) | |
| MEK1 | S218D, S222D mutations | Western (P-ERK) | |
| PI3K-AKT-mTOR | PIK3CA | myr-FLAG tag | Western (P-AKT) |
| AKT1 | myr-FLAG tag | Western (P-AKT, P-S6K1) | |
| Rheb | Q64L mutation | Western (P-S6K1) | |
| NF-κB | IKKα | S176E, S180E mutations | Reporter (NF-κB_Luc) |
| IKKβ | S177E, S181E mutations | Reporter (NF-κB_Luc) | |
| Jak/Stat | JAK2 | V617F mutation | Reporter (Stat_Luc) |
| Stat3 | A662C, N664C, V667L mutations | Reporter (Stat_Luc) | |
| Wnt/b-catenin | β-catenin | S33A, S37A, T41A, S45A mutations | Reporter (TCF-LEF_Luc) |
| GSK3β | K85A mutation | Reporter (TCF-LEF_Luc) | |
| β-catenin | S33Y mutation | Reporter (TCF-LEF_Luc) | |
| JNK | JNK2 | WT overexpression | Reporter (AP1_Luc) |
| JNK2 | Mkk7 fusion | Reporter (AP1_Luc) | |
| ERK5 | MEK5 | S311D, T315D mutations | Western (ERK5 laddering) |
| MEK5 | myr-FLAG tag | Western (ERK5 laddering) | |
| Notch | Notch1 | intracellular domain only | Reporter (HES1_Luc) |
| Notch3 | intracellular domain only | Reporter (HES1_Luc) | |
| p38 | p38 (MAPK14) | WT overexpression | Western (P-p38) |
| MKK6 | S207E, T211E mutations | Western (P-p38) | |
| Hedgehog | Gli2 | truncation | Reporter (Gli_Luc) |
| SmoM2 | W535L mutation | Reporter (Gli_Luc) | |
| TGFβ | TGFβR1 | WT overexpression | Immunofluorescence (P-Smad2/3) |
| Mitochondrial apoptosis (intrisic pathway) | BCL2 | WT overexpression | Western (cleaved caspase 9) |
| BCL-XL | WT overexpression | Western (cleaved caspase 9) | |
| Death receptor apoptosis (extrisic pathway) | Caspase-8 | C360A mutation | Western (cleaved caspase 8) |
| All apoptosis | Caspase-3 | C163A mutation | Western (cleaved caspase 3/7) |
| Estrogen receptor | Erα | Y537S mutation | Reporter (ERE_Luc) |
| Androgen receptor | AR | V7 variant | Western (ARE_Luc) |
| Hippo | YAP2 | FLAG-YAP2 (5SA) | Immunofluorescence (nuclear YAP) |
| Lats2 | kinase dead (K697R) | Immunofluorescence (nuclear YAP) | |
| p53 | p53 | dominant negative R175H mutant | Reporter (p53_Luc) |
| Ral | Hras | G12V, E37G mutations | |
| Rgl2 | Rgl2-CAAX | ||
| RalA | G23V (two forms - full and mature peptide) |
In all, our library was composed of 36 constructs capable of modulating 17 major signaling pathways. All constructs were fully sequenced to confirm fidelity to the original source and 86% (31/36) of constructs were functionally validated by immunoblotting, reporter assay, or immunofluorescence to confirm proper engagement of each signaling pathway (Figure 1 and Table 1). These constructs can be used in arrayed or pooled screening formats. To investigate the utility of this library in the context of drug resistance, we first examined the setting of BRAF-mutant melanoma, one in which acquired resistance to RAF and MEK inhibitors has been well studied. Using a positive selection, pooled screening approach, we infected the BRAF-mutant melanoma cell line UACC-62 with the pooled library and treated populations of the cells with either vehicle control or the MEK1/2 inhibitor AZD6244 at multiple doses yielding between 20% to 80% growth inhibition (150 nM, 750 nM, and 1.5 μM). After 2-3 weeks of treatment, genomic DNA was isolated from each population and the construct barcode sequences were amplified and quantified by Illumina deep sequencing. By isolating pathway constructs that were over-represented in the drug treated populations versus the control populations, we could identify pathways that conferred a survival advantage to the cells expressing them under the selective pressure of MEK inhibition. This screen identified 5 pathways capable of conferring resistance (Figure 2). Three of these pathways, RAS-MAPK, PI3K and NF-κB, had previously been implicated as resistance mechanisms to RAF and MEK inhibitors, validating the ability of the method to identify biologically relevant resistance pathways. In addition, we found two novel pathways that were capable of conferring resistance, the Notch1 and ERα pathways. On the basis of the strength of this finding, we followed the preliminary screen with additional screens of 15 targeted therapies in relevant oncogene-driven cancer cell line models at multiple doses of each drug. While indicating novel resistance mechanisms in melanoma, breast cancer, colorectal cancer, myelofibrosis, and acute myeloid leukemia [46–49], overall, the screens provided several important observations. First, among the pathways screened, a few, such as RAS-MAPK, PI3K and Notch1, are capable of driving resistance to a broad number of inhibitors and in many cancer types. In contrast, other pathways were only observed to confer resistance in a few contexts. For example, NF-κB pathway activation could confer resistance to MAPKi in BRAF-mutant melanoma, but did not score as a resistance pathway in many other cancer types. Next, our evidence suggests that the further down a linear pathway one inhibits (e.g., inhibiting ERK vs. RAF), the more likely it is that the spectrum of resistance mechanisms will be dominated by alternative signaling pathways, while reactivation of the inhibited pathway is often the dominant mechanism of resistance to inhibitors higher up on the pathway. Put differently, in a competitive, pooled screen format, we find that resistance driven by simple pathway reactivation is favored over alternative survival pathways when possible, and that blocking driver pathways at lower nodes tends to favor the emergence of alternative resistance pathways. Finally, our screen provided some hope for the problem of managing resistance because, in general, fewer than 5 pathways was typically capable of providing resistance in a given context.
Figure 1. Example of pathway activating construct activity validation.
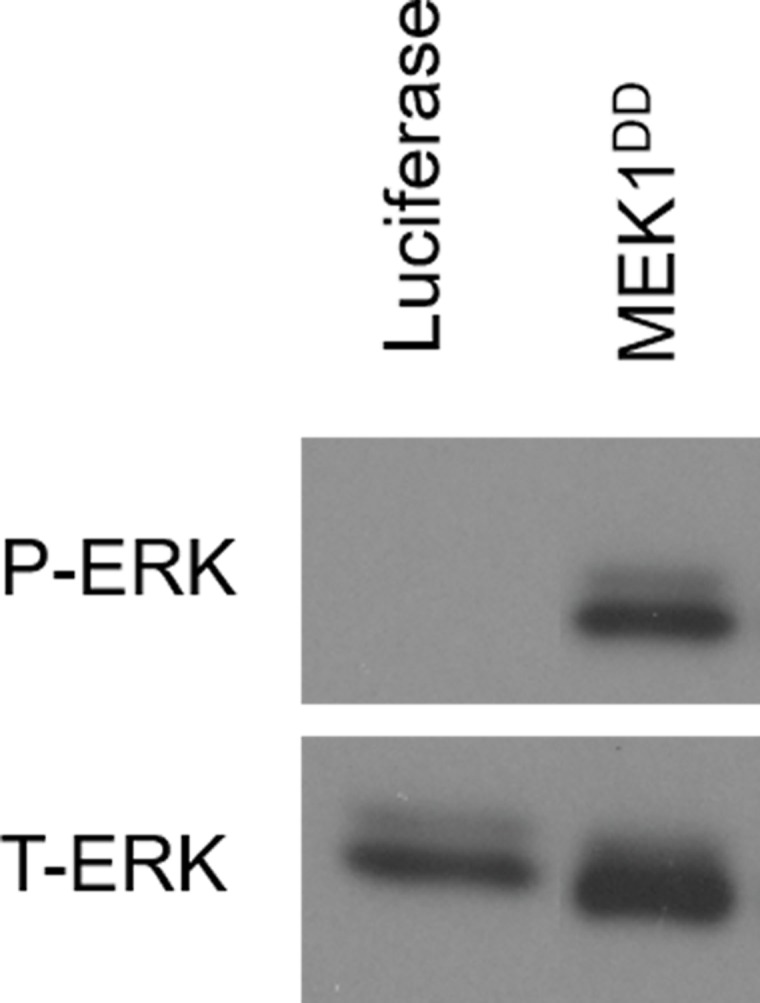
Target cells (shown here, 293T) were infected with constructs encoding individual pathway activating constructs. Following selection with puromycin, whole cell lysates were collected and immunoblotted for the appropriate markers of pathway activation. For the MAPK pathway activator, constitutively active mutant of MEK1, phospho-ERK was used as an indicator of proper pathway activation.
Figure 2. Pathway activating screen results and example validation methods.

A. Results of a pathway activating screen on UACC-62 cells for signaling pathways that provide resistance to the MEK inhibitor, AZD6244, at several doses. Abundance of scoring (MAPK, PI3K, NFκB, Notch1 and ER) and control (HcRed, luciferase, MEK1) pathway activators in drug-treated cells relative to diluent- treated cells is shown. B. Screen results can be used to guide investigation of naturally occurring mechanisms of the phenotype of interest. In the setting of resistance, evolved or intrinsically resistant cell lines can have the screen hits inhibited genetically or pharmacologically to investigate whether this reverses resistance. In this example, knock down of Notch1 sensitized MAPKi-resistant cells to the inhibitor. C. The screen hit can be blocked to determine if this prevents the emergence of the phenotype of interest. Here, cells were treated with the screen inhibitor, and inhibitor of the candidate pathway and the combo. Cells were counted weekly and it was observed that only the combination of inhibitors significantly delayed the emergence of resistance. D. Screen results can direct examination of tumor samples. In a survey of relapsed tumor biopsies, a most show evidence of known resistance mechanisms but a subset show activity, mutually exclusive with known mechanisms, of the candidate pathway.
While offering an intriguing global picture of the oncogenic pathways that are capable of regulating a particular phenotype, it is essential to recognize that the hits uncovered in a pathway activation screen, like other gain-of-function screens, are merely candidates until further confirmatory validation experiments are performed. The screen is capable of guiding researchers to targets with a good probability of physiological and clinical relevancy, however, the limitations of the method necessitate a robust and thorough confirmatory workflow. We have found that the validation of the screens is best approached using a combination of methods in three general categories (Figure 2). First, using the example of drug resistance, cells can be interrogated for a natural propensity of the nominated pathway to be selected for in an unbiased setting. For example, cell lines with either evolved or intrinsic resistance can be assessed for the capacity of inhibitors of the nominated pathway to reverse resistance. We validated the finding that Notch1 activation can drive resistance to MAPK pathway inhibitors in BRAF mutant melanomas by evolving resistance to RAF (PLX4720), MEK (AZD6244) and ERK (VX-11E) inhibitors in 6 BRAF mutant melanoma cell lines, ultimately developing 18 pooled and 84 clonal resistant derivatives. Among these, 39% of the pooled and 29% of the clonal populations could be resensitized to MAPK inhibition through shRNA mediated knockdown of Notch1. Similarly, we validated the finding that MCL-1 and BCL-XL up-regulation can drive resistance to the potential to be used to address many other questions in selective BCL-2 inhibitor ABT-199 (venetoclax) in acute myeloid leukemia (AML) by demonstrating the resensitization of cells with acquired resistance through knockdown or small molecule inhibition of these candidate resistance proteins [48]. Finally, we validated Ras effector pathways PI3K and MAPK as drivers of acquired resistance to JAK inhibitors in JAK2 mutant cells using a similar approach in cells with acquired JAKi resistance [47]. A second validation approach is to demonstrate that by blocking the pathway in question, evolution of the phenotype it drives is either delayed or prevented entirely. For example, using this approach, we have substantiated screening hits by showing, for example, cancer biology. For example, Weinberg and Hanahan have famously implicated a defined set of cellular and tissue functions as the ‘hallmarks of cancer” [52]. Some of these hallmarks remain poorly understood from a signaling pathway and drug targeting perspective. It is straightforward therefore to envision the use of pathway activating screens to determine the relative impact of various pathways on hallmark phenotypes in diverse cancer models. Additionally, while library-based screening methods have thus far been mostly limited to in vitro cancer cell line applications, it is exciting to consider the additional information that could be gleaned from an in vivo application of these approaches [53–55]. Expanded that inhibition of BCL-XL and MCL-1 can prevent the pathway focused screens could shed light on enduring development of resistance to BCL-2 inhibition in AML [48], and inhibition of the MAPK pathway with MEK inhibitors can prevent the development of resistance to EGFR inhibitors in colorectal cancer (CRC) [49]. Finally, one can validate screening results by identifying evidence that the pathway in question is activated selectively in phenotype-positive tumors, particularly if the pattern of activation in a population of samples is mutually exclusive with the occurrence of other known mechanisms triggering the same phenotype. For example, in a fraction of human melanoma tumors with acquired resistance to RAF or RAF+MEK inhibition, we found evidence of Notch1 activation through elevated expression of the protein and its transcriptional targets. Importantly, tumors with evidence of Notch1-driven resistance did not overlap with those that showed evidence of MAPK reactivation- or PI3K activation-based resistance mechanisms, suggesting that Notch1 may functionally drive resistance in these tumors. [46]. In a similar fashion, we have found evidence of activating mutations in a candidate resistance pathway member, RAS, co-occurring with activating mutations in JAK2 in neoplastic cells from patients with myeloproliferative neoplasms [47]. Tumors bearing this mutation often show intrinsic resistance to JAK inhibition [50, 51], consistent with our results suggesting that concurrent RAS mutations may contribute to this resistance. Overall, by using the set of general validation principles described above to provide physiological validation of screening results, we have found that the results of several pathway screens have been confirmed mechanistically and in clinical samples.
FURTHER APPLICATIONS
To date, we have utilized pathway activating screens to uncover pathways driving resistance to therapy in a broad array of cancer types, and much of the work in our group and others is now leveraging findings from these screens to design more robust therapeutic strategies, some of which are being explored clinically [47, 49]. More broadly, however, this approach has the questions in cancer biology such as the seemingly preferential mutation of certain pathway members, signaling network crosstalk, and the functional impacts of intratumoral heterogeneity. Along with the ability to screen for drug dependencies and mechanism of action of poorly understood therapies [25], it is clear that there exist many applications for this technology outside the field of drug resistance.
Importantly, limitations of the current technology exist. First, while we attempted to include many of the pathways most heavily implicated in tumor biology, the library is nevertheless restricted to pathways of known importance. Thus, it is best viewed as a tool for mapping these pathways to phenotypes of interest rather than as an approach to define new signaling pathways. Moving forward, it will be important to expand the library to include additional pathways such as those controlling epigenetic, metabolic, and transcriptional processes. Also, commonly occurring mutations and alternative pathway nodes may affect the function of a gene and hence, a pathway, differently than the methods of activation or inhibition that are already included in the library. To address these limitations, we have partnered with the National Cancer Institute's Ras Program to expand the current library several times over and make these reagents broadly available to the scientific community. We hope this resource will make further discoveries possible with the ultimate goal of improving our understanding of cancer biology and, hence, therapies for cancer patients.
Functional genomics has proven to be a valuable complement to large-scale sequencing and traditional hypothesis-driven cancer research, particularly as it becomes more clear that a personalized medicine approach will be critical not only for first line therapy, but also for the continued management of patient tumor heterogeneity and evolving resistance. Regardless of the challenges of managing diverse disease types, research has shown that cells remain dependent on a core set of signaling pathways for their continued growth and survival. Harnessing the power of screen-based functional genomics techniques to interrogate the relative importance of these pathways in a multitude of settings will be an important component of the development of effective personalized medicine.
MATERIALS AND METHODS
Library construction and validation
To construct the library, cDNA templates for each construct were obtained, barcoded, and cloned into a common expression vector using the Gateway system. Using polymerase chain reaction (PCR), barcodes and relevant Gateway cloning sites were added to each cDNA sequence. The attB1 primers contained the attB1 sequence, a 4-nucleotide (nt) barcode assigned to individual constructs followed by a 14-nt common linker sequence containing a Kozak sequence and ~21 nt of the open reading frame (ORF) of interest. The reverse, attB2 primer contained the attB2 sequence, a C-terminal V5 epitope tag if the cDNA was lacking an epitope tag and the final 21 nt of the ORF (no stop codon) or only the attB2 sequence and the final 24 nt (including the stop codon) if no tag was desired. Where applicable, both tagged and untagged versions of each ORF were functionally validated (Table 1). The resultant PCR fragment was gel purified and transferred to the entry vector, pDONR223, using the BP recombination reaction (Invitrogen). The generated entry clones were sequence verified with the primers, M13-F and M13-R. We ensured the correct sequence of the entire ORF, proper integration of the barcode sequence and the in-frame translation of all elements. If the proper mutations were not present in the original cDNA (as was the case for SMO and LATS2), they were added to the ORF in the entry clone using the QuikChange II XL Site Directed Mutagenesis Kit (Agilent). All mutations were sequence verified. Verified entry clones were submitted to the LR reaction using LR clonase (Invitrogen) to transfer the ORF to a suitable expression vector. All ORFs were transferred to the vector, pcw107-V5, which contains the promoter, PKG, to achieve physiological-like expression levels of the ORF. Expression vectors were fully sequenced with the primers, PGK-F and WPRE-R, and ORF specific internal primers as necessary.
In order to functionally validate the members of the library, lentivirus particles containing the clones were made using a three plasmid system of the expression clone, VSV-G and δVPR, to transfect 293T cells as previously described [23]. Viral particles were titered by limiting dilution in UACC-62 cells. To measure expression of each ORF, immunoblot of the epitope tag, V5, or the encoded protein itself, was performed on whole cell lysates from 293T cells stably and individually expressing each ORF. The pathway activating ability of the constructs was validated using the assays described in Table 1 after puromycin selection of transduced 293T cells. All infections were done by adding a 1:10 to 1:20 dilution of lentivirus particle-containing culture media and 7.5μg/mL polybrene to 293T cells in 6-well plates. Plates were centrifuged at 1200xg for 1 hr at 37°C. After 24 hours, 2μg/mL puromycin was added and cells were incubated for an additional 48 hours before validation assays were performed.
Primary screens
A pooled lentiviral library was constructed by titering all pathway activating constructs and control constructs individually and then combining them for approximate equal representation. The pooled library was aliquoted and stored at −80°C for use in all primary screens. To screen a particular drug/cell line combination, cells were seeded at 500,000 cells per well in 6-well plates and infected at a multiplicity of infection (MOI) of 0.3 as described above. This MOI predicts that the majority of cells will only receive one construct. After puromycin selection, the surviving cells were lifted and divided into 7 equal populations. One group, representing the t = 0 infected pool, was frozen at −80°C. The other six were plated in 6-well plates. Three wells received drug in the range of GI20 to GI80 while the other received diluent only (typically, dimethyl sulfoxide (DMSO)). Drug, vehicle and media were changed every 3 days for 2 to 4 weeks. Cells were split at 1:10 as they became confluent. Samples were then trypsinized and washed and genomic DNA was collected using the Qiagen DNeasy Blood and Tissue Kit.
In order to prepare samples for Illumina Sequencing, we PCR amplified construct barcodes using a common P5 Illumina adapter primer, PGK-Illumina-F, and a unique P7 Illumina barcoded adapter primer, P7- Illumina-RIP-Index-X (where X is a unique numerical identifier). Each screen replicate was paired with a unique P7-reverse primer containing a 6-nt index primer in order to allow sequencing of several samples per lane of Illumina sequencing. Producing a 250-nt fragment, the P5-forward primer is specific for the PGK primer and the P7-reverse primer binds the ATG region of the ORF. These sequences flank the ORF specific barcode. The DNA fragment were purified and size confirmed by 2% agarose gel electrophoresis (Qiagen). Normalized sample pools were submitted to sequencing using band intensities to quantify relative sample amounts. Targeting the region of DNA upstream of the 4-nt ORF barcode, the Illumina Sequencing Primer, ISP, was used to generate a 27-nt Illumina HiSeq read containing the 4-nt barcode, a 17-nt linker sequence and ATG site and the 6-nt index primer. For technical replication, each sample was prepared with two different P7-reverse primers. We found that both the technical and biological replicates yielded comparable data with r2 = 0.9. For each unique sequence representing the 4-nt ORF barcode and the 6-nt index primer, the number of reads was counted and the fractional representation in the screen was determined by dividing by the total number of reads of each index primer (i.e. total number of reads for each sample). Fractional representation in each technical and biological replicate was then calculated and averaged. Finally, the fractional representation of each drug treated sample was normalized to the fractional representation of each ORF in the vehicle treated samples. To identify hits, constructs whose representation was enriched in drug treated versus vehicle treated samples were identified. The cutoff for hit calling was defined as the presence of at least one activating construct per pathway conferring greater than 50% enrichment above controls across at least two drug concentrations, as constructs scoring at or above this level were found to be reliably validated in secondary, eight-point growth inhibition 50% (GI50 assays).
Acknowledgments
The Gateway vectors, pDONR223 and pcw107, were obtained from J. Doench (Broad Institute). We thank Kathleen Ottina, Colin Martz, and David Sabatini for critical material and intellectual contributions to the pathway activating screening methodology.
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
GRANT SUPPORT
Our work is supported by the Duke University School of Medicine, the NIH Building Interdisciplinary Research Careers in Women's Health Program (K12HD043446), the Liz Tilberis Award from the Ovarian Cancer Research Fund Alliance, a research award from Golfers Against Cancer, a Stewart Trust Fellowship, a V Scholar Award from the V Foundation for Cancer Research, a DoD Breast Cancer Research Program Breakthrough Award (BC151664), and NIH award R01CA207083, all to K.C.W.
REFERENCES
- 1.International Cancer Genome C, Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, Bernabe RR, Bhan MK, Calvo F, Eerola I, Gerhard DS, Guttmacher A, Guyer M, Hemsley FM, Jennings JL, Kerr D, et al. International network of cancer genome projects. Nature. 2010;464(7291):993–998. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153(1):17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 4.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, Kris M, Varmus H. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101(36):13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 6.Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, Cortes ML, Fernandez-Lopez JC, Peng S, Ardlie KG, Auclair D, Bautista-Pina V, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486(7403):405–409. doi: 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van ‘t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 9.Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45(10):1127–1133. doi: 10.1038/ng.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brummelkamp TR, Bernards R. New tools for functional mammalian cancer genetics. Nat Rev Cancer. 2003;3(10):781–789. doi: 10.1038/nrc1191. [DOI] [PubMed] [Google Scholar]
- 11.Kittler R, Pelletier L, Heninger AK, Slabicki M, Theis M, Miroslaw L, Poser I, Lawo S, Grabner H, Kozak K, Wagner J, Surendranath V, Richter C, Bowen W, Jackson AL, Habermann B, et al. Genome-scale RNAi profiling of cell division in human tissue culture cells. Nat Cell Biol. 2007;9(12):1401–1412. doi: 10.1038/ncb1659. [DOI] [PubMed] [Google Scholar]
- 12.Mukherji M, Bell R, Supekova L, Wang Y, Orth AP, Batalov S, Miraglia L, Huesken D, Lange J, Martin C, Sahasrabudhe S, Reinhardt M, Natt F, Hall J, Mickanin C, Labow M, et al. Genome-wide functional analysis of human cell-cycle regulators. Proc Natl Acad Sci U S A. 2006;103(40):14819–14824. doi: 10.1073/pnas.0604320103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tyner JW, Deininger MW, Loriaux MM, Chang BH, Gotlib JR, Willis SG, Erickson H, Kovacsovics T, O'Hare T, Heinrich MC, Druker BJ. RNAi screen for rapid therapeutic target identification in leukemia patients. Proc Natl Acad Sci U S A. 2009;106(21):8695–8700. doi: 10.1073/pnas.0903233106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mullenders J, Bernards R. Loss-of-function genetic screens as a tool to improve the diagnosis and treatment of cancer. Oncogene. 2009;28(50):4409–4420. doi: 10.1038/onc.2009.295. [DOI] [PubMed] [Google Scholar]
- 15.Berns K, Hijmans EM, Mullenders J, Brummelkamp TR, Velds A, Heimerikx M, Kerkhoven RM, Madiredjo M, Nijkamp W, Weigelt B, Agami R, Ge W, Cavet G, Linsley PS, Beijersbergen RL, Bernards R. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature. 2004;428(6981):431–437. doi: 10.1038/nature02371. [DOI] [PubMed] [Google Scholar]
- 16.Gobeil S, Zhu X, Doillon CJ, Green MR. A genome- wide shRNA screen identifies GAS1 as a novel melanoma metastasis suppressor gene. Genes Dev. 2008;22(21):2932–2940. doi: 10.1101/gad.1714608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Firestein R, Bass AJ, Kim SY, Dunn IF, Silver SJ, Guney I, Freed E, Ligon AH, Vena N, Ogino S, Chheda MG, Tamayo P, Finn S, Shrestha Y, Boehm JS, Jain S, et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature. 2008;455(7212):547–551. doi: 10.1038/nature07179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF, Sjostrom SK, Garraway LA, Weremowicz S, Richardson AL, Greulich H, Stewart CJ, Mulvey LA, Shen RR, Ambrogio L, Hirozane-Kishikawa T, et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129(6):1065–1079. doi: 10.1016/j.cell.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 19.De S, Cipriano R, Jackson MW, Stark GR. Overexpression of kinesins mediates docetaxel resistance in breast cancer cells. Cancer research. 2009;69(20):8035–8042. doi: 10.1158/0008-5472.CAN-09-1224. [DOI] [PubMed] [Google Scholar]
- 20.Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, Caponigro G, Hieronymus H, Murray RR, Salehi-Ashtiani K, Hill DE, Vidal M, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468(7326):968–972. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johannessen CM, Johnson LA, Piccioni F, Townes A, Frederick DT, Donahue MK, Narayan R, Flaherty KT, Wargo JA, Root DE, Garraway LA. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature. 2013;504(7478):138–142. doi: 10.1038/nature12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wood KC, Konieczkowski DJ, Johannessen CM, Boehm JS, Tamayo P, Botvinnik OB, Mesirov JP, Hahn WC, Root DE, Garraway LA, Sabatini DM. MicroSCALE screening reveals genetic modifiers of therapeutic response in melanoma. Science signaling. 2012;5:rs4. doi: 10.1126/scisignal.2002612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, Lubonja R, Thomas SR, Alkan O, Bhimdi T, Green TM, Johannessen CM, Silver SJ, Nguyen C, Murray RR, Hieronymus H, et al. A public genome-scale lentiviral expression library of human ORFs. Nat Methods. 2011;8(8):659–661. doi: 10.1038/nmeth.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muellner MK, Uras IZ, Gapp BV, Kerzendorfer C, Smida M, Lechtermann H, Craig-Mueller N, Colinge J, Duernberger G, Nijman SM. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat Chem Biol. 2011;7(11):787–793. doi: 10.1038/nchembio.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martins MM, Zhou AY, Corella A, Horiuchi D, Yau C, Rakshandehroo T, Gordan JD, Levin RS, Johnson J, Jascur J, Shales M, Sorrentino A, Cheah J, Clemons PA, Shamji AF, Schreiber SL, et al. Linking tumor mutations to drug responses via a quantitative chemical-genetic interaction map. Cancer discovery. 2015;5(2):154–167. doi: 10.1158/2159-8290.CD-14-0552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilson FH, Johannessen CM, Piccioni F, Tamayo P, Kim JW, Van Allen EM, Corsello SM, Capelletti M, Calles A, Butaney M, Sharifnia T, Gabriel SB, Mesirov JP, Hahn WC, Engelman JA, Meyerson M, et al. A functional landscape of resistance to ALK inhibition in lung cancer. Cancer Cell. 2015;27(3):397–408. doi: 10.1016/j.ccell.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, Qi LS, Kampmann M, Weissman JS. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 2014;159(3):647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zalatan JG, Lee ME, Almeida R, Gilbert LA, Whitehead EH, La Russa M, Tsai JC, Weissman JS, Dueber JE, Qi LS, Lim WA. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell. 2015;160(1-2):339–350. doi: 10.1016/j.cell.2014.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, Nureki O, Zhang F. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517(7536):583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483(7387):100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 31.Gregory MA, Phang TL, Neviani P, Alvarez-Calderon F, Eide CA, O'Hare T, Zaberezhnyy V, Williams RT, Druker BJ, Perrotti D, Degregori J. Wnt/Ca2+/NFAT signaling maintains survival of Ph+ leukemia cells upon inhibition of Bcr-Abl. Cancer Cell. 2010;18(1):74–87. doi: 10.1016/j.ccr.2010.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whitehurst AW, Bodemann BO, Cardenas J, Ferguson D, Girard L, Peyton M, Minna JD, Michnoff C, Hao W, Roth MG, Xie XJ, White MA. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature. 2007;446(7137):815–819. doi: 10.1038/nature05697. [DOI] [PubMed] [Google Scholar]
- 33.Turner NC, Lord CJ, Iorns E, Brough R, Swift S, Elliott R, Rayter S, Tutt AN, Ashworth A. A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. 2008;27(9):1368–1377. doi: 10.1038/emboj.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fotheringham S, Epping MT, Stimson L, Khan O, Wood V, Pezzella F, Bernards R, La Thangue NB. Genome-wide loss-of-function screen reveals an important role for the proteasome in HDAC inhibitor-induced apoptosis. Cancer Cell. 2009;15(1):57–66. doi: 10.1016/j.ccr.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 35.Singleton KR, Hinz TK, Kleczko EK, Marek LA, Kwak J, Harp T, Kim J, Tan AC, Heasley LE. Kinome RNAi Screens Reveal Synergistic Targeting of MTOR and FGFR1 Pathways for Treatment of Lung Cancer and HNSCC. Cancer research. 2015;75(20):4398–4406. doi: 10.1158/0008-5472.CAN-15-0509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singleton KR, Kim J, Hinz TK, Marek LA, Casas-Selves M, Hatheway C, Tan AC, DeGregori J, Heasley LE. A receptor tyrosine kinase network composed of fibroblast growth factor receptors, epidermal growth factor receptor, v-erb-b2 erythroblastic leukemia viral oncogene homolog 2, and hepatocyte growth factor receptor drives growth and survival of head and neck squamous carcinoma cell lines. Molecular pharmacology. 2013;83(4):882–893. doi: 10.1124/mol.112.084111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Casas-Selves M, Kim J, Zhang Z, Helfrich BA, Gao D, Porter CC, Scarborough HA, Bunn PA, Jr, Chan DC, Tan AC, DeGregori J. Tankyrase and the canonical Wnt pathway protect lung cancer cells from EGFR inhibition. Cancer research. 2012;72(16):4154–4164. doi: 10.1158/0008-5472.CAN-11-2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, Chodon T, Nelson SF, McArthur G, Sosman JA, Ribas A, Lo RS. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, Kehoe SM, Johannessen CM, Macconaill LE, Hahn WC, Meyerson M, Garraway LA. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29(22):3085–3096. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, Salton M, Dahlman KB, Tadi M, Wargo JA, Flaherty KT, Kelley MC, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480(7377):387–390. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Montagut C, Sharma SV, Shioda T, McDermott U, Ulman M, Ulkus LE, Dias-Santagata D, Stubbs H, Lee DY, Singh A, Drew L, Haber DA, Settleman J. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer research. 2008;68(12):4853–4861. doi: 10.1158/0008-5472.CAN-07-6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu XW, Gimotty PA, Kee D, Santiago-Walker AE, Letrero R, D'Andrea K, Pushparajan A, Hayden JE, Brown KD, et al. Acquired Resistance to BRAF Inhibitors Mediated by a RAF Kinase Switch in Melanoma Can Be Overcome by Cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18(6):683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paraiso KH, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC, Wood E, Fedorenko IV, Sondak VK, Anderson AR, Ribas A, Palma MD, Nathanson KL, Koomen JM, Messina JL, Smalley KS. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer research. 2011;71(7):2750–2760. doi: 10.1158/0008-5472.CAN-10-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shi H, Hong A, Kong X, Koya RC, Song C, Moriceau G, Hugo W, Yu CC, Ng C, Chodon T, Scolyer RA, Kefford RF, Ribas A, Long GV, Lo RS. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov. 2014;4(1):69–79. doi: 10.1158/2159-8290.CD-13-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weinberg RA. The biology of cancer. New York: Garland Science, Taylor & Francis Group; 2014. [Google Scholar]
- 46.Martz CA, Ottina KA, Singleton KR, Jasper JS, Wardell SE, Peraza-Penton A, Anderson GR, Winter PS, Wang T, Alley HM, Kwong LN, Cooper ZA, Tetzlaff M, Chen PL, Rathmell JC, Flaherty KT, et al. Systematic identification of signaling pathways with potential to confer anticancer drug resistance. Science signaling. 2014;7:ra121. doi: 10.1126/scisignal.aaa1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Winter PS, Sarosiek KA, Lin KH, Meggendorfer M, Schnittger S, Letai A, Wood KC. RAS signaling promotes resistance to JAK inhibitors by suppressing BAD- mediated apoptosis. Science signaling. 2014;7(357):ra122. doi: 10.1126/scisignal.2005301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin KH, Winter PS, Xie A, Roth C, Martz CA, Stein EM, Anderson GR, Tingley JP, Wood KC. Targeting MCL-1/BCL-XL Forestalls the Acquisition of Resistance to ABT-199 in Acute Myeloid Leukemia. Sci Rep. 2016;6:27696. doi: 10.1038/srep27696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Misale S, Bozic I, Tong J, Peraza-Penton A, Lallo A, Baldi F, Lin KH, Di Nicolantonio F, Nowak MA, Zhang L, Wood KC, Bardelli A. Vertical suppression of the EGFR pathway delays onset of resistance in colorectal cancer models. Nature Communications. 2015;6:8305–8314. doi: 10.1038/ncomms9305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pardanani A, Gotlib JR, Jamieson C, Cortes JE, Talpaz M, Stone RM, Silverman MH, Gilliland DG, Shorr J, Tefferi A. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J Clin Oncol. 2011;29(7):789–796. doi: 10.1200/JCO.2010.32.8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, Estrov Z, Fridman JS, Bradley EC, Erickson-Viitanen S, Vaddi K, Levy R, Tefferi A. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363(12):1117–1127. doi: 10.1056/NEJMoa1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 53.Meacham CE, Lawton LN, Soto-Feliciano YM, Pritchard JR, Joughin BA, Ehrenberger T, Fenouille N, Zuber J, Williams RT, Young RA, Hemann MT. A genome-scale in vivo loss-of-function screen identifies Phf6 as a lineage- specific regulator of leukemia cell growth. Genes Dev. 2015;29(5):483–488. doi: 10.1101/gad.254151.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pallasch CP, Leskov I, Braun CJ, Vorholt D, Drake A, Soto- Feliciano YM, Bent EH, Schwamb J, Iliopoulou B, Kutsch N, van Rooijen N, Frenzel LP, Wendtner CM, Heukamp L, Kreuzer KA, Hallek M, et al. Sensitizing protective tumor microenvironments to antibody-mediated therapy. Cell. 2014;156(3):590–602. doi: 10.1016/j.cell.2013.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, Chen WW, Barrett FG, Stransky N, Tsun ZY, Cowley GS, Barretina J, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476(7360):346–350. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]