

Abstract
Background
The ability to establish root nodule symbioses is restricted to four different plant orders. Soil actinobacteria of the genus Frankia can establish a symbiotic relationship with a diverse group of plants within eight different families from three different orders, the Cucurbitales, Fagales and Rosales. Phylogenetically, Frankia strains can be divided into four clusters, three of which (I, II, III) contain symbiotic strains. Members of Cluster II nodulate the broadest range of host plants with species from four families from two different orders, growing on six continents. Two Cluster II genomes were sequenced thus far, both from Asia.
Results
In this paper we present the first Frankia cluster II genome from North America (California), Dg2, which represents a metagenome of two major and one minor strains. A phylogenetic analysis of the core genomes of 16 Frankia strains shows that Cluster II the ancestral group in the genus, also ancestral to the non-symbiotic Cluster IV. Dg2 contains the canonical nod genes nodABC for the production of lipochitooligosaccharide Nod factors, but also two copies of the sulfotransferase gene nodH. In rhizobial systems, sulfation of Nod factors affects their host specificity and their stability.
Conclusions
A comparison with the nod gene region of the previously sequenced Dg1 genome from a Cluster II strain from Pakistan shows that the common ancestor of both strains should have contained nodABC and nodH. Phylogenetically, Dg2 NodH proteins are sister to rhizobial NodH proteins. A glnA-based phylogenetic analysis of all Cluster II strains sampled thus far supports the hypothesis that Cluster II Frankia strains came to North America with Datisca glomerata following the Madrean-Tethyan pattern.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-3140-1) contains supplementary material, which is available to authorized users.
Keywords: Frankia, Uncultured, Datisca glomerata, nodABC, nodH, mce
Background
Nitrogen is the element that most often limits plant growth. Some prokaryotes can form the enzyme complex nitrogenase to reduce atmospheric dinitrogen and introduce it into the biosphere. Plants can only access this source of nitrogen by entering symbioses with nitrogen-fixing prokaryotes. Root nodule symbioses, where bacteria fix nitrogen while being hosted inside plant cells within special organs, the root nodules, belong to the most efficient of such symbioses. All plants able to enter root nodule symbioses belong to a single dicotyledonous clade, known as the nitrogen-fixing clade made up from the orders Fabales, Fagales, Rosales and Cucurbitales [1, 2]. Two groups of nitrogen-fixing soil bacteria can induce root nodules: rhizobia, a polyphyletic group of proteobacteria, enter symbioses with legumes (Fagales) and with one non-legume genus, Parasponia (Cannabaceae, Rosales). Actinobacteria of the genus Frankia can induce nodules on a diverse group of plants from 23 genera from eight different families belonging to the orders Fagales (Betulaceae, Casuarinaceae and Myricaceae), Rosales (Elaeagnaceae, Rhamnaceae and Rosaceae) and Cucurbitales (Coriariaceae and Datiscaceae). The distribution of symbiotic species within this clade suggests a common origin of the predisposition to evolve root nodule symbioses assumed to have arisen only once ca. 100 million years ago [1, 2]. Subsequently nitrogen-fixing root nodule symbioses evolved several times independently among the plants with the common predisposition, their independent origins reflected by differences in infection pathways as well as nodule structure and physiology [3].
Based on 16S rDNA phylogeny, the genus Frankia has been divided into four clusters, three of which contain symbiotic strains [4]. The non-symbiotic cluster (Cluster IV) represents a heterologous, very diverse group of strains that were isolated from nodules but cannot induce nodule formation. This group is closely related to rhizosphere strains previously detected only by direct amplification of 16S rDNA [5]. The three symbiotic clusters more or less correspond to host-specificity groups. The members of Cluster I can nodulate species of the three actinorhizal families of the order Fagales except for the genus Gymnostoma (Casuarinaceae). Cluster III strains nodulate the actinorhizal Elaeagnaceae (Rosales), and all actinorhizal genera of the Rhamnaceae (Rosales) except for the genus Ceanothus, and with the genera Gymnostoma (Casuarinaceae) and Morella (Myricaceae, Fagales). Cluster II strains form nitrogen-fixing root nodule symbioses with members of the families Coriariaceae and Datiscaceae (Cucurbitales) as well as all the actinorhizal members of the Rosaceae (i.e., the Dryadoideae tribe) and Ceanothus (Rosales). In contrast with strains of Clusters I, III and IV, so far only one strain of Cluster II could be cultured, and this strain represents an alkaliphile [6].
Analysis of Frankia genomes has shown a surprising variation with regard to genome sizes. Strong differences exist within Cluster I, 5.0 − 5.6 Mb for Casuarina-infective strains; [7–10] and ca. 7.5 − 7.7 Mb for the others [7, 11–14]). So far, Cluster II genomes are consistently in the lower size range (5.3–5.6 Mb; [6, 15]). Cluster III genomes range from 7.6 to 10.5 Mb [7, 16–18], similar to Cluster IV genomes (6.9 – 10 Mb; [19, 20]). These size variations have been suggested to reflect differences in saprotrophic potential [4]. Cluster II strains display a large host range and, at least in western North America, low genetic diversity [21] which might indicate a recent evolutionary bottleneck. This is interesting since its host genera from the Cucurbitales, Datisca sp. and Coriaria sp., show a disjunct distribution [22, 23]. In the case of the genus Datisca, the Eurasian species Datisca cannabina is found in Pakistan and northwestern India and in the east Mediterranean, while the American species Datisca glomerata grows in California and northern Baja California, Mexico [15, 22].
As stated above, Frankia Cluster II strains can enter symbioses with members of the Cucurbitales and of the Rosales. The most recent common ancestor of the actinorhizal Cucurbitales, Coriariaceae and Datiscaceae has been molecular-clock dated to 73 +/- 3 mya [24]. Even if we assume that the symbiosis arose not in the common ancestor, but in oldest of both families, the Coriariaceae, the symbiosis would have preceded the diversification of the crown group of Coriaria. According to Yokoyama et al. [23], the divergence between the Coriaria species from the Northern and the Southern Hemisphere can be dated to 63 or 59 million years ago using fossil-calibrated rbcL and matK molecular clocks. As for the North American host plants of Frankia Cluster II, [25] date the origin of Ceanothus in California to 16.6–34.7 mya. The origin of the Dryadoideae tribe of the Rosaceae is dated to the younger Oligocene (23.03–28.1 mya; [26]). These data suggest that North American Cluster II symbioses are significantly younger than the Cucurbitales symbiosis.
Frankia Cluster II strains could have reached North America either from South America with Coriaria ruscifolia [23, 27], or from Asia when Datisca spread from what is now northern India/Pakistan/Nepal to North-East Asia and then to western North America following the Madrean-Tethyan pattern [28]. According to [29, 30] a continuous, sclerophyllous, dry-adapted flora existed between western North America (“Madrean”) and Eurasia (“Tethyan”) across a North Atlantic land bridge during the Ecocene and Oligocene (55 − 25 mya). This is consistent with the timing of Datisca speciation: [31] estimated the time of allopatric speciation that led to the distinction between the Eurasian D. cannabina and the American D. glomerata to 36.5–50.5 mya and 42 mya based on allozyme data and rbcL sequences, respectively. In North America, the Frankia Cluster II microsymbiont of Coriaria ruscifolia or Datisca sp. could have extended its host range to Ceanothus sp. and the Dryadoideae.
In all phylogenies including different Cluster II strains, strains from New Zealand have always been sister to all other Cluster II strains [16, 21, 32, 33]. Cross-infection studies showing that Ceanothus sp. and Purshia sp. could not be nodulated in New Zealand [34] suggest strong divergence between Frankia Cluster II strains in New Zealand and in North America. There seems to be less divergence between Asian and North American strains, in that a Frankia Cluster II strain sampled from soil or nodules of Coriaria nepalensis in an area in Pakistan where both C. nepalensis and D. cannabina are distributed, can nodulate the Californian Datisca species D. glomerata [15]. This makes it more likely that Cluster II Frankia strains in North America originated from nodules of Datisca sp. which arrived from Asia via the Bering Strait.
The first genome of a Frankia Cluster II strain to be sequenced, Candidatus Frankia datiscae Dg1, originated from nodules of C. nepalensis in Pakistan [15, 35]. The second sequenced genome of a Cluster II strain, BMG5.1, originated from nodules of Coriaria japonica growing in Japan [6]. Thus, the analysis of a Frankia Cluster II genome from an area devoid of host plants from the genus Coriaria could promise novel insights into the diversity within Cluster II and the differences between the three symbiotic Frankia clusters. Specifically, the genome of a North American strain promises to reveal information on the evolution and distribution of Frankia Cluster II from Asia/New Zealand/South America to North America.
The evolution of root nodule symbioses involved the recruitment of mechanisms from the evolutionarily older arbuscular mycorrhizal (AM) symbioses [36]. In legume/rhizobia symbioses, flavonoids in the root exudates of the host plants induce the expression of bacterial nodulation (nod) genes leading to the synthesis of lipochitooligosaccharide (LCO) signal molecules, the Nod factors. Also AM fungi produce LCO signal factors [37]. These LCOs are perceived by plant kinases of the LysM family and activate the common symbiotic pathway (CSP) that controls both legume/rhizobia and AM symbioses ([38]; reviewed by [39]). This CSP is also used for microsymbiont signaling in actinorhizal symbioses, as shown for D. glomerata [40] and Casuarina glauca [41, 42]. In this context, it seems likely that symbiotic signaling of Frankia strains also involved LCO-like compounds that are perceived by LysM receptor kinases. However, so far only the genome of Candidatus Frankia datiscae Dg1 has been shown to contain the canonical nod genes nodABC which in rhizobia are responsible for the synthesis of the common backbone of the LCO Nod factors [15, 35]. The second Cluster II strain to be sequenced, BMG5.1 did not contain homologs of the canonical nod genes in the draft genome [6].
In order to gain more insight into the evolution of North American Cluster II Frankia strains and to answer the question whether the presence of the canonical nod genes in Dg1 typical for Cluster II, or represents an exception, we sequenced the genome of a Cluster II inoculum from D. glomerata in California.
Methods
Plant and bacterial material
Datisca glomerata (C. Presl) Baill. seeds originating from plants growing at Gates Canyon in Vacaville, California, USA. Plant seedlings were grown under sterile condition to avoid contamination of Frankia strain Dg1 which were growing in the greenhouse. Seedlings were then transferred to pot filled with autoclaved mixture of soil and sand, ratio 1:1. Plants were grown in a growth chamber under a 15 h/9 h light dark cycle and day/night temperature of 23 °C/19 °C, relative humidity 65 % and light intensity of 60–100 μEm-2s-1. A soil sample containing the uncultured Frankia strain was acquired under a growing D. glomerata at Gates Canyon, Vacaville, California. Nine-week old D. glomerata plants in the growth chamber were inoculated with this infected soil. Inoculated plants were fertilized with 1-quarter-strength Hoagland’s medium without nitrogen [43].
Isolation of genomic DNA from isolated vesicle clusters
To isolate the Frankia vesicle clusters from root nodules, the protocol from Lundquist and Huss-Danell [44] was applied with some modifications. Root nodules harvested from D. glomerata were surfaced sterilized in 1 % sodium hypochlorite for 10 min. Then they were washed with sterile dd H2O. Nodules were homogenized in a mortar in homogenization buffer (0.05 M Tris-HCl pH 7.9–8, 4 % (w/v) PVP, 0.1 M KCl, 5 mM EDTA, 0.6 M sucrose, 10 mM Na2S2O4). The homogenized tissue was poured over an 80 μm nylon filter and the flow through was poured over a 16 μm nylon filter. The 16 μm filter was washed several times with a wash buffer (50 mM HEPES pH 7.8, 4 % (w/v) PVP, 10 mM EGTA, 10 mM EDTA, 2 mM Na2S2O4) and the cells obtained on the filter were transferred to a new tube containing pectinase buffer (10 mM Tris-HCl pH 6, 10 mM NaCl). The cells were incubated with pectinase (Macerozyme R10; Saveen-Werner, Sweden) at 37 °C for 2 h, shaking slowly (110 rpm). The vesicle clusters were spun down and resuspend in TES buffer (0.3 M sucrose, 25 mM EDTA pH 8.0, 25 mM Tris/HCl pH 8.0). The vesicle clusters were broken using the ultrasonic homogenizer Sonoplus HD 2070 (Bandelin Electronic) at 90 % amplitude and 30 % pulsing for 30 s. The genomic DNA then was extracted using CN solution (10 % CTAB in 0.7 M NaCl) and phenol/chloroform (1:1).
Sequencing and genome assembly of the Candidatus Frankia datiscae Dg2 metagenome
The quality of the DNA was assessed by gel-electrophoresis and the quantity was estimated using the Quant-iT PicoGreen dsDNA kit (Invitrogen) and the Tecan Infinite 200 Microplate Reader (Tecan Deutschland GmbH, Mainz, Germany). To obtain the complete genome sequence, a whole-genome-shotgun PCRfree (Nextera DNA Sample Prep Kit; Illumina, Munich, Germany) and an 8 K mate pair library (Nextera Mate Pair Sample Preparation Kit; Illumina) were generated based on the manufacturer’s protocol. After sequencing and processing of the raw data, a de novo assembly was performed using the GS De Novo Assembler software release version 2.8 (Roche, Mannheim, Germany) with default settings. In our approach, we used a 2 x 300 bp paired end sequencing run. For quality-control and filtering, a pipeline including trimmomatic (Bolger et al. [45]), r2cat [46] and contig-length vs. read-count analysis [47, 48] was implemented. The resulting data set was manually inspected and improved.
Reference assembly on the Candidatus F. datiscae Dg2 metagenome from the metagenome dataset
Based on contig length vs. read count analysis [47–49] and on taxonomic profiling by applying LCA [50], the established dataset was classified as a metagenome dataset including different Frankia strains, Datisca glomerata and a low amount of a diverse bacterial strains. To identify related species of Frankia in the metagenome dataset, a reference assembly was applied. For reference assembly, metagenomic contigs were mapped onto the reference genome Frankia sp. Dg1 [15] by means of r2cat [46]. Mapped contigs and their corresponding reads were extracted and de novo assembled using the GS Assembler (version 2.8; Roche) with default settings. Furthermore, automatic gene prediction and annotation of coding sequences on metagenomic Frankia contigs was performed within the genome annotation system GenDB 2.0 [51] as described previously [47, 52, 53]. An in silico gap closure approach was performed to close the gap between the two nodC contigs [49, 52, 54, 55].
Nucleotide sequence data for the reconstructed Frankia sp. Dg2 strains were deposited in the EBI database (accession numbers FLUV01000001-FLUV01002738). The nod region was finished manually and therefore separately deposited at EMBL (accession number LT622247).
Identification of single-nucleotide polymorphisms in the Dg2 metagenome
SNP and DIP detection is 1 of the key analyses to estimate the amount of included strains in a reconstructed reference sequence out of a metagenome. Therefore metagenomic reads were mapped on the reconstructed reference sequence of Frankia sp. Dg2 by applying gsMapper (Roche) using strict settings (90 % sequence, 100 bp minimal overlap). SNP and DIP detection was performed by means of ReadXplorer [56]. The implemented SNP and DIP detection in ReadXplorer not only reveals small-scale evolutionary differences, but also allows analysis of the resulting functional differences emerging from polymorphisms. ReadXplorer detects SNPs and DIPs based on a user-definable minimum percentage of variation and a minimum count of mismatching bases in the mappings at the examined position. This allows the detection of small amounts of underrepresented strains in a reconstructed reference sequence.
Phylogenetic analysis and comparison of the reconstructed Dg2 metagenome to the genomes of completely sequenced and annotated members of the genus Frankia
To compare and phylogenetically classify Frankia sp. Dg2 in relation to completely sequenced and annotated members of the genus Frankia, the comparative genomics platform EDGAR was applied [57]. The core genome of all selected strains was calculated by means of EDGAR and based on all core genes for each Frankia strain, phylogenetic distances were calculated from multiple sequence alignments. A phylogenetic tree was deduced from concatenated core gene alignments using PHYLIP [58].
Identification of Frankia genes involved in secondary metabolite synthesis by applying antiSMASH
antiSMASH [59, 60] is the first comprehensive pipeline enabling identification of biosynthetic loci covering the whole range of known secondary metabolite compound classes. This tool was used to search for secondary metabolite synthesis clusters in the different available Frankia genomes. For this approach, all Frankia genomes were selected and imported in antiSMASH. The output was in detail manually analyzed for secondary metabolite synthesis clusters and compared with related clusters in our bacteria.
Read-based comparative analysis based on fragment recruitment
Fragment recruitment approaches were performed as described previously [61]. Briefly, reads that led to low abundant SNPs to the Dg2 metagenome sequence were exported and aligned to Frankia sp. Dg1 genome and Dg2 metagenome sequences by applying blastN [62]. Reads were used for further evaluation if at least 80 % of a read were aligned to one of the target sequences and if the identity was at least 55 %. Sequence homology to Dg1 and Dg2 was compared to get better insight into the taxonomical relationship of this low abundant Frankia strain.
Average nucleotide identity and genome alignment of Frankia Cluster II strains
Average nucleotide identity (ANI) was analysed as described previously [63, 64, 65] to determine the relationship between different Frankia genomes.
MAUVE [66] was used to align and to visualize rearrangements in the chromosomes of Candidatus Frankia datiscae Dg1 vs. Frankia sp. Dg2 and Frankia sp. BMG5.1, Frankia alni ACN14a vs. Frankia sp. ACN1ag, Frankia alni ACN14a vs. Frankia sp. QA3 and in Frankia sp. CcI3 vs. Frankia sp. BMG5.23, respectively. The bioinformatics tool MAUVE including the progressiveMauve algorithm was applied with default settings for the comparative analysis [67]. Before applying Mauve, r2cat [46] was used to adapt order and direction of contings.
Protein phylogeny
The sequences encoded by the open reading frames identified as putative nodH genes from the assembled Dg2 metagenome (Dg2nodH1, FDG2_3270; Dg2nodH2, FDG2_3293) were each used as a query for a separate BLASTX search against two databases, the non-redundant (nr) dataset of GenBank (searched on 2015.12.17), and all genomes available in JGI-IMG (searched on 2015.12.17). A total of 204 amino acid sequences from ten different genera were identified with similarity of 1e-20 or above to at least one of the two putative orthologs. A total of 25 sequences were collected by selecting, from each genus, up to three sequences from unique species with the best e-values. These sequences were combined with Dg2nodH1 and Dg2nodH2 to generate a dendrogram.
The collected amino acid sequences were first aligned using MAFFT [68]. Aligned sequences were then analyzed using ProtTest3 [69] to estimate the best amino acid substitution model. The estimated parameters were then used in GARLI 2.0 [70] under default settings to generate a maximum likelihood tree. Three parallel searches were conducted in order to avoid selecting a tree lodged on a local optimum. Bootstrap analysis was conducted with 100 replicates. For each bootstrap replicate, parameters were estimated by GARLI 2.0 and two parallel searches were conducted.
GlnA-based phylogeny of Frankia strains
Field samples were collected and specimen vouchers were deposited in the herbarium of UC Davis (CA, USA). DNA was extracted from single nodule lobes using a modified CTAB procedure [71] after the nodule lobes had been washed with ddH2O, TEA buffer, and the periderm had been removed. PCR of a partial sequence of the glnA gene using primers DB41 and DB44 [33] was conducted. Fifty microliter PCR reactions (10 ng template DNA, each primer at 0.5 μM, 2 mM MgCl2, 1 unit of Taq DNA polymerase, 20 μM of each dNTP, and 5 μl of 10X buffer) were run on thermocycler (Perkin-Elmer manual, Perkin-Elmer Corp., Norwalk, CT) programmed for a hot start (95 ° C, 2 min.) and 30 cycles of 94 ° C for 30 s, 57 ° C for 45 s, and 72 ° C for 1 min with a final extension for 7 min. A negative control, lacking template DNA, was also run. PCR amplifications were analyzed in a 1.5 % agarose gel run in 0.5 X TBE buffer and visualized with ethidium bromide under UV light. PCR products were cloned using a TOPO TA cloning kit (Invitrogen) and sequenced using Big Dye reagents (Applied Biosystems, Foster City, CA) and analyzed on an ABI Prism 3100 automated sequencer (Applied Biosystems) according to the manufacturers’ instructions.
Sequences were proofread and assembled using SeqMan (1998, DNAstar, Madison, WI.) and then Blast searched at the NCBI Home page (www.ncbi.nlm.nih.gov/BLAST/) to identify and categorize sequences. Cluster II Frankia sequences generated were then aligned with existing Cluster II Frankia glnA sequences.
All alignments were created using MUSCLE (multiple sequence comparison by log-expectation [72]; Edgar 2004) at the EMBL-EBI website. Maximum Likelihood analyses were performed using PhyML 3.0 [73] on the ATGC bioinformatics server. The substitution model was GTR, with estimated invariant sites, and rate variation sampled from a gamma distribution. Support for branches was evaluated using bootstrap analysis as above.
Real time quantitative reverse transcription PCR (RT-qPCR) for the analysis of gene expression in nodules
RNA was isolated as described by Persson et al. [35], but using the Spectrum Plant Total RNA Kit (Sigma-Aldrich, Stockholm Sweden) from young D. glomerata nodules (i.e., nodules with two to three lobes) harvested eight weeks after infection. RT-qPCR analysis was performed as described by Persson et al. [35]. Reverse transcription was done using the SuperScript IV First Strand Synthesis System (Invitrogen). Primers were designed using Primer3Plus [74].
Results
Sequencing of the Candidatus Frankia datiscae Dg2 metagenome
DNA isolated from vesicle clusters originating from a Datisca glomerata plant growing at Gates Canyon, Vacaville, CA, USA, was de novo sequenced by applying a strategy combining whole-genome-shotgun and mate pair sequencing on the Illumina MiSeq platform. Sequencing on MiSeq platform resulted in 8,217,565 reads with a total of about 2.1 Gb sequence information. Assembly of the data by applying of the gsAssembler 2.8 resulted in 52,586 contigs, 1441 scaffolds and a size of 28.3 Mb. First, all assembled reads were taxonomically classified by applying LCA [50]. In total, about 1,700,000 reads were classified to the domain Bacteria. Mainly these reads were classified to the genus Frankia (ca. 1,150,000 reads), but also to Streptomyces (ca. 28,000 reads) and Mycobacterium (ca. 4,534 reads). In addition, ca. 210,000 reads were classified to Eukaryota. To gain deeper insights into the sample composition, a ’contig-length vs. read-count‘plot was calculated (Additional file 1; [47–49]). Obtained results represented a point cloud, but two contig groups at 1x and at 10x were apparent. Contigs of both groups were analyzed by means BLASTn [62] to deduce further taxonomical information on these contigs. The underrepresented contigs of the 1x group were assigned to Datisca glomerata and to diverse bacterial strains, whereas the other group represents contigs belonging to Frankia. The data were filtered by means of a reference mapping to Frankia sp. Dg1 applying r2cat. This mapping approach was aimed to reconstruct the Frankia genome from the identified metagenome. In total, 3,269,398 reads (40.68 % of all metagenome sequence reads) amounting to 576.12 Mb sequence information were extracted for further analysis. Further genome assembly of extracted reads related to the Frankia Dg1 genome applying the GS assembler (version 2.8) yielded 8,283 contigs (1066 scaffolds) and amounting a total length of 5.92 Mb.
Detection of different Frankia strains in a reconstructed draft genome by means of SNP and sequence analysis
The relatively high number of assembled contigs and scaffolds suggested that the sample contains more than one Frankia strain. To determine the degree of conservation of the Frankia sp. Dg2 sequence, all sequence reads of the project were mapped on the reconstructed Dg2 metagenome. The mapping approach was aimed at the estimation of Frankia species in the metagenome. In total, 6,234,666 reads (75.87 % of all metagenome sequence reads) yielding approximately 1.59 Gb sequence information were mapped on the Dg2 metagenome sequence. Based on SNP calling by applying ReadXplorer [56], 158,053 SNPs were predicted. More than 80 % of the SNPs show a low coverage <10 % of all bases at a certain variable position. The low abundant SNPs were always represented by a particular base and no additional variants were found. That may indicate a low abundant, related Frankia strain in the metagenomic sample.
To get taxonomical information about this strain, 44,620 reads leading to the low abundant SNPs were exported and used for a fragment recruitment using Dg1 and Dg2 as reference sequence. In both fragment recruitment approaches, 99 % of all reads were mapped to the reference sequences. Mapped reads to the reference genomes showed an average sequence identity of 96.6 % to Dg1, whereas the average sequence identity to Dg2 was somewhat lower (93.8 %). Therefore this putative strain is more closely related to Dg1.
In combination with the SNP approach, a visual inspection of the assembly data was performed. In a few cases, a 50−50 distribution of SNPs in comparison to the consensus sequence was observed. In addition, conserved Frankia contigs are followed by two different contig variants, detected in 267 cases. Local similarities were detected frequently within such contig pairs. It is very likely that these contig pairs contain variants of Frankia genes/regions that are significantly different to each other and therefore were not assembled into one contig. Occurrence of sequence differences is not evenly distributed over the aligned and matching contig segments indicating that some regions are more conserved than others, e.g. regions with housekeeping genes are highly conserved in both Frankia strains. Therefore, such local similarities between contigs in first instance confirm the existence of two closely related Frankia sp. Dg2 strains. These genomes share conserved regions without a SNP, but also contain some individual regions. In total, 198 of 267 contig variants represent variations in intergenic regions, whereas the remaining variants mainly refer to insertions/deletions of mobile genetic elements or changes in genes encoding hypothetical proteins.
The reconstructed Frankia sp. Dg2 metagenome contains sequence information of three different Frankia strains originating from D. glomerata nodules. Based on a SNP calling approach in ReadXplorer [56] including a manual inspection of the assembly and mapping result, these three different Frankia strains were detected. With bioinformatic methods, it was not possible to split the two abundant Frankia sp. Dg2 strains, Dg2a and Dg2b, in separate datasets because of their close relatedness (ca. 98–99 %) and a similar abundance in the sample (ca. 55 % Dg2a, ca. 45 % Dg2b). Therefore, the reconstructed sequence will be called “metagenome” in the next chapters, because it contains sequence information of 2 closely related Frankia strains. The third strain, Dg2c, was so underrepresented in the sample (ca. 1 %) that its effect on the consensus sequence could be ignored.
Features of the reconstructed Dg2 metagenome
The final sequence of the Frankia sp. Dg2 metagenome was established as described above. Based on scaffold information it could be proven that the Frankia sp. Dg2 metagenome only comprises a circular chromosome. Additional Frankia plasmids were not detected in the remaining metagenome data. Considering the final size of the Frankia sp. Dg2 metagenome, a 93-fold coverage was achieved after different filter steps. Relevant data of the Frankia sp. Dg2 metagenome project are summarized in Table 1. The circular chromosome of the Frankia sp. Dg2 metagenome has a size of 5,929,312 bp and features a GC content of 67.90 %. Gene prediction and annotation of the assembled metagenome sequence were performed within the GenDB system [51]. This approach resulted in the prediction of 6,536 coding sequences, 36 tRNA genes, and two rrn operons. The Frankia sp. Dg2 metagenome sequence contained a typical origin of replication oriC. Housekeeping genes, e.g. genes involved in glycolysis, essential for the survival of the bacterium, were identified on the circular chromosome of Frankia sp. Dg2. Due to this fact, the reconstructed Frankia sp. Dg2 sequence represents a characteristic first chromosome. In addition, a region related to the plasmid pFSYMDG01 of Dg1 was found on a contig originating from the Dg2 chromosome. It seems that Frankia plasmids can integrate into the chromosome, which is not surprising because the parBA gene system, encoding a chromosome/plasmid partitioning system, was found in both the Dg1 chromosome (FsymDg_4545 and FsymDg_4546) and in the Dg2 metagenome (FDG2_6558 and DG2_6557; parA/parB).
Table 1.
Sequencing and assembly statistics of the reconstructed Candidatus F. datiscae Dg2 metagenome
| Features | Frankia sp. Dg2 |
|---|---|
| All Reads | 8,217,565 |
| All Bases | 2,163,821,348 |
| Filtered Reads for genome reconstruction | 3,269,398 |
| Filtered Bases for genome reconstruction | 576,128,786 |
| Assembled Reads for genome reconstruction | 3,123,854 |
| Assembled Bases for genome reconstruction | 550,550,122 |
| Contigs | 8283 |
| Largest Contig | 36,613 bp |
| Scaffolds | 1066 |
| Largest Scaffold | 368,548 bp |
| Reconstructed genome size | 5.929.312 bp |
| Predicted genes | 6536 |
| Genes with predicted function | 3248 |
| tRNA | 36 |
| rrn-Operons | 2 |
| GC content | 67.90 % |
Core genome comparison of Dg2 with other Frankia genomes
Using EDGAR [57] a phylogenetic tree of Frankia strains was calculated using core genome comparison (Fig. 1). In this tree, Cluster II is the basal symbiotic Frankia cluster as previously described [6, 35, 75]. Consistent with the data presented by Gtari et al. [6], Cluster II is not only sister to the other symbiotic Clusters I and III, but also to the non-symbiotic Cluster IV.
Fig. 1.
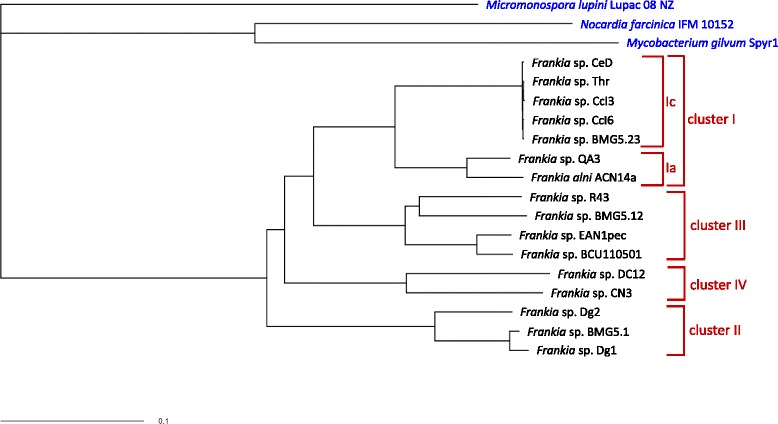
Comparison of core genomes of 16 sequenced Frankia strains [5, 7–11, 14–20, 94] using EDGAR [57]. Outgroups (given in blue) were three actinobacterial genomes, Micromonospora lupini Lupac 08 [95], Nocardia farcinica [96] and Mycobacterium gilvum Spyr1 [97]. The phylogenetic tree was deduced from concatenated core gene alignments using PHYLIP [58]. Bootstrap values were 100 for every branch
Within Cluster II, Dg2 (California) is sister to Dg1 (Pakistan; [15]) and BMG5.1 (Japan; [6]). However, given that this phylogeny does not contain a strain from New Zealand which are the ancestral strains in Cluster II in all other phylogenies, this cannot be interpreted to mean that North American Cluster II strains are ancestral to Eurasian ones. At any rate, the comparison shows that in contrast with North American Cluster II Frankia strains [21], Eurasian ones do not show low genetic diversity.
Average nucleotide identity (ANI) and alignment of genomes of Frankia cluster II strains
To determine similarities between the different Frankia genomes of the Clusters I and II, pairwise Average Nucleotide Identies (ANI) were calculated. Usually genomes of prokaryotic isolates, which belong to the same species, have ANI values above 95 % [63].
Analysis of ANI showed that while Candidatus Frankia datiscae Dg1 and Frankia sp. BMG5.1 belong to the same species (96 %; see also [6]), Dg2 belongs to a different species (ca. 88 % ANI vs. Dg1/BMG5.1). To better understand the evolution of Cluster II strains, genome alignments were performed for Dg1/Dg2 (Fig. 2a) and Dg1/BMG5.1 (Fig. 2b). It should be taken into account that the genomes of Dg2 and BMG5.1 are draft genome the scaffolds of which (1066 scaffolds for Dg2, 116 for BMG5.2) were aligned with the Dg1 genome so that not all rearrangements are visible. For comparison, genome alignments were performed for two strains from the same species from Cluster Ic, i.e., the Casuarina-infective strains CcI3 (fully assembled chromosome; [7]) and BMG5.23 (166 scaffolds; [8]) with ANI 99 % (Fig. 2c) and for two strains from different species from Frankia Cluster Ia, i.e., the Alnus-infective strains ACN14a (fully assembled chromosome; [7]) and ACN1ag (90 scaffolds; [13]) with ANI 92 % (Fig. 2d). Thus, a high amount of genome rearrangements is observed in Cluster II strains compared to the Cluster I strains, which would seem consistent with the high amount of complete IS elements in Dg1 compared to other Frankia strains [35]. However, a genome alignment of two Alnus-infective strains from different continents, ACN14a (Canada) and QA3 (Pakistan; fully assembled chromosome; [11]) with ANI 91 % shows a far higher amount of genome rearrangements than between Dg1 and Dg2 (Fig. 2e). To some extent, this can be explained by the fact that the genomes of both ACN14a and QA3 represent fully assembled chromosomes.
Fig. 2.
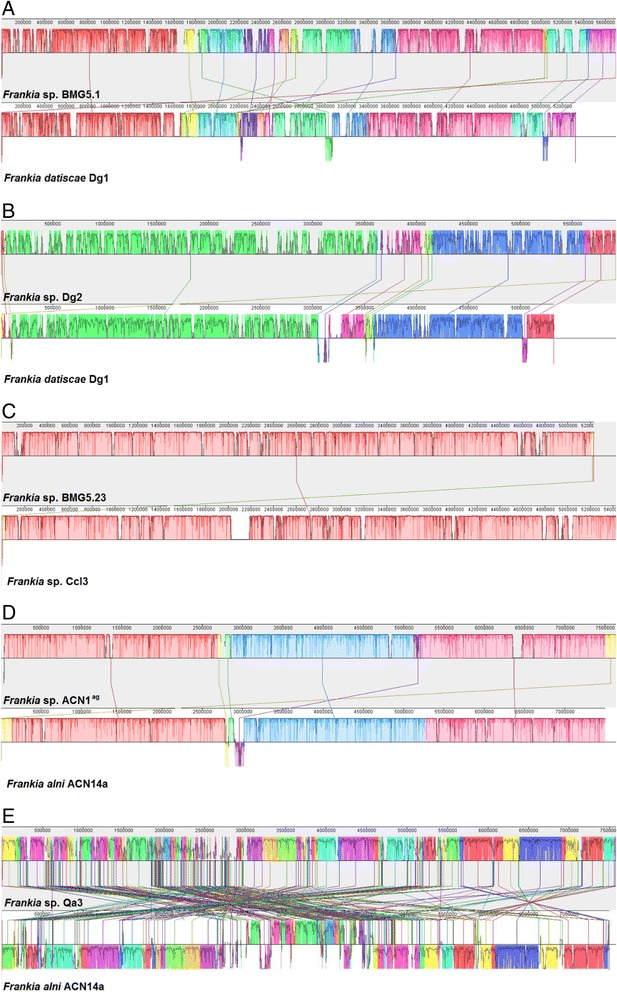
Frankia genome alignments using MAUVE [66]. Chromosome coordinates are plotted on the x-axis and the y-axis denotes the percentage of sequence identity. Colored areas appear above and possibly below the center line. Each of these areas surrounds a region of the genome sequence that aligned to part of another genome, and is presumably homologous and internally free from genomic rearrangement. When an area lies above the center line the aligned region is in the forward orientation relative to the first genome sequence. Areas below the center line indicate regions that align in the reverse complement orientation. Regions outside the colored areas lack detectable homology among the input genomes. Inside each area, Mauve draws a similarity profile of the genome sequence. The height of the similarity profile corresponds to the average level of conservation in that region of the genome sequence. Regions that are completely white were not aligned and probably contain sequence elements specific to a particular genome. The height of the similarity profile is calculated to be inversely proportional to the average alignment column entropy over a region of the alignment. a Pairwise alignment of the genomes of Candidatus Frankia datiscae Dg1 (chromosome) and Candidatus Frankia datiscae Dg2 (1066 scaffolds) (88 % ANI). b Pairwise alignment of the genomes of Candidatus Frankia datiscae Dg1 (chromosome) and Frankia sp. BMG5.1 (116 scaffolds) (96 % ANI). c Pairwise alignment of the genomes of Frankia sp. CcI3 (chromosome) and Frankia sp. BMG5.23 (166 scaffolds) (99 % ANI). d Pairwise alignment of the genomes of Frankia alni ACN14a (chromosomes) and Frankia sp. ACN1ag (90 scaffolds) (92 % ANI). e Pairwise alignment of the genomes of F. alni ACN14a (chromosome) and Frankia sp. QA3 (chromosome) (91 % ANI)
Dg2 is predicted to have greater saprotrophic capabilities than Dg1
A comparison between the ORFs found in the genomes of Dg1 and BMG5.1 versus Dg2, respectively, shows that Dg2 has the ability to use nitrate as N-source in that it can form assimilatory nitrate reductase and nitrite reductase (FDG2_0181, _0191, _0192, _0193) which are also encoded on the BMG5.1 genome but lacking in Dg1. Several transporters are present in the Dg2 metagenome that also occur in other Frankia strains, but are missing in Dg1 and BMG5.1, e.g., a putative ABC sugar transporter (FDG2_6040, _6041, _6042). Dg2 also contains an acetone carboxylase putatively involved in the use of acetone as carbon source which are missing in Dg1 and BMG5.1 (FDG2_0380, _0381, _0382; Clark and Ensign 1999). So Dg2 should be better equipped for saprotrophic growth than Dg1.
Furthermore, the Dg2 metagenome contains more operons for the production of secondary metabolites than either the genome of Dg1 or of BMG5.1 (Additional file 2).
Dg2 contains the canonical nod genes nodABC linked to 2 copies of nodH
Like Dg1, Dg2 contains the canonical nod genes nodABC, distributed over several transcriptional units, and in a configuration very similar to that in Dg1 except that in Dg2, all nod genes are clustered in one area of the chromosome (Fig. 3). Sequence alignments of the encoded proteins are shown in Additional file 3. Like in Dg1, a nodBC operon (DG2_3264-DG2_3265; here, containing a 5’-truncated nodB gene) is followed by nodIJ homologs (nltI and nltJ, DG2_3266 and DG2_3267) and like in Dg1, the distance between the open reading frames makes it questionable whether nodBCnltIJ are forming an operon [35].
Fig. 3.
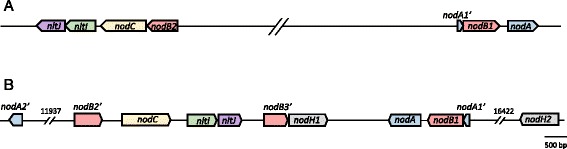
Arrangement of the canonical nod genes and the nodIJ homologs nltIJ in the genomes of Dg1 (a) vs. Dg2 (b). In Dg2, the canonical nod genes are clustered in a single part of the chromosome. The nodABC gene region of Dg2 also contains two copies of nodH. nodA genes are given in blue, nodB in red, nodC in yellow, nodH in grey, nltI in green and nltJ in purple
The presence of several truncated copies indicates a number of transposition events in the past. The configuration of the Dg2 nodA1’nodBnodA operon (DG2_3275 – DG2_3273) matches that in Dg1; furthermore, the 3’-truncated nodA1’ genes at the beginning of the Dg1 and the Dg2 operon are missing the same parts and are nearly identical (Fig. 3; Additional file 3). Thus, the transpositions that led to the nodA1’nodBnodA configuration must have occurred in a common ancestor of Dg1 and Dg2. Dg2 contains an additional 5’-truncated copy of nodA, Dg2nodA2’ (DG2_3248) which is nearly identical with Dg2nodA1 (Fig. 3; Additional file 3) and presumably arose by incomplete duplication of Dg2nodA1. NodB sequence alignment shows that Dg1NodB2, encoded by the nodB gene of the nodBC(nltIJ) operon, contains an insertion in its N-terminal part while its orthologue Dg2NodB2’ (DG2_3264) is truncated in the 5’- and 3’-part (Additional file 3). Thus, a comparison of the nod gene region of Dg1 and Dg2 indicates that the nodA1’nodB1nodA operon was present in the common ancestor of both strains. In Dg1, half of the nod region was transferred over more than 1 Mb to another part of the chromosome. In Dg2, a partial duplication of nodA took place, yielding nodA2’, and the nodB2 gene (part of the nodB2C operon) acquired a 5’- and a 3’-deletion.
In Dg2, the nodABC genes are linked to two complete copies of an additional nod gene, nodH, encoding a sulfotransferase (DG2_3270 and DG2_3293; Fig. 3). Similar to the nodABC genes (nodA, 59.10 % GC; nodA1’, 59.92 % GC; nodA2’, 60.36 % GC; nodB1, 66.08 % GC; nodB2’, 66.66 % GC; nodB3’, 62.76 % GC; nodC, 64.60 % GC; nltI, 60.6 %; nltJ, 66.5 %), both nodH genes have a GC content below the average of the Dg2 metagenome (nodH1, 60.64 % GC; nodH2, 61.04 % GC).
NodH phylogeny
The amino acid sequences from the two putative NodH proteins encoded by Dg2 were used in a maximum likelihood phylogenetic analysis. PROTTEST found JTT + G (α = 0.824) to be the best model based on the Bayesian information criterion (BIC) and on the corrected Akaike information criterion (AICc). GARLI2.0 generated the most likely phylogeny with a likelihood score of -6545.95. A putative sulfotransferase from Cyanothece sp. PCC7424 (NCBI, GI: 218173889), the only cyanobacterial sequence in the tree with the most distantly related taxa with by far the longest branch, was used to root the tree. The resulting tree (Fig. 4) shows that the two Dg2 NodH proteins form a sister group to rhizobial NodH proteins.
Fig. 4.
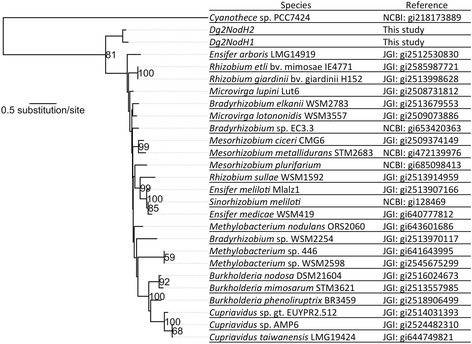
The most likely phylogeny of nodH genes based on amino acid sequences. Nodes with bootstrap support of 50 or higher have their values indicated at each node. The scale bar indicates the number of amino acid substitutions per site
NodH expression could not be detected in nodules
NodH is clearly not essential to nodulate Coriaria nepalensis, the original host for Dg1, or D. glomerata, in whose nodules Dg1 was maintained for over a decade, or D. cannabina which can be nodulated by Dg1 as well as by Dg2, respectively (Additional file 4). To address the question about the expression of Dg2 nodH in nodules, D. glomerata nodules induced by nodules induced by Dg2 were examined for nodA, nodB, nodC and nodH mRNA using quantitative RT-PCR. While nodABC were expressed, expression of nodH could not be detected in Dg2-induced nodules of D. glomerata (Additional file 5).
Phylogeny of Cluster II Frankia strains – North America vs. Eurasia
Previously, Vandenheuvel et al. [21] had found low genetic diversity of Cluster II Frankia strains from western North America while using a glnA fragment as phylogenetic marker. Our core genome comparison shows that while Cluster II strains are not as genetically diverse as Cluster III strains, there are strong differences between Cluster II strains from Asia (represented by Dg1 and BMG5.1) and from North America (represented by Dg2; Fig. 1). Among the three Cluster II genomes available, Dg2 is sister to the two genomes from Asia, Dg1 from Pakistan and BMG5.1 from Japan [6]. In Cluster II phylogenies involving more strains, the New Zealand strains were always ancestral [32]. Therefore, without a genome from a Cluster II strain from New Zealand included, this core genome tree cannot be used as the basis to determine the earliest divergent lineage of Cluster II Frankia.
When the glnA-based phylogeny of Vandenheuvel et al. [21] was extended to include all Cluster II strains analysed thus far (the Coriaria-infective strains from Nouioui et al. [27], as well as a new collection of sequences amplified from nodules of North American Dryadoideae), the diversity of the Eurasian Cluster II strains was as low as that of the North American Cluster II strains (Fig. 5; Additional file 6), a phylogenetic grouping which contradicts our core genome comparison of the two Asian strains Dg1 and BMG5.1 with Dg2 (Fig. 1). Furthermore, the glnA-based phylogeny did not result in an unambiguous separation of Eurasian and North American Cluster II strains. No greater diversity was found when 16S rRNA sequences were included in the comparison (data not shown). So glnA, while a suitable marker for the phylogeny of the whole Frankia genus [33], does not seem to show enough sequence diversity to reflect the phylogeny of Cluster II Frankia strains.
Fig. 5.
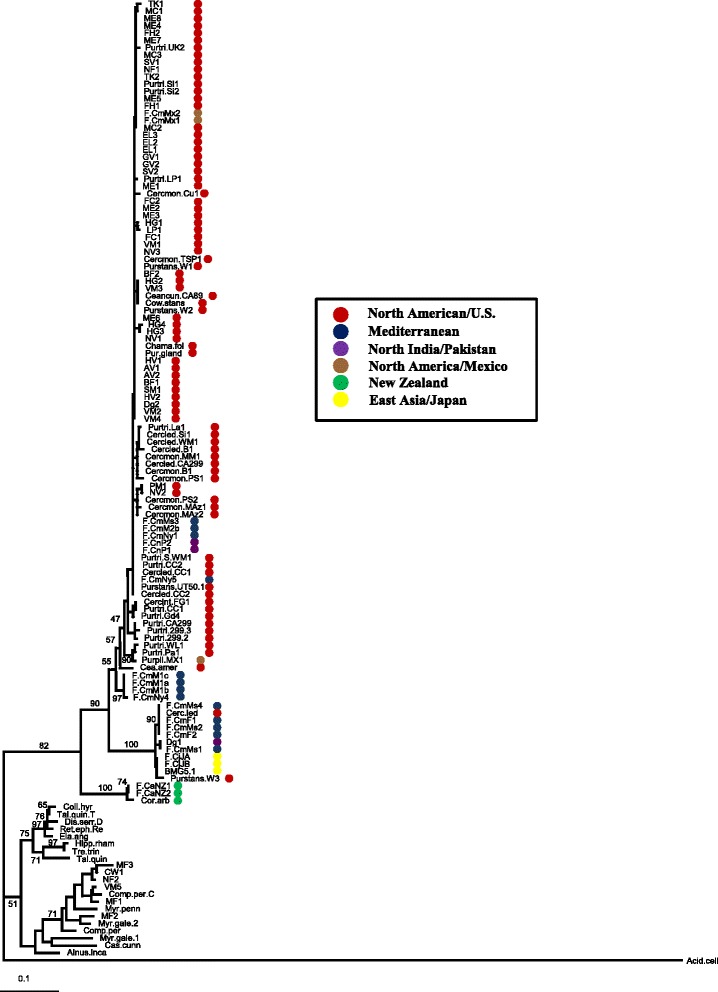
Phylogeny of Cluster II Frankia strains based on glnA. All DNA sequences used in this figure are referenced in Additional file 6. Acidothermus cellulolyticus [93] and several Cluster I and Cluster III strains were used as outgroups. Sequences were taken from Vanden Heuvel et al. [21], Clawson et al. [33] and Nouioui et al. [27] and from the three Cluster II genomes available; sequences of 34 field samples were contributed by this study (see Additional file 6)
Discussion
In this study, we sequenced the DNA from Frankia vesicle clusters isolated from nodules of Datisca glomerata inoculated using a field sample of soil from Gates Canyon, Vacaville, CA, USA. It turned out to be a metagenome (Dg2) representing two dominant (Dg2a, Dg2b) and one minor (Dg2c) Frankia Cluster II strains. Analysis of average nucleotide identity (ANI) revealed that Dg2 represents a new species in Frankia Cluster II, while the previously sequenced genomes of Candidatus Frankia datiscae Dg1 (originating in Pakistan; [35]) and of Frankia sp. BMG5.1 (originating in Japan; [6]) belong to the same species [6]. For comparison with the diversity of Cluster I Frankia strains, all Casuarina-infective strains the genomes of which were published so far belong to the same species (ANIs between 99.4 and 99.9 %), while the three Alnus-infective Frankia strains from Canada the genomes of which were sequenced represent two species: ACN14a [7] and AvcI1 [14] have 99 % ANI, while ACN1ag has 92 % and 91.44 % ANI, respectively, with ACN14a and AvcI1 [14]. The Alnus-infective Frankia strain from Pakistan, QA3 [11], represents a third species, showing 91 % ANI with ACN14a, 92.97 % ANI with AvcI1 and 92 % ANI with ACN1ag.
Frankia genome comparisons have shown that members of the non-symbiotic Cluster IV and of the symbiotic Cluster III have the largest genomes with ca. 10 Mb. Genome reduction took place in the symbiotic Cluster I – strongly in Casuarina-infective strains where genome size is reduced to 5–6 Mb [7–10], and less strongly in Alnus-infective strains where genome sizes are between 7 and 8 Mb [7, 11, 13, 14]. This differential genome reduction has been correlated with the saproptrophic potential of the corresponding strains and with the phytogeographical distribution of their hosts [7], or with their host range [76, 77]. The fact that for decades, Cluster II strains could not be cultured, indicating a low saprotrophic potential, would lead to the prediction of a small genome size, while the low host specificity of Cluster II strains [21] would predict a large genome size. Since all three Cluster II genomes sequenced so far are in the range of 5–6 Mb, the saprotrophic potential seems to be the decisive factor here.
A phylogenetic tree based on core genome comparison of 16 Frankia strains and three actinobacterial outgroups showed that Frankia Cluster II is not only paraphyletic to the other symbiotic Frankia strains, but is also also paraphyletic to Cluster IV which contains the non-symbiotic strains (Fig. 1). While Cluster II symbioses may be the oldest root nodule symbioses [2], they cannot precede the acquisition of the predisposition for developing root nodules by the precursor of the symbiotic plant clade which is dated to ca. 100 mya.
The only cultured Cluster II strain reported to date is an alkaliphile [6]. It is highly unlikely for a root symbiont that colonizes the proton-rich rhizosphere and acidic plant endosphere, to secondarily gain alkaliphily making it intolerant of pH 6 in culture. However, this perplexing character is apparently not limited to a single strain; Canizo et al. [78] showed that while Cluster II Frankia nodulated Coriaria myrtifolia at a range of pH between 5 and 9, higher rates of nodulation and host growth correlated directly with increasing pH. Furthermore, in New Zealand, Coriaria sp. grows on volcanic slopes (see, e.g., [79]), and soils of active volcanic areas tend to be alkaline [80]. Considering the genome phylogeny which shows Cluster II Frankia as sister to all other Frankia, this raises the possibility that alkaliphiles predates symbiosis in Frankia. As the alkaliphilic and symbiotic crown group of the genus diverged into Cluster II and its sister, the sister lost its alkaliphily.
Ultimately, more Cluster II strains have to be cultured to determine whether this scenario is correct. If, however, the ancestral Frankia strains were alkaliphiles, Cluster II strains would not necessarily represent examples of genome reduction. Contrarily, the evolutionary more derived Frankia strains might represent examples of genome expansion during the adaptation to diverse moderate environments.
How many strains are present in a typical Frankia Cluster II-induced nodule?
Studies on the diversity of Frankia in root nodules have usually relied on the PCR amplification of 16S rRNA fragments (see, e.g., [81]). Since actinorhizal nodules have thick periderms, sequences amplified that way do not necessarily derive from the microsymbionts of said nodules, but may be derived from bacteria living in the dead cells of the periderm and having escaped surface sterilization. This may also be exemplified by the fact that not all strains isolated from nodules of a particular plant can re-infect that plant species (see, e.g., [82]).
The genomes of two strains have now been sequenced based on DNA derived from vesicle cluster preparations from nodules of several plants. Earlier results based on the analysis of OTUs from nodules had shown that greenhouse-grown nodules of Datisca glomerata contained more than one Frankia strain, although only one strain, Candidatus Frankia datiscae Dg1, was represented in vesicle clusters [35]. Our study shows the first Frankia metagenome isolated from D. glomerata nodules induced by a field sample from D. glomerata, Dg2. The difference with Dg1 may have been due to the fact that Dg1 originated from soil below a nodulated Coriaria nepalensis plant and was propagated for more than ten years in D. glomerata plants in a greenhouse [35], while Dg2 is based on soil collected from below a nodulated D. glomerata plant in the field that was used to nodulate a single series of D. glomerata plants in a growth chamber before the genome was analysed. Thus, Dg2 is likely to better represent the situation in the field. Nevertheless, further studies on vesicle clusters isolated from Frankia Cluster II nodules in particular, and Frankia nodules in general, are necessary to find out how many strains are normally represented as symbionts in the nodules of an individual actinorhizal plant. Another question to be analysed in the future is whether the wide host range of Frankia Cluster II is due to the fact that different members of an assemblage dominate in nodules from different host species.
Dg2 contains the canonical nod genes and also the sulfotransferase gene nodH: does it produce sulfated Nod factors?
Bacterial signaling in rhizobial symbioses as well as fungal signaling in arbuscular mycorrhizal symbioses [39], and also Frankia signaling in actinorhizal symbioses [40, 41, 83] involves the common symbiotic signal transduction pathway (CSSP). This pathway has been well examined in legumes, where rhizobial or fungal lipochitooligosaccharide (LCO) signal factors bind to modified chitin receptors in the plasma membrane of root epidermal cells which signal to a common symbiotic receptor kinase SymRK/DMI2 [38, 39]. The modified chitin receptors belong to a protein family that meanwhile has been shown to perceive rhizobial LCO Nod factors, LCO Myc factors from arbuscular mycorrhizal fungi, peptidoglycans and exopolysaccharides [84]. Studies on Alnus glutinosa and Casuarina glauca have shown that in symbioses of Frankia Cluster I, non-LCO signal substances are signaling via the CSSP [83, 85, 86]. This is consistent with the fact that Cluster I (and Cluster III) Frankia strains do not contain the canonical nod genes nodABC which encode the enzymes that in rhizobia are responsible for the biosynthesis of LCO Nod factors [7]. Similarly, some rhizobial strains that do not contain the canonical nod genes and cannot form LCO Nod factors can induce nodules on Aeschynomene sp., and also this process involves signaling via the CSSP [87].
So far, one Frankia genome was known to contain the canonical nod genes nodABC, Dg1 [35]. Dg2a and Dg2b not only contained the canonical nod genes but also two copies of the nodH gene encoding a sulfotransferase which in rhizobia is responsible for the production of sulfated LCO Nod factors. As in case of Dg1, the canonical nod genes of Dg2 could be shown to be expressed in nodules, but neither nodH copy was transcribed under these conditions. A comparative analysis of the nod operons showed that the common ancestor of Dg1 and Dg2 – which should also be the ancestor of BMG5.1 – is likely to have contained the canonical nod genes nodABC and probably also a copy of nodH (Fig. 3; Additional file 3).
While the presence of a diverse NodA protein family in actinobacteria suggested that the corresponding gene evolved in actinobacteria and was later picked up by rhizobia, maybe as an operon together with nodB and nodC [35], the situation is not clear for nodH. In the absence of a NodH protein family in non-symbiotic relatives of either Frankia sp. or rhizobia, the fact that phylogenetically, Dg2 NodH proteins are forming a sister group of rhizobial NodH proteins (Fig. 4) merely indicates either that a gene transfer took place between rhizobia and Frankia strains, or that both groups obtained nodH genes from the same donor.
The high number of full size insertion elements in Dg1 [35] and the clear signs of transposition in the nod gene regions of Dg1 and Dg2, respectively, indicate significant genome instability. Under these circumstances, the fact that both Dg1 and Dg2 contain an intact copy of nodA, nodB and nodC each seems to indicate selection pressure, and thus a physiological function of nodABC. This does not necessarily lead to the conclusion that nodulation by Dg1 and Dg2, or by one of them, requires LCO Nod factors. Nevertheless, the fact that nodABC are expressed in nodules induced by Dg1 and Dg2 suggests that LCOs do play a role in the symbiosis.
Sulfation of LCO Nod factors, apart from playing a role in host specificity, can increase the stability of Nod factors by interfering with their digestion by chitinases [88]. This is important in the rhizosphere but may not be relevant for Nod factors in planta. So if sulfation of Nod factors does not play a role in binding Nod factor receptors within D. glomerata nodules, there might be a rationale for not expressing nodH genes in planta, only ex planta. It is also possible that the nodH genes are not expressed in D. glomerata nodules because they are not necessary to nodulate this particular host species, as exemplified by the fact that D. glomerata is nodulated by Dg1 which does not contain nodH.
Other unique features of Frankia Cluster II strains: mammalian cell entry genes
With three genomes of Cluster II available, it becomes interesting to do phylogenetic profiling to identify genes that only occur in Cluster II, not in any other Frankia cluster. Most strikingly, what is exclusive to Cluster II genomes are large sets of mce (mammalian cell entry) genes. They represent the actinobacterial paralogues of the Mla systems (ABC-type transporters; mlaD encodes the periplasmic substrate-binding proteins, mlaE encodes the permease and mlaF the ATP-binding protein) of Gram-negative bacteria that maintain asymmetry in the outer membrane [89]. Mce genes were first analysed in Mycobacterium tuberculosis. Mce systems seem to represent steroid transporters in that Mycobacterium spp. Mce4 facilitates the uptake of cholesterol and also in Rhodococcus jostii RHA1, mce4 encodes an ATP-dependent steroid transporter necessary for growth on, e.g., beta-sitosterol [90]. They are involved in virulence as could be shown since peptides that strongly bind, and thereby block, the periplasmic component of the mce4 system from M. tuberculosis could prevent the mycobacterial entry to type II pneumocytes [91]. In Streptomyces coelicolor, the mce locus is involved in mediating the interactions with plants and amoebae. Mce genes are present across the actinobacteria and presumably represent an ancient locus; niche specialization has led to divergence of the mce clusters in each actinobacterial genus throughout evolution [92].
Analysis showed that the mce systems in the three representatives of Frankia Cluster II are highly diverse (Additional file 7). Several Frankia Cluster II mce operons contain truncated genes. One operon is present in BMG5.1 and Dg2, but not in Dg1, although it is conserved in other actinobacteria, i.e. in Saccharomonospora marina, suggesting that Dg1 lost the entire operon. In Dg2, this operon mostly contains truncated genes (FDG2_3167 – FDG2_3173; Additional file 7). Furthermore, Dg1 does not contain a mlaF homolog but this component might be provided by another ABC transporter system. Every Cluster II strain contains at least two mlaE genes (encoding permeases) and more than 10 mlaD genes (encoding the periplasmic substrate-binding proteins). Given that the mce systems are involved in the colonization of plants in case of S. coelicolor, it is tempting to speculate that they might be involved in the infection process of Frankia Cluster II strains.
How did Frankia Cluster II invade the North American continent?
A glnA-based phylogenetic tree of all Cluster II Frankia sequences available to date with Cluster I and III sequences and the glnA sequence from Acidothermus cellulolyticus [93] as outgroup (Fig. 5) had the three sequences from New Zealand in the ancestral position but the Eurasian and North American strains did not form separate clades in that support for the separation was weak and several strains ended up in the ‘wrong’ group (American strains in the Eurasian group and vice versa). However, if the Cluster II strains nodulating the North American host plants (Dryadoideae, Ceanothus sp. and D. glomerata) were derived from South American strains which originally came from New Zealand [16], we would not expect the separation between the New Zealand strains and the Northern Hemisphere strains we see in Fig. 5. So the currently available are more consistent with the hypothesis that Frankia Cluster II strains reached the North American continent with D. glomerata via the Bering strait.
Conclusions
The first metagenome of an assemblage of North American Frankia Cluster II strains, Dg2, represents a different species than the two Asian strains. A phylogenetic tree based on the core genomes of 16 Frankia strains puts Cluster II in the basal position of the entire genus, and shows that the Cluster II genomes available thus far display more diversity than the Casuarina-infective strains from Cluster I. The Dg2 metagenome contains not only the canonical nod genes nodABC, but also the LCO sulfotransferase gene nodH, and a comparison between the nod regions of Candidatus Frankia datiscae Dg1 from Pakistan and Dg2 from California shows that both strains have a common ancestor the genome of which probably contained nodH.
Acknowledgements
We want to thank Peter Lindfors and Anna Pettersson for taking care of the plants, and Dr. Susanne Renner for helpful discussions on the evolution of Cucurbitales. This project was supported by a grant from the Swedish research council Vetenskapsrådet to KP (VR 2012-17840-97281-33), and USDA-NIFA-CA-D*-PLS-7688-H to AMB. The bioinformatics support of the BMBF-funded project “Bielefeld-Gießen Center for Microbial Bioinformatics”-BiGi within the German Network for Bioinformatics Infrastructure (deNBI.de) is gratefully acknowledged.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files. Nucleotide sequence data for the reconstructed Frankia sp. Dg2 strains were deposited in the EBI database under Bioproject PRJEB12692.
Authors’ contributions
TVN and KP conceived and designed the study. TVN isolated the DNA, DW and JK were responsible for sequencing and assembly. TVN, KP, AMB, KB, JB, DW and JK performed the genome analysis. BVH performed the glnA-based phylogeny (data and analysis). TVN, DW, KB, AMB, BVH and KP wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- ANI
Average nucleotide identity
- CSSP
Common symbiotic signal transduction pathway
- LCA
Lowest common ancestor (algorithm)
- LCO
Lipochitooligosaccharide
- Mya
million years ago
- Nod factors
Nodulation factors
- PCR
Polymerase chain reaction
- RT-qPCR
Reverse transcription quantitative real-time PCR
- SNP
Single nucleotide polymorphism
Additional files
Contig-length vs. Read-count plot of the Dg2 metagenome. (DOCX 348 kb)
Secondary metabolite production encoded on genomes of Frankia Cluster II strains. (XLSX 12 kb)
Alignments of Nod DNA and protein sequences from Cluster II Frankia strains Dg1 and Dg2. (DOCX 42 kb)
Candidatus Frankia datiscae Dg2-induced nodules on roots of D. glomerata and D. cannabina. (PPTX 5298 kb)
Expression of nod genes in nodules induced by Dg2 on roots of D. glomerata. (DOCX 13 kb)
GlnA sequences used for the phylogenetic tree depicted in Fig. 5. (XLSX 26 kb)
MCE-related proteins encoded by the genomes of BMG5.1, Dg1 and Dg2. (XLSX 26 kb)
References
- 1.Soltis DE, Soltis PS, Morgan DR, Swensen SM, Mullin BC, Dowd JM, et al. Chloroplast gene sequence data suggest a single origin of the predisposition for symbiotic nitrogen fixation in angiosperms. Proc Natl Acad Sci U S A. 1995;92:2647–2651. doi: 10.1073/pnas.92.7.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Werner GDA, Cornwell WK, Sprent JI, Kattge J, Kiers ET. A single evolutionary innovation drives the deep evolution of symbiotic N2-fixation in angiosperms. Nat Commun. 2014;5:4087. doi: 10.1038/ncomms5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swensen SM. The Evolution of actinorhizal symbioses: Evidence for multiple origins of the symbiotic association. Am J Bot. 1996;83:1503–1512. doi: 10.2307/2446104. [DOI] [Google Scholar]
- 4.Normand P, Orso S, Cournoyer B, Jeannin P, Chapelon C, Dawson J, et al. Molecular phylogeny of the genus Frankia and related genera and emendation of the family Frankiaceae. Int J Syst Bacteriol. 1996;46:1–9. doi: 10.1099/00207713-46-1-1. [DOI] [PubMed] [Google Scholar]
- 5.Normand P, Chapelon C. Direct characterization of Frankia and of close phyletic neighbors from an Alnus viridis rhizosphere. Physiol Plant. 1997;99:722–731. doi: 10.1111/j.1399-3054.1997.tb05377.x. [DOI] [Google Scholar]
- 6.Gtari M, Ghodhbane-Gtari F, Nouioui I, Ktari A, Hezbri K, Mimouni W, et al. Cultivating the uncultured: growing the recalcitrant cluster-2 Frankia strains. Sci Rep. 2015;5:13112. doi: 10.1038/srep13112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Normand P, Lapierre P, Tisa LS, Gogarten JP, Alloisio N, Bagnarol E, et al. Genome characteristics of facultatively symbiotic Frankia sp. strains reflect host range and host plant biogeography. Genome Res. 2007;17:7–15. doi: 10.1101/gr.5798407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghodhbane-Gtari F, Hurst SG, Oshone R, Morris K, Abebe-Akele F, Thomas WK, et al. Draft Genome sequence of Frankia sp. strain BMG5.23, a salt-tolerant nitrogen-fixing actinobacterium isolated from the root nodules of Casuarina glauca grown in Tunisia. Genome Announc. 2014;2(3). doi: 10.1128/genomeA.00520-14. [DOI] [PMC free article] [PubMed]
- 9.Hurst SG, Oshone R, Ghodhbane-Gtari F, Morris K, Abebe-Akele F, Thomas WK, et al. Draft Genome sequence of Frankia sp. strain Thr, a nitrogen-fixing actinobacterium isolated from the root nodules of Casuarina cunninghamiana grown in Egypt. Genome Announc. 2014;2(3). doi: 10.1128/genomeA.00493-14. [DOI] [PMC free article] [PubMed]
- 10.Mansour SR, Oshone R, Hurst SG, Morris K, Thomas WK, Tisa LS. Draft genome sequence of Frankia sp. strain CcI6, a salt-tolerant nitrogen-fixing actinobacterium isolated from the root nodule of Casuarina cunninghamiana. Genome Announc. 2014;2(3). doi: 10.1128/genomeA.01205-13. [DOI] [PMC free article] [PubMed]
- 11.Sen A, Beauchemin N, Bruce D, Chain P, Chen A, Walston Davenport K, et al. Draft genome sequence of Frankia sp. strain QA3, a nitrogen-fixing actinobacterium isolated from the root nodule of Alnus nitida. Genome Announc. 2013;1(2). doi: 10.1128/genomeA.00103-13. [DOI] [PMC free article] [PubMed]
- 12.Oshone R, Hurst SG, Abebe-Akele F, Simpson S, Morris K, Thomas WK, et al. Permanent draft genome sequences for two variants of Frankia sp. strain CpI1, the first Frankia strain isolated from root nodules of Comptonia peregrina. Genome Announc. 2016;4(1). doi: 10.1128/genomeA.01588-15. [DOI] [PMC free article] [PubMed]
- 13.Swanson E, Oshone R, Simpson S, Morris K, Abebe-Akele F, Thomas WK, et al. Permanent draft genome sequence of Frankia sp. strain AvcI1, a nitrogen-fixing actinobacterium isolated from the root nodules of Alnus viridis subsp. crispa grown in Canada. Genome Announc. 2015;3(6). doi: 10.1128/genomeA.01511-15. [DOI] [PMC free article] [PubMed]
- 14.Swanson E, Oshone R, Simpson S, Morris K, Abebe-Akele F, Thomas WK, et al. Permanent draft genome sequence of Frankia sp. strain ACN1ag, a nitrogen-fixing actinobacterium isolated from the root nodules of Alnus glutinosa. Genome Announc. 2015;3(6). doi: 10.1128/genomeA.01483-15. [DOI] [PMC free article] [PubMed]
- 15.Persson T, Benson DR, Normand P, Vanden Heuvel B, Pujic P, Chertkov O, et al. Genome sequence of “Candidatus Frankia datiscae” Dg1, the uncultured microsymbiont from nitrogen-fixing root nodules of the dicot Datisca glomerata. J Bacteriol. 2011;193:7017–7018. doi: 10.1128/JB.06208-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nouioui I, Beauchemin N, Cantor MN, Chen A, Detter JC, Furnholm T, et al. Draft genome sequence of Frankia sp. strain BMG5.12, a nitrogen-fixing actinobacterium isolated from Tunisian soils. Genome Announc. 2013;1(4). doi: 10.1128/genomeA.00468-13. [DOI] [PMC free article] [PubMed]
- 17.Wall LG, Beauchemin N, Cantor MN, Chaia E, Chen A, Detter JC, et al. Draft genome sequence of Frankia sp. strain BCU110501, a nitrogen-fixing actinobacterium isolated from nodules of Discaria trinevis. Genome Announc. 2013;1(4). doi: 10.1128/genomeA.00503-13. [DOI] [PMC free article] [PubMed]
- 18.Pujic P, Bolotin A, Fournier P, Sorokin A, Lapidus A, Richau KH, et al. Genome Sequence of the atypical symbiotic Frankia R43 Strain, a nitrogen-fixing and hydrogen-producing actinobacterium. Genome Announc. 2015;3(6). doi: 10.1128/genomeA.01387-15. [DOI] [PMC free article] [PubMed]
- 19.Ghodhbane-Gtari F, Beauchemin N, Bruce D, Chain P, Chen A, Walston Davenport K, et al. Draft genome sequence of Frankia sp. strain CN3, an atypical, noninfective (Nod–) Ineffective (Fix–) Isolate from Coriaria nepalensis. Genome Announc. 2013;1(2). doi: 10.1128/genomeA.00085-13. [DOI] [PMC free article] [PubMed]
- 20.Tisa LS, Beauchemin N, Cantor MN, Furnholm T, Ghodhbane-Gtari F, Goodwin L, et al. Draft fenome sequence of Frankia sp. strain DC12, an atypical, noninfective, ineffective isolate from Datisca cannabina. Genome Announc. 2015;3(4). doi: 10.1128/genomeA.00889-15. [DOI] [PMC free article] [PubMed]
- 21.Vanden Heuvel BD, Benson DR, Bortiri E, Potter D. Low genetic diversity among Frankia spp. strains nodulating sympatric populations of actinorhizal species of Rosaceae, Ceanothus (Rhamnaceae) and Datisca glomerata (Datiscaceae) west of the Sierra Nevada (California) Can J Microbiol. 2004;50:989–1000. doi: 10.1139/w04-079. [DOI] [PubMed] [Google Scholar]
- 22.Liston A, Rieseberg LH, Elias TS. Morphological stasis and molecular divergence in the intercontinental disjunct genus Datisca (Datiscaceae) Aliso. 1989;12:525–542. [Google Scholar]
- 23.Yokoyama J, Suzuki M, Iwatsuki K, Hasebe M. Molecular phylogeny of Coriaria, with special emphasis on the disjunct distribution. Mol Phylogenet Evol. 2000;14:11–19. doi: 10.1006/mpev.1999.0672. [DOI] [PubMed] [Google Scholar]
- 24.Schaefer H, Renner SS. Phylogenetic relationships in the order Cucurbitales and a new classification of the gourd family (Cucurbitaceae) Taxon. 2011;60:122–138. [Google Scholar]
- 25.Onstein RE, Carter RJ, Xing Y, Richardson JE, Linder HP. Do Mediterranean-type ecosystems have a common history?--Insights from the Buckthorn family (Rhamnaceae) Evol Int J Org Evol. 2015;69:756–771. doi: 10.1111/evo.12605. [DOI] [PubMed] [Google Scholar]
- 26.Töpel M, Antonelli A, Yesson C, Eriksen B. Past climate change and plant evolution in Western North America: a case study in Rosaceae. PLoS ONE. 2012;7:e50358. doi: 10.1371/journal.pone.0050358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nouioui I, Ghodhbane-Gtari F, Fernandez MP, Boudabous A, Normand P, Gtari M, et al. Absence of cospeciation between the uncultured Frankia microsymbionts and the disjunct actinorhizal Coriaria species. BioMed Res Int. 2014;2014:924235. doi: 10.1155/2014/924235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wen J, Ickert-Bond SM. Evolution of the Madrean–Tethyan disjunctions and the North and South American amphitropical disjunctions in plants. J Syst Evol. 2009;47:331–348. doi: 10.1111/j.1759-6831.2009.00054.x. [DOI] [Google Scholar]
- 29.Axelrod DI. Mediterranean type ecosystems: origin and structure. Hist. Mediterran. Ecosyst. Calif. Berlin, Heidelberg: Springer Berlin Heidelberg; 1973. pp. 225–277. [Google Scholar]
- 30.Axelrod DI. Evolution and biogeography of Madrean-Tethyan sclerophyll vegetation. Ann Mo Bot Gard. 1975;62:280–334. doi: 10.2307/2395199. [DOI] [Google Scholar]
- 31.Liston A. Biogeographic relationships between the Mediterranean and North American floras: insights from molecular data. Lagascalia. 1997;19:323–330. [Google Scholar]
- 32.Benson DR, Stephens DW, Clawson ML, Silvester WB. Amplification of 16S rRNA genes from Frankia strains in root nodules of Ceanothus griseus, Coriaria arborea, Coriaria plumosa, Discaria toumatou, and Purshia tridentata. Appl Environ Microbiol. 1996;62:2904–2909. doi: 10.1128/aem.62.8.2904-2909.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clawson ML, Bourret A, Benson DR. Assessing the phylogeny of Frankia-actinorhizal plant nitrogen-fixing root nodule symbioses with Frankia 16S rRNA and glutamine synthetase gene sequences. Mol Phylogenet Evol. 2004;31:131–138. doi: 10.1016/j.ympev.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 34.Clawson ML, Benson DR, Resch SC, Stephens DW, Silvester WB. Typical Frankia infect actinorhizal plants exotic to New Zealand. N Z J Bot. 1997;35:361–367. doi: 10.1080/0028825X.1997.10410161. [DOI] [Google Scholar]
- 35.Persson T, Battenberg K, Demina IV, Vigil-Stenman T, Vanden Heuvel B, Pujic P, et al. Candidatus Frankia datiscae Dg1, the actinobacterial microsymbiont of Datisca glomerata, expresses the canonical nod genes nodABC in symbiosis with its host plant. PLoS ONE. 2015;10:e0127630. doi: 10.1371/journal.pone.0127630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parniske M. Arbuscular mycorrhiza: the mother of plant root endosymbioses. Nat Rev Microbiol. 2008;6:763–775. doi: 10.1038/nrmicro1987. [DOI] [PubMed] [Google Scholar]
- 37.Maillet F, Poinsot V, André O, Puech-Pagès V, Haouy A, Gueunier M, et al. Fungal lipochitooligosaccharide symbiotic signals in arbuscular mycorrhiza. Nature. 2011;469:58–63. doi: 10.1038/nature09622. [DOI] [PubMed] [Google Scholar]
- 38.Op den Camp R, Streng A, De Mita S, Cao Q, Polone E, Liu W, et al. LysM-type mycorrhizal receptor recruited for rhizobium symbiosis in nonlegume Parasponia. Science. 2011;331:909–912. doi: 10.1126/science.1198181. [DOI] [PubMed] [Google Scholar]
- 39.Oldroyd GED. Speak, friend, and enter: signalling systems that promote beneficial symbiotic associations in plants. Nat Rev Microbiol. 2013;11:252–263. doi: 10.1038/nrmicro2990. [DOI] [PubMed] [Google Scholar]
- 40.Markmann K, Giczey G, Parniske M. Functional adaptation of a plant receptor-kinase paved the way for the evolution of intracellular root symbioses with bacteria. PLoS Biol. 2008;6:e68. doi: 10.1371/journal.pbio.0060068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gherbi H, Markmann K, Svistoonoff S, Estevan J, Autran D, Giczey G, et al. SymRK defines a common genetic basis for plant root endosymbioses with arbuscular mycorrhiza fungi, rhizobia, and Frankia bacteria. Proc Natl Acad Sci U S A. 2008;105:4928–4932. doi: 10.1073/pnas.0710618105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Svistoonoff S, Benabdoun FM, Nambiar-Veetil M, Imanishi L, Vaissayre V, Cesari S, et al. The independent acquisition of plant root nitrogen-fixing symbiosis in Fabids recruited the same genetic pathway for nodule organogenesis. PLoS ONE. 2013;8:e64515. doi: 10.1371/journal.pone.0064515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoagland DR, Arnon DI. The water-culture method for growing plants without soi. Berkeley, California: University of California, College of Agriculture, Agricultural Experiment Station; 1938. [Google Scholar]
- 44.Lundquist P-O, Huss-Danell K. Response of nitrogenase to altered carbon supply in a Frankia-Alnus incana symbiosis. Physiol Plant. 1991;83:331–338. doi: 10.1111/j.1399-3054.1991.tb00102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Husemann P, Stoye J. r2cat: synteny plots and comparative assembly. Bioinforma Oxf Engl. 2010;26:570–571. doi: 10.1093/bioinformatics/btp690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heinl S, Wibberg D, Eikmeyer F, Szczepanowski R, Blom J, Linke B, et al. Insights into the completely annotated genome of Lactobacillus buchneri CD034, a strain isolated from stable grass silage. J Biotechnol. 2012;161:153–166. doi: 10.1016/j.jbiotec.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 48.Schwientek P, Szczepanowski R, Rückert C, Kalinowski J, Klein A, Selber K, et al. The complete genome sequence of the acarbose producer Actinoplanes sp. SE50/110. BMC Genomics. 2012;13:112. doi: 10.1186/1471-2164-13-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wibberg D, Jelonek L, Rupp O, Hennig M, Eikmeyer F, Goesmann A, et al. Establishment and interpretation of the genome sequence of the phytopathogenic fungus Rhizoctonia solani AG1-IB isolate 7/3/14. J Biotechnol. 2013;167:142–155. doi: 10.1016/j.jbiotec.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 50.Huson DH, Auch AF, Qi J, Schuster SC. MEGAN analysis of metagenomic data. Genome Res. 2007;17:377–386. doi: 10.1101/gr.5969107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meyer F, Goesmann A, McHardy AC, Bartels D, Bekel T, Clausen J, et al. GenDB--an open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 2003;31:2187–2195. doi: 10.1093/nar/gkg312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wibberg D, Blom J, Jaenicke S, Kollin F, Rupp O, Scharf B, et al. Complete genome sequencing of Agrobacterium sp. H13-3, the former Rhizobium lupini H13-3, reveals a tripartite genome consisting of a circular and a linear chromosome and an accessory plasmid but lacking a tumor-inducing Ti-plasmid. J Biotechnol. 2011;155:50–62. doi: 10.1016/j.jbiotec.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 53.Maus I, Stantscheff R, Wibberg D, Stolze Y, Winkler A, Pühler A, et al. Complete genome sequence of the methanogenic neotype strain Methanobacterium formicicum MF(T.) J Biotechnol. 2014;192(Pt A):40–41. doi: 10.1016/j.jbiotec.2014.09.018. [DOI] [PubMed] [Google Scholar]
- 54.Küberl A, Schneider J, Thallinger GG, Anderl I, Wibberg D, Hajek T, et al. High-quality genome sequence of Pichia pastoris CBS7435. J Biotechnol. 2011;154:312–320. doi: 10.1016/j.jbiotec.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 55.Maus I, Wibberg D, Stantscheff R, Cibis K, Eikmeyer F-G, König H, et al. Complete genome sequence of the hydrogenotrophic archaeon Methanobacterium sp. Mb1 isolated from a production-scale biogas plant. J Biotechnol. 2013;168:734–736. doi: 10.1016/j.jbiotec.2013.10.013. [DOI] [PubMed] [Google Scholar]
- 56.Hilker R, Stadermann KB, Doppmeier D, Kalinowski J, Stoye J, Straube J, et al. ReadXplorer--visualization and analysis of mapped sequences. Bioinforma Oxf Engl. 2014;30:2247–2254. doi: 10.1093/bioinformatics/btu205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Blom J, Albaum SP, Doppmeier D, Pühler A, Vorhölter F-J, Zakrzewski M, et al. EDGAR: a software framework for the comparative analysis of prokaryotic genomes. BMC Bioinformatics. 2009;10:154. doi: 10.1186/1471-2105-10-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Felsenstein J. PHYLIP (Phylogeny Inference Package). 2005. Available from: http://evolution.genetics.washington.edu/phylip.html
- 59.Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, et al. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011;39:W339–W346. doi: 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E, et al. antiSMASH 2.0--a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013;41:W204–W212. doi: 10.1093/nar/gkt449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eikmeyer FG, Köfinger P, Poschenel A, Jünemann S, Zakrzewski M, Heinl S, et al. Metagenome analyses reveal the influence of the inoculant Lactobacillus buchneri CD034 on the microbial community involved in grass ensiling. J Biotechnol. 2013;167:334–343. doi: 10.1016/j.jbiotec.2013.07.021. [DOI] [PubMed] [Google Scholar]
- 62.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57:81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 64.Konstantinidis KT, Tiedje JM. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci U S A. 2005;102:2567–2572. doi: 10.1073/pnas.0409727102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wibberg D, Rupp O, Blom J, Jelonek L, Kröber M, Verwaaijen B, et al. Development of a Rhizoctonia solani AG1-IB specific gene model enables comparative genome analyses between phytopathogenic R. solani AG1-IA, AG1-IB, AG3 and AG8 isolates. PLoS ONE. 2015;10:e0144769. doi: 10.1371/journal.pone.0144769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Darling ACE, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Darling AE, Mau B, Perna NT. ProgressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinforma Oxf Engl. 2011;27:1164–1165. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zwickl DJ. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. 2006. Available from: https://repositories.lib.utexas.edu/handle/2152/2666. Accessed 18 May 2012.
- 71.Doyle J, Doyle J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1987;19:11–15. [Google Scholar]
- 72.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 74.Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JAM. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007;35:W71–W74. doi: 10.1093/nar/gkm306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sen A, Daubin V, Abrouk D, Gifford I, Berry AM, Normand P. Phylogeny of the class Actinobacteria revisited in the light of complete genomes. The orders 'Frankiales' and Micrococcales should be split into coherent entities: proposal of Frankiales ord. nov., Geodermatophilales ord. nov., Acidothermales ord. nov. and Nakamurellales ord. nov. Int J Syst Evol Microbiol. 2014;64:3821–32. doi: 10.1099/ijs.0.063966-0. [DOI] [PubMed] [Google Scholar]
- 76.Clark DD, Ensign SA. Evidence for an inducible nucleotide-dependent acetone carboxylase in Rhodococcus rhodochrous B276. J Bacteriol. 1999;181:2752–2758. doi: 10.1128/jb.181.9.2752-2758.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kucho K, Yamanaka T, Sasakawa H, Mansour SR, Uchiumi T. Different dynamics of genome content shuffling among host-specificity groups of the symbiotic actinobacterium Frankia. BMC Genomics. 2014;15:609. doi: 10.1186/1471-2164-15-609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cañizo A, Miguel C, Rodriguez-Barrueco C. The effect of pH on nodulation and growth of Coriaria myrtifolia L. Plant Soil. 1978;49:195–198. doi: 10.1007/BF02149922. [DOI] [Google Scholar]
- 79.McGlone MS, Neall VE, Clarkson BD. The effect of recent volcanic events and climatic changes on the vegetation of Mt Egmont (Mt Taranaki) N Z J Bot. 1988;26:123–144. doi: 10.1080/0028825X.1988.10410105. [DOI] [Google Scholar]
- 80.Kimber J, Ping C, Summer M, Wilding L. Handbook of Soil Science. New York: CRC Press; 2000. [Google Scholar]
- 81.Clawson ML, Benson DR. Natural diversity of Frankia strains in actinorhizal root nodules from promiscuous hosts in the family Myricaceae. Appl Environ Microbiol. 1999;65:4521–4527. doi: 10.1128/aem.65.10.4521-4527.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fernandez MP, Meugnier H, Grimont PA, Bardin R. Deoxyribonucleic acid relatedness among members of the genus Frankia. Int J Syst Bacteriol. 1989;39:424–429. doi: 10.1099/00207713-39-4-424. [DOI] [Google Scholar]
- 83.Granqvist E, Sun J, Op den Camp R, Pujic P, Hill L, Normand P, et al. Bacterial-induced calcium oscillations are common to nitrogen-fixing associations of nodulating legumes and nonlegumes. New Phytol. 2015;207:551–558. doi: 10.1111/nph.13464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wong JEMM, Midtgaard SR, Gysel K, Thygesen MB, Sørensen KK, Jensen KJ, et al. An intermolecular binding mechanism involving multiple LysM domains mediates carbohydrate recognition by an endopeptidase. Acta Crystallogr D Biol Crystallogr. 2015;71:592–605. doi: 10.1107/S139900471402793X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cérémonie H, Debellé F, Fernandez MP. Structural and functional comparison of Frankia root hair deforming factor and rhizobia Nod factor. Can J Bot. 1999;77:1293–1301. [Google Scholar]
- 86.Chabaud M, Gherbi H, Pirolles E, Vaissayre V, Fournier J, Moukouanga D, et al. Chitinase-resistant hydrophilic symbiotic factors secreted by Frankia activate both Ca2+ spiking and NIN gene expression in the actinorhizal plant Casuarina glauca. New Phytol. 2016;209:86–93. doi: 10.1111/nph.13732. [DOI] [PubMed] [Google Scholar]
- 87.Fabre S, Gully D, Poitout A, Patrel D, Arrighi J-F, Giraud E, et al. Nod factor-independent nodulation in Aeschynomene evenia required the common plant-microbe symbiotic toolkit. Plant Physiol. 2015;169:2654–2664. doi: 10.1104/pp.15.01134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Staehelin C, Schultze M, Kondorosi É, Mellor RB, Boiler T, Kondorosi A. Structural modifications in Rhizobium meliloti Nod factors influence their stability against hydrolysis by root chitinases. Plant J. 1994;5:319–330. doi: 10.1111/j.1365-313X.1994.00319.x. [DOI] [Google Scholar]
- 89.Malinverni JC, Silhavy TJ. An ABC transport system that maintains lipid asymmetry in the gram-negative outer membrane. Proc Natl Acad Sci U S A. 2009;106:8009–8014. doi: 10.1073/pnas.0903229106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mohn WW, van der Geize R, Stewart GR, Okamoto S, Liu J, Dijkhuizen L, et al. The actinobacterial mce4 locus encodes a steroid transporter. J Biol Chem. 2008;283:35368–35374. doi: 10.1074/jbc.M805496200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rodríguez DC, Ocampo M, Varela Y, Curtidor H, Patarroyo MA, Patarroyo ME. Mce4F Mycobacterium tuberculosis protein peptides can inhibit invasion of human cell lines. Pathog Dis. 2015;73. [DOI] [PubMed]
- 92.Clark LC, Seipke RF, Prieto P, Willemse J, van Wezel GP, Hutchings MI, et al. Mammalian cell entry genes in Streptomyces may provide clues to the evolution of bacterial virulence. Sci Rep. 2013;3:1109. doi: 10.1038/srep01109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Barabote RD, Xie G, Leu DH, Normand P, Necsulea A, Daubin V, et al. Complete genome of the cellulolytic thermophile Acidothermus cellulolyticus 11B provides insights into its ecophysiological and evolutionary adaptations. Genome Res. 2009;19:1033–1043. doi: 10.1101/gr.084848.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ngom M, Oshone R, Hurst SG 4th, Abebe-Akele F, Simpson S, Morris K, Sy MO, Champion A, Thomas WK, Tisa LS. Permanent draft genome sequence for Frankia sp. strain CeD, a nitrogen-fixing actinobacteria isolated from the root nodules of Casuarina equistifolia grown in Senegal. Genome Announc. 2016;4:e00265-16. [DOI] [PMC free article] [PubMed]
- 95.Trujillo ME, Bacigalupe R, Pujic P, Igarashi Y, Benito P, Riesco R, Médigue C, Normand P. Genome features of the endophytic actinobacterium Micromonospora lupini strain Lupac 08: on the process of adaptation to an endophytic life style? PLoS ONE. 2014;9:e108522. doi: 10.1371/journal.pone.0108522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ishikawa J, Yamashita A, Mikami Y, Hoshino Y, Kurita H, Hotta K, Shiba T, Hattori M. The complete genomic sequence of Nocardia farcinica IFM 10152. Proc Natl Acad Sci U S A. 2004;101:14925–14930. doi: 10.1073/pnas.0406410101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kallimanis A, Karabika E, Mavromatis K, Lapidus A, Labutti KM, Liolios K, Ivanova N, Goodwin L, Woyke T, Velentzas AD, Perisynakis A, Ouzounis CC, Kyrpides NC, Koukkou AI, Drainas C. Complete genome sequence of Mycobacterium sp. strain (Spyr1) and reclassification to Mycobacterium gilvum Spyr1. Stand Genomic Sci. 2011;5:144–153. doi: 10.4056/sigs.2265047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets supporting the conclusions of this article are included within the article and its additional files. Nucleotide sequence data for the reconstructed Frankia sp. Dg2 strains were deposited in the EBI database under Bioproject PRJEB12692.