

Abstract
Neuraminidase deficiency (mucolipidosis I, sialidosis types I and II, cherry-red spot myoclonus syndrome) is a lysosomal storage disorder with an expanding clinical phenotype. Here, we report the striking diagnostic history of late-onset neuraminidase deficiency in two sisters, currently aged 14 (patient 1) and 15 (patient 2).
Patient 1 was referred for evaluation of her vision after a traffic accident. During this examination, nummular cataract, macular cherry-red spot, and optic nerve atrophy were seen. Furthermore, tremors were noticed in her arms and legs. This combination suggested a lysosomal storage disorder. Her family history revealed an older sister, patient 2, who had a long history of unexplained neurologic symptoms; she was under unsuccessful treatment for conversion disorder. Patient 2 showed identical ophthalmological findings. In retrospect, she had presented with avascular osteonecrosis of the right femur head at age 9.
Urinary oligosaccharide patterns and enzyme activity revealed neuraminidase deficiency in both patients. Urinary-bound sialic acid levels were normal. Sequencing of NEU1 demonstrated two known compound heterozygous mutations (c.1195_1200dup p.His399_Tyr400dup; c.679G>A, p.Glu227Arg).
The substantial time window between onset of typical symptoms and diagnosis in patient 2 suggests inadequate awareness of lysosomal storage disorders among clinicians. Of special interest is the observation that normal urinary sialic acid levels do not exclude neuraminidase deficiency. Urinary oligosaccharide screening is essential to diagnosis in such cases. In addition, patient 2 is the fourth case in the literature with a history of femur head necrosis. Bone defects might therefore be an early manifestation of late-onset neuraminidase deficiency.
Introduction
Neuraminidase deficiency (mucolipidosis I, sialidosis types I and II, sialidase deficiency, cherry-red spot myoclonus syndrome, OMIM 256550) is an autosomal recessive lysosomal storage disorder (LSD) caused by mutations in the NEU1 gene (Bonten et al. 2000). NEU1 mutations result in diminished function of the enzyme exo-α-sialidase (EC 3.2.1.18), which leads to the buildup of sialic acid-containing glycoconjugates in tissues and oligosacchariduria. Based on the clinical presentation, two extremes of the phenotypic spectrum can be distinguished. The severe, early onset variant (type II) is characterized by coarse facial features, skeletal dysplasia, severe ataxia, and mental retardation (Lowden and O’Brien 1979). The late onset variant (type I) generally presents between the age of 8 and 25 with progressive myoclonus, ataxia, and visual impairment. A macular cherry-red spot is often seen. Over time, some patients develop seizures and mild cognitive impairment. The clinical spectrum of this disorder is still expanding as genome-wide screening now allows identification of patients with a mild or atypical phenotype (Canafoglia et al. 2014).
Here, we report the diagnosis of late-onset neuraminidase deficiency in two sisters, currently aged 14 (patient 1) and 15 (patient 2). We describe their remarkable diagnostic history, which illustrates pitfalls in the diagnostic process and confirms osteonecrosis as an early manifestation of late-onset neuraminidase deficiency.
Case Reports
Patient 1, the index case, is a 14-year-old girl who first experienced loss of visual acuity at the age of 12. Specifically, impaired perception of contrast and depth led to frequent falls and recurrent trauma. At 13, her worsening eyesight led to a traffic accident resulting in fractures of the lower leg. Ensuing ophthalmological examination revealed impaired best-corrected visual acuity of 0.5, poor stereoscopic vision, macular cherry-red spot (Fig. 1a), partial paleness of the optic disk (Fig. 1a), nummular cataract (Fig. 1b), and upbeat nystagmus in both eyes. Optical coherence tomography of the macula showed clearly hyperreflective and thickened ganglion cell layer of the macula. Based on her history and clinical symptoms, a lysosomal storage disorder was suspected.
Fig. 1.
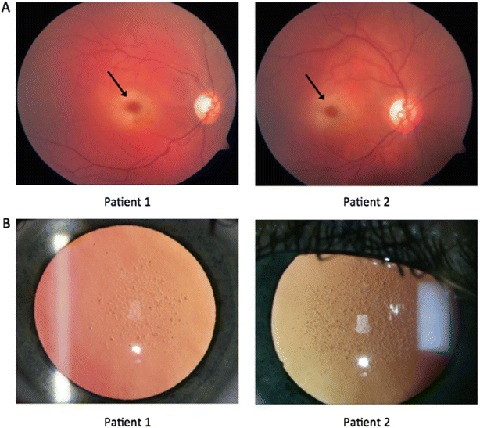
(a) Fundoscopy in patients 1 and 2, at the age of 13 and 14, respectively, showed bilateral (only the right eye is shown) cherry-red spot in the macula (arrow) and atrophy of the optic nerve. (b) Slit lamp examination revealed bilateral nummular cataract in both patients
Her family history revealed an older sister, patient 2 (now 15 years old), who was under treatment for conversion disorder. Subsequent examination of patient 2 showed ophthalmological findings identical to her sister (Fig. 1a, b). Her best-corrected visual acuity was limited to 0.3 and 0.5. In retrospect, she had presented with avascular osteonecrosis of the right femur head at the age of 9. Spontaneous revascularization occurred gradually, resulting in mild femur head deformation only (Fig. 2). Her hip pain, weakness of the right leg, unsteady gait, and tiredness, however, worsened gradually. From the age of 13, she experienced progressive loss of visual acuity because of cataract and myopia. Concurrently, she developed mild ataxia and high-frequency intention tremor, affecting all four limbs. A cerebellar cause was suspected but could not be confirmed by neuroimaging or laboratory testing. In the absence of a somatic explanation, her neurologic symptoms were considered to be of psychosomatic etiology. The recent divorce of her parents and her history of osteonecrosis were together considered etiological for her movement disorder. Treatment for conversion disorder was started but proved ineffective. Within a year, walking without support and writing by hand became impossible because of worsening ataxia and myoclonus. Strikingly, a metabolic cause of her symptoms was only suspected upon mentioned ophthalmological examination of her younger sister. At evaluation in our center, neurologic examination of patient 2 showed downbeat nystagmus, severe ataxia, severe intention tremor, and sporadic myoclonus of the limbs and eyelids. The combination of typical ocular and neurologic signs prompted further investigation in both patients.
Fig. 2.
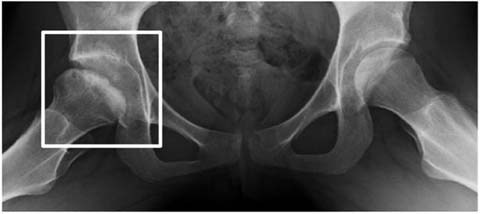
X-ray of the pelvis in patient 2 at age 12 showed mild flattening and mild sclerosis of the right femur head as a result of avascular osteonecrosis (Legg–Calvé–Perthes syndrome) that started at age 9. Revascularization occurred spontaneously within 3 years, but pain and unsteadiness of the right hip remained
Biochemical Phenotype and Genotype
Brain MRI showed no intracranial pathology in either. Urinary-bound sialic acid levels measured by mass spectrometry were in the normal range (57 and 36 mmol/creatinine M for patient 1 and 2, respectively; normal range: 1.5–57) (van der Ham et al. 2007). However, urinary analysis by thin-layer chromatography revealed oligosaccharide patterns characteristic for (galacto)sialidosis in both patients (Fig. 3) (Holmes and O’Brien 1979). This finding prompted enzyme activity testing in cultured fibroblast. Activity of N-acetyl-neuraminidase was severely impaired (0.5 and 0.4 mmol/mg/h for patient 1 and 2, respectively; normal range: 15–45). Galactosialidosis was excluded by normal galactosidase activity (1,246 and 1,257 nmol/mg/h for patient 1 and 2, respectively; normal range: 600–1,650). Targeted next-generation sequencing (Nijman et al. 2014) of NEU1 revealed two previously described compound heterozygous mutations (c.1195_1200dup p.His399_Tyr400dup; c.679G>A, p.Glu227Arg) in both patients, confirming neuraminidase deficiency (Bonten et al. 2000; Lukong et al. 2000). Nomenclature is according to HGVS guidelines and is based upon transcript NM 000434.3.
Fig. 3.
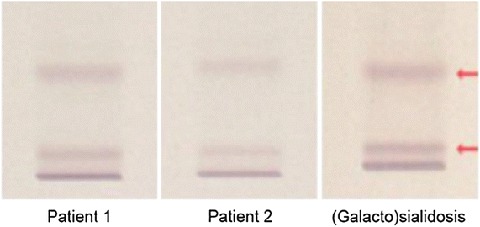
Thin-layer chromatography of urinary oligosaccharides showed patterns compatible with galactosialidosis or neuraminidase deficiency (sialidosis) in both patients (Holmes and O’Brien 1979). The third panel serves as pathologic standard for both disorders. This method cannot distinguish galactosialidosis from neuraminidase deficiency. Enzyme activity testing in cultured fibroblasts is required for diagnosis
Clinical Management
Our current management focuses on adjustment and rehabilitation. As symptoms worsen during menstruation, both patients receive continuous oral contraception. Furthermore, both patients use clonazepam to reduce myoclonic jerks. No myoclonic seizures have occurred in either.
At present, both patients suffer from worsening eyesight, ataxia, tremor, and action myoclonus, threatening independent functioning.
Discussion
The combination of gait disturbance and loss of vision is a typical presentation for late-onset (type I) neuraminidase deficiency. Despite this typical presentation, the diagnostic delay in patient 2 was long and might have been even longer if distinctive ophthalmological signs such as macular cherry-red spot had not been found in the index patient. During these years, patient 2 was assessed by multiple specialists and underwent therapy for conversion disorder without success. Her diagnostic history shows that accepting a psychosomatic explanation for neurologic symptoms can postpone identification of a possible somatic etiology. Besides, it suggests inadequate awareness of metabolic disorders among clinicians.
Thin-layer chromatography of urinary oligosaccharides can be labor intensive (van der Ham et al. 2007) and difficult to interpret (Xia et al. 2013). Measurement of urinary sialic acid levels could be a convenient alternative. However, urinary sialic acid levels were not elevated in our patients, which has been reported before (van der Ham et al. 2007; Canafoglia et al. 2014). This underlines the fact that neuraminidase deficiency should be suspected even if sialic acid excretion in the urine is normal. In our patients, thin-layer chromatography of urinary oligosaccharides did show an abnormal pattern, which was key to diagnosis. This traditional diagnostic test should thus remain part of screening panels for storage disorders.
Avascular osteonecrosis is well described in a variety of LSDs, including Gaucher’s disease (Aldenhoven et al. 2009) and to a lesser extent Fabry disease (Lien and Lai 2005; Sacre et al. 2010) and Niemann-Pick type B (Wasserstein et al. 2013). Thus far, it has not been considered a clinical sign of neuraminidase deficiency. Recent reports describe three neuraminidase-deficient patients with a history of osteonecrosis of the femur (Urbanski et al. 2014; Canafoglia et al. 2014). Patient 2 also presented with avascular femoral head osteonecrosis. Together this indicates that osteonecrosis can be an early clinical sign of late-onset neuraminidase deficiency.
Synopsis
Diagnosis of neuraminidase deficiency in two sisters, first misdiagnosed with conversion disorder, shows importance of urinary oligosaccharide screening when sialic acid excretion is normal and suggests osteonecrosis as an early symptom.
Compliance with Ethics Guidelines
Conflict of Interest
Imre Schene, Viera Ayuso, Koen van Gassen, Monique de Sain- van der Velden, Inge Cuppen, Peter M. van Hasselt, and Gepke Visser declare that they have no conflict of interest.
This article does not contain any studies with human or animal subjects performed by any of the authors.
Details of the Contributions of Individual Authors
Imre Schene drafted the first manuscript.
Viera Kalinina Ayuso is the ophthalmologist who executed the ophthalmological examinations, contributed Fig. 1, and revised the manuscript.
Koen van Gassen executed the genetic investigation that demonstrated the compound heterozygous mutations in NEU1 and revised the manuscript.
Monique de Sain- van der Velden executed the metabolic investigations, contributed Fig. 3 and revised the manuscript.
Inge Cuppen is the pediatric neurologist who executed the neurological examination in our center and revised the manuscript.
Peter M. van Hasselt contributed to the follow-up of the patients and revised the manuscript.
Gepke Visser is the metabolic pediatrician of the patients and coordinated and revised the manuscript.
Footnotes
Competing interests: None declared
References
- Aldenhoven M, Sakkers RJB, Boelens J, de Koning TJ, Wulffraat NM. Review: musculoskeletal manifestations of lysosomal storage disorders. Ann Rheum Dis. 2009;68:1659–1665. doi: 10.1136/ard.2008.095315. [DOI] [PubMed] [Google Scholar]
- Bonten EJ, Arts WF, Beck M, et al. Novel mutations in lysosomal neuraminidase identify functional domains and determine clinical severity in sialidosis. Hum Mol Genet. 2000;9:2715–2725. doi: 10.1093/hmg/9.18.2715. [DOI] [PubMed] [Google Scholar]
- Canafoglia L, Robbiano A, Pareyson D, et al. Expanding sialidosis spectrum by genome-wide screening: NEU1 mutations in adult-onset myoclonus. Neurology. 2014;82:2003–2006. doi: 10.1212/WNL.0000000000000482. [DOI] [PubMed] [Google Scholar]
- Holmes EW, O’Brien JS. Separation of glycoprotein-derived oligosaccharides by thin-layer chromatography. Anal Biochem. 1979;93:167–170. doi: 10.1016/S0003-2697(79)80131-1. [DOI] [PubMed] [Google Scholar]
- Lien YH, Lai LW. Bilateral femoral head and distal tibial osteonecrosis in a patient with Fabry disease. Am J Orthop. 2005;34:192–194. [PubMed] [Google Scholar]
- Lowden JA, O’Brien JS. Sialidosis: a review of human neuraminidase deficiency. Am J Hum Genet. 1979;31:1–18. [PMC free article] [PubMed] [Google Scholar]
- Lukong KE, Elsliger MA, Chang Y, et al. Characterization of the sialidase molecular defects in sialidosis patients suggests the structural organization of the lysosomal multienzyme complex. Hum Mol Genet. 2000;9:1075–1085. doi: 10.1093/hmg/9.7.1075. [DOI] [PubMed] [Google Scholar]
- Nijman IJ, van Montfrans JM, Hoogstraat M, et al. Targeted next-generation sequencing: a novel diagnostic tool for primary immunodeficiencies. J Allergy Clin Immunol. 2014;133:529–534. doi: 10.1016/j.jaci.2013.08.032. [DOI] [PubMed] [Google Scholar]
- Sacre K, Lidove O, Giroux Leprieur B, Ouali N, Laganier J, Caillaud C, Papo T. Bone and joint involvement in Fabry disease. Scand J Rheumatol. 2010;39:171–174. doi: 10.3109/03009740903270631. [DOI] [PubMed] [Google Scholar]
- Urbanski G, Bekri S, Barth M, Verny C, Lavigne C. A case of type I sialidosis with osteonecrosis revealing a new mutation in NEU1. J Inborn Errors Metab Screening. 2014;2:1–3. doi: 10.1177/2326409814543468. [DOI] [Google Scholar]
- van der Ham M, Prinsen BH, Huijmans JG, et al. Quantification of free and total sialic acid excretion by LC-MS/MS. J Chromatogr. 2007;848:251–257. doi: 10.1016/j.chroma.2006.12.038. [DOI] [PubMed] [Google Scholar]
- Wasserstein M, Godbold J, McGovern MM. Skeletal manifestations in pediatric and adult patients with Niemann Pick disease type B. J Inherit Metab Dis. 2013;36:123–127. doi: 10.1007/s10545-012-9503-0. [DOI] [PubMed] [Google Scholar]
- Xia B, Asif G, Arthur L, Pervaiz MA, Li X, Liu R, Cummings RD, He M. Oligosaccharide analysis in urine by maldi-tof mass spectrometry for the diagnosis of lysosomal storage diseases. Clin Chem. 2013;59:1357–1368. doi: 10.1373/clinchem.2012.201053. [DOI] [PubMed] [Google Scholar]