

Abstract
Venous thromboembolism occurs frequently in cancer patients. Two variants in the factor 5 gene (F5), rs6025 encoding for the factor V Leiden mutation R506Q, and rs4524 encoding K858R, have been found to be associated with venous thromboembolism. We assessed the joint effect of active cancer and these two F5 variants on venous thromboembolism risk in a case-cohort study. Cases with a first venous thromboembolism (n=609) and a randomly selected age-weighted cohort (n=1,691) were sampled from the general population in Tromsø, Norway. Venous thromboembolism was classified as cancer-related if it occurred in the period 6 months before to 2 years after a diagnosis of cancer. Active cancer was associated with an 8.9-fold higher risk of venous thromboembolism (95% CI 7.2–10.9). The risk of cancer-related venous thromboembolism was 16.7-fold (95% CI 9.9–28.0) higher in subjects heterozygous for rs6025 compared with non-carriers of this variant without active cancer. In subjects with active cancer the risk of venous thromboembolism was 15.9-fold higher (95% CI 9.1–27.9) in those with one risk allele at rs4524, and 21.1-fold (95% CI 12.4–35.8) higher in those with two risk alleles compared with non-carriers without active cancer. A synergistic interaction was observed between active cancer and factor V Leiden (relative excess risk due to interaction 7.0; 95% CI 0.5–14.4) and rs4524 (relative excess risk due to interaction 15.0; 95% CI 7.5–29.2). The incidence of venous thromboembolism during the initial 6 months following a diagnosis of cancer was particularly high in subjects with risk alleles at these loci. This implies that the combination of cancer and F5 variants synergistically increases venous thromboembolism risk.
Introduction
Venous thromboembolism (VTE), consisting of deep vein thrombosis (DVT) and pulmonary embolism (PE), is a common and potentially fatal condition.1 The overall incidence is 1–2 per 1,000 person-years in adults and increases sharply with age.2,3 The association between VTE and cancer is well established and dates back to the 19th century when Bouillaud4 and later Trousseau5 described a relationship between certain malignancies, hypercoagulability and thrombosis. Cancer is associated with a 4- to 7-fold increased risk of VTE, and it is estimated that 15% of cancer patients develop symptomatic VTE during the course of their disease.6–8 The risk of death in cancer patients with VTE is two times greater than in those with cancer alone, and cancer-related VTE is the second leading cause of death in cancer patients.9–11
Family and twin studies have shown that 60% of the VTE risk can be attributed to genetic factors.12,13 In the last two decades, knowledge in this field has expanded and several single nucleotide polymorphisms have been identified that, if present, can affect an individual’s risk of VTE.14 These polymorphisms are single base-pair variations that have the potential to alter the function (e.g. factor V Leiden) and plasma levels (e.g. prothrombin 20210A) of proteins and are predominately located in or near genes encoding for proteins in the coagulation or fibrinolytic pathways.15
Activated factor V (FV) acts as an important cofactor in the coagulation cascade by facilitating conversion of prothrombin to thrombin and by promoting degradation of activated factor VIII together with activated protein C (APC) and protein S.16,17 Variations in the factor 5 gene (F5) commonly result in attenuated down-regulation of activated FV by APC.18 The rs4524 single nucleotide polymorphism (169542517T>C) is the result of a missense mutation (lysine to arginine) at position 2684 in the B-domain of F5 which is thought to result in APC resistance.19 A missense mutation (arginine to glutamine) at position 506 results in the well-known factor V Leiden (FVL) (1691G>A, rs6025) single nucleotide polymorphism. FVL is pro-thrombotic by two mechanisms: its APC-resistant properties and its abnormal factor VIII degradation by APC.18 Cohort studies have found that among white Americans and Europeans approximately 5–8% of the population are heterozygous for FVL.20–23
In recent years, major advances have been made in understanding the role of genetic factors in the risk of VTE. However, the majority of genetic studies have excluded individuals with cancer-related thrombosis, and the few studies that have been performed on inherited risk factors for VTE (e.g. FVL, prothrombin mutation) in cancer patients have shown inconsistent and conflicting results, likely because of insufficient statistical power due to small populations of patients.24–27 As a consequence, knowledge on the biological interaction between cancer and inherited risk factors for VTE is limited. The aim of the present study was to investigate the joint effect of inherited risk variants of F5, FVL and rs4524, and active cancer on the absolute and relative risks of VTE in a population-based case-cohort study.
Methods
Study population
Participants were recruited from the fourth (n=27,158) and sixth (n=12,984) surveys of the Tromsø Study, a single-center population-based cohort study, conducted in 1994–1995 and 2007–2008, respectively. All (Tromsø 4) or parts (Tromsø 6) of the population of the Tromsø municipality over the age of 24 were invited to participate in these surveys. The attendance rate was high, with 77% of the eligible population participating in Tromsø 4 and 66% participating in Tromsø 6. A total of 29,128 subjects aged 25 to 97 years participated in at least one of the studies. A detailed description of the Tromsø Study has previously been published elsewhere.28 The registration and validation of VTE is described in the Online Supplementary Methods.
From the date of inclusion in the Tromsø 4 (1994/95) or Tromsø 6 (2007/08) survey until the end of follow-up on December 31, 2012, there were 660 incident VTE events. The individuals who had these events were cases in our study. A sub-cohort of age-weighted subjects (n=1,793) was randomly selected from the fourth and sixth surveys of the Tromsø study (n=29,128). Subjects with a previous history of cancer (n=152) or with missing values for FVL (n=5) and F5 rs4524 (n=13) variants were excluded. Our final case-cohort therefore consisted of a total of 2,300 subjects: 609 VTE cases and 1,691 subjects in the sub-cohort. The constitution of the case-cohort study is summarized in Figure 1.
Figure 1.
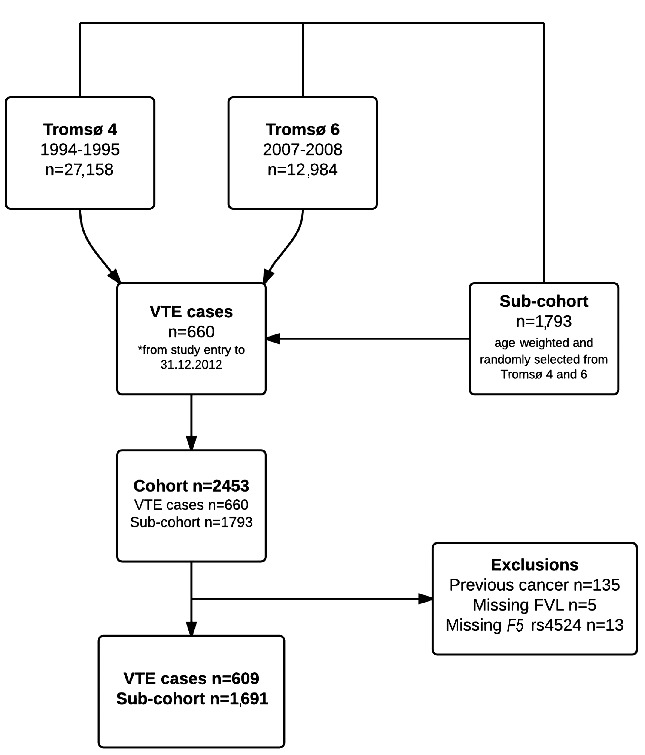
Flowchart illustrating the composition of the case–cohort study.
This study was approved by the Regional Committee for Medical and Health Research Ethics in Northern Norway (REC North) and all participants provided informed written consent to participation.
Baseline measurements
Baseline measures are described in the Online Supplementary Methods.
Sequenom genotyping and quality control
Genotyping methods are described in the Online Supplementary Methods.
Cancer assessment
Cancer assessment is described in the Online Supplementary Methods.
Definition of active cancer
Temporal proximity to cancer diagnosis is a strong predictor of VTE risk. Our study and previous studies have found that up to 50% of cancer-related VTE events occur in the 2.5-year period starting from 6 months prior to a diagnosis of cancer until 2 years after the diagnosis of cancer.29–31 The risk of VTE by time since cancer diagnosis in our cohort is illustrated in Figure 2. For the purpose of our study, a VTE event was classified as related to active cancer if it occurred within 6 months prior to a diagnosis of cancer until 2 years following the cancer diagnosis. Extending the observation period of cancer increases the likelihood of dilution due to inclusion of VTE not necessarily caused by cancer. Following the period of active cancer, subjects with cancer were classified as having a previous cancer, as the risk of VTE remains marginally increased even after the period of active cancer.
Figure 2.
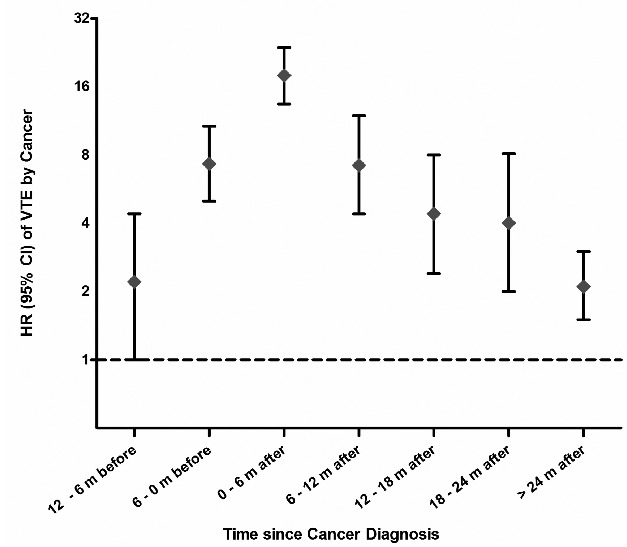
Hazard ratios for VTE at 6-month intervals starting from 1 year before cancer diagnosis.
Statistical analysis
The statistical analysis was carried out using STATA version 13.0 (Stata Corporation, College Station, TX, USA). Cox proportional hazards regression models were used to calculate age- and sex-adjusted hazard ratios (HR) with 95% confidence interval (CI) for VTE across categories of cancer (no, active and previous) and gene variants. Subjects were considered heterozygous if one risk allele was present at the locus of interest and homozygous if two risk alleles were present. Cancer-free subjects without the risk alleles were used as the reference group. In addition to calculating hazard ratios for VTE, sub-group analyses were performed separately for DVT and PE. The proportional hazards assumption was confirmed by the use of the Schoenfeld global test.
Cancer was entered as a time-varying co-variate in the model. The data were split with respect to the date of cancer diagnosis to distinguish those related to active and previous cancer. Hence, subjects who developed cancer during follow-up contributed with “non-exposed” person-time from the baseline inclusion date to 6 months before cancer diagnosis, with “active cancer exposed” person-time during the active cancer period (−6 to 24 months around the date of cancer diagnosis), and with “previous cancer exposed” person-time in the period >24 months following a diagnosis of cancer. We used the number of person-years from the original cohort (n=29,128) as a basis for the calculation of absolute risks.
Methods for assessing synergism between the F5 variants and active cancer on VTE risk are described in detail in the Online Supplementary Methods.
We used Kaplan-Meier curves to estimate cumulative incidence to illustrate time to VTE in subjects with active cancer and the presence of risk alleles. The Fine-Gray model32 was applied for sensitivity analysis to account for mortality as a competing event.
Results
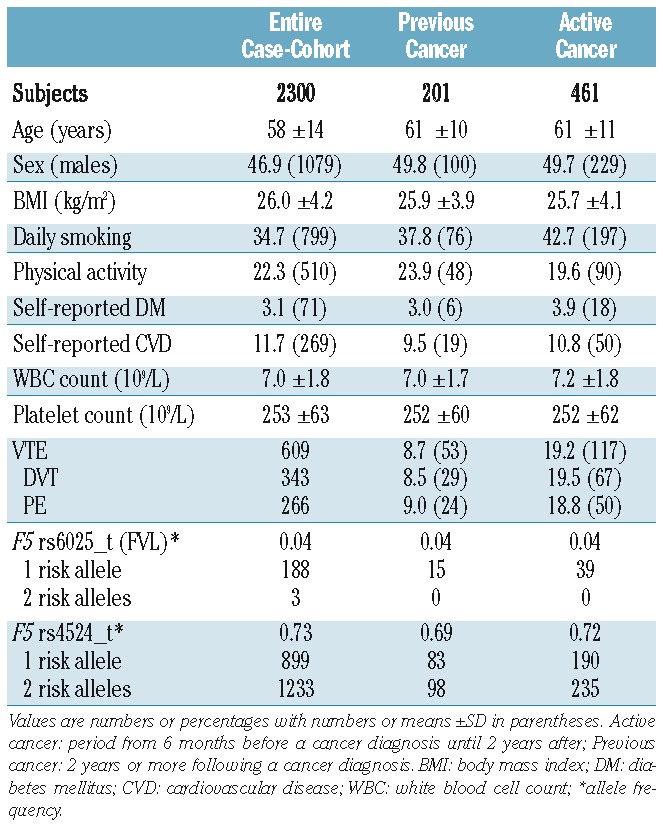
There were 461 subjects diagnosed with cancer during a median of 12.9 years follow-up. The distribution of cancer by cancer site is presented in Online Supplementary Table S1. The mean age at cancer diagnosis was 69 years. The baseline characteristics of subjects according to cancer status are presented in Table 1. The median age of all subjects was 59 years. The proportion of current smokers was higher in the cancer population (41.2% versus 34.7%).
Table 1.
Baseline characteristics of the study population according to cancer status.
Of the 609 subjects with VTE, 343 (56.3%) had DVT and 266 (43.7%) had PE; 117 of the VTE events were cancer-related. A VTE event was classified as related to active cancer if it occurred within 6 months prior to the diagnosis of cancer until 2 years following a cancer diagnosis. There were 53 events in the previous cancer group and the remaining 439 events occurred in the non-cancer group.
Allele frequencies for FVL and F5 rs4524 are presented in Table 1. The FVL variant was heterozygous in 188 of 2,300 subjects (8.2%) and homozygous in three (0.1%). Similar to a previously cited reference population (allele frequency 0.05),33 the overall allele frequency of FVL was 0.042 in our cohort. The allele frequency was substantially higher in cases than in the sub-cohort (0.073 versus 0.031, respectively). The allele frequency of the F5 rs4524 variant was 0.732 in our population, which is similar to that in a reference population of European ancestry (0.736).34 The frequency of this allele was also higher in VTE cases than in the sub-cohort (0.775 versus 0.732, respectively). There were 899 heterozygous individuals (39.1%) and 1,233 homozygous individuals (53.6%). With the exception of FVL and urological cancers (allele frequency 0.104, P=0.009) and FVL and pancreatic cancer (allele frequency 0.167, P=0.001), which has been previously described,35 the allele frequencies of FVL and F5 rs4524 in subjects with various different types of cancer were similar to those in non-cancer subjects.
Active cancer was associated with an overall 8.9-fold (95% CI 7.2–10.9) higher risk of VTE, with an age- and sex-adjusted hazard ratio of 8.5 (95% CI 6.9–10.5). In subjects with active cancer, the risk of DVT was increased 9.4-fold (95% CI 7.1–12.3) and the risk of PE 8.3-fold (95% CI 6.1–11.4), when compared to the risks in subjects without active cancer. Based on person-year information from the full cohort, the incidence rate of VTE was 1.6 per 1,000 person-years in the total population and 13.6 per 1,000 person-years in subjects with active cancer. The incidence rate of VTE was 29.4 per 1,000 person-years in subjects with FVL and active cancer, and 12.5 and 16.7 per 1,000 person-years in subjects with active cancer and one or two risk alleles at F5 rs4524, respectively.
The risk estimates for VTE by categories of FVL and active cancer are shown in Table 2. Among the subjects with active cancer, 39 (8.5%) were heterozygous for FVL; there were no homozygous individuals. Among the non-cancer subjects, the risk of VTE was increased 2.1-fold (95% CI 1.6–2.7) in the presence of one FVL risk allele and 3.3-fold (95% CI 0.8–13.2) in the presence of two risk alleles. The risk of an active cancer-related VTE was considerably higher in subjects heterozygous for FVL (HR 16.7, 95% CI 9.9–28.0) than in non-carriers without active cancer. The risk of VTE in heterozygous subjects was increased 3.4-fold (95% CI 1.7–6.8) in the group with previous cancer. Heterozygous individuals with active cancer had a 25.3-fold (95% CI 14.1–45.4) higher risk of DVT and 7-fold (95% CI 2.2–22.0) higher risk of PE. When non-carriers with active cancer were used as the reference group, there was a 1.9-fold (95% CI 1.1–3.3) and a 2.7-fold (95% CI 1.5–5.1) higher risk of VTE and DVT, respectively, in those with one FVL risk allele and active cancer (Table 3). When adjusting for the F5 rs4524 single nucleotide polymorphism in the model, the risk estimates were essentially the same (data not shown). The additive interaction (relative excess risk due to interaction, RERI) and attributable proportion due to interaction (AP) between FVL and active cancer on VTE risk are shown in Table 4. We observed a supra-additive interaction between FVL and active cancer for VTE risk (RERI; 7.0 95% CI 0.5–14.4), demonstrating a synergistic effect, as the joint effect of active cancer and FVL was stronger than the sum of the individual effects. The AP was 0.42, demonstrating that 42% of the cases in the combined group could be attributed to the interaction between the two exposures. This synergistic effect was driven mainly by the synergistic interaction on DVT risk, as a positive additive interaction was not seen for PE in subjects heterozygous for FVL with active cancer. An interaction was not, however, observed on a multiplicative scale, when fitting the interaction terms into the Cox regression model (HR 0.9, 95% CI 0.5–1.7).
Table 2.
Age- and sex-adjusted hazard ratios for VTE according to categories of F5 rs6025 (FVL) and F5 rs4524 risk alleles and cancer.
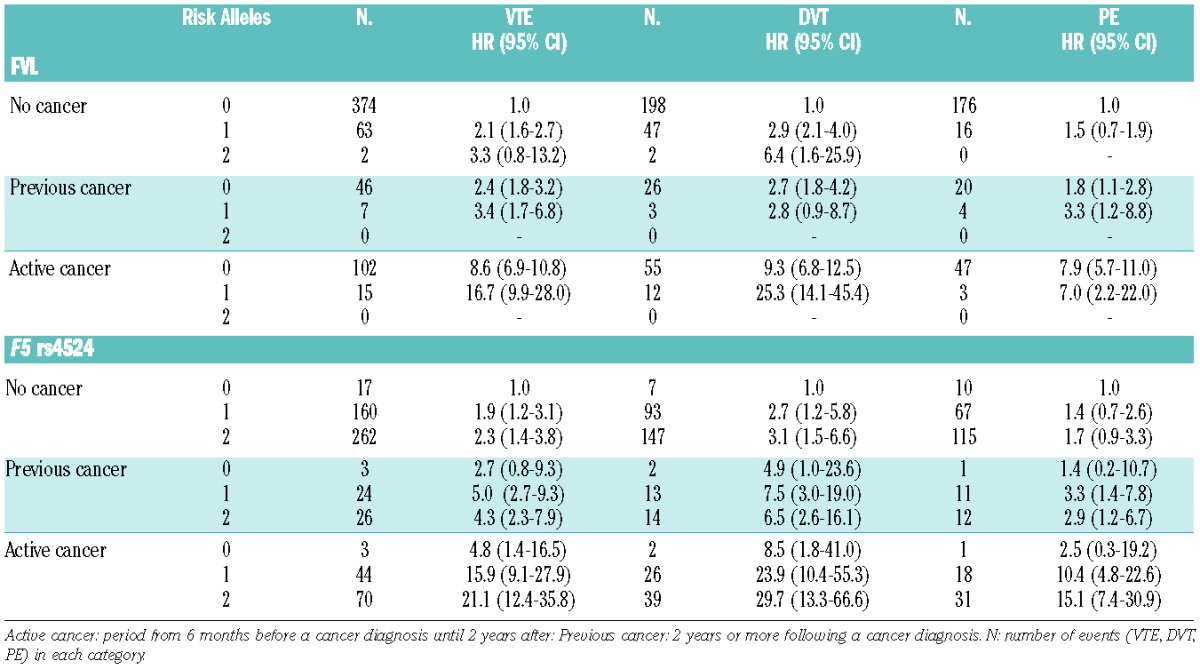
Table 3.
Age- and sex-adjusted hazard ratios for VTE by categories of FVL and F5 rs4524 risk alleles among those with active cancer.
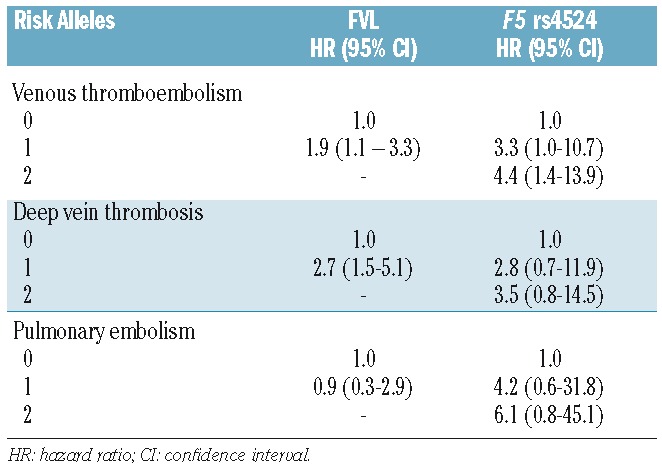
Table 4.
Additive interaction between F5 variants and active cancer.
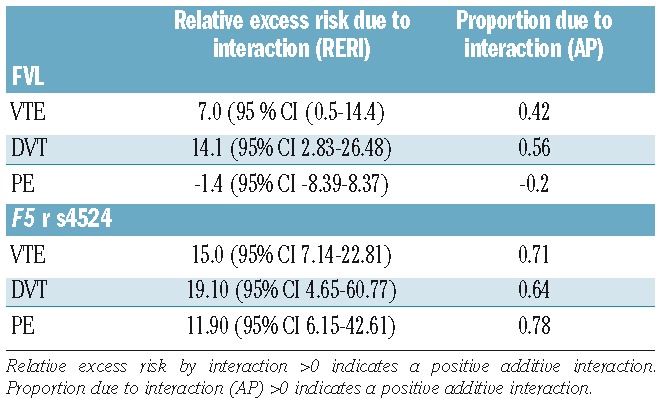
In cancer-free subjects, the risk of VTE in individuals with the F5 rs4524 variant was similar to that of subjects with FVL with a 1.9-fold (95% CI 1.2–3.1) and 2.3-fold (95% CI 1.4–3.8) higher VTE risk in subjects with one and two risk alleles, respectively, compared to non-carriers (Table 2). In the presence of active cancer, the risk of VTE increased from 15.9-fold (95% CI 9.1–27.9) in F5 rs4524 heterozygous subjects to 21.1-fold (95% 12.4–35.8) in homozygous subjects. The risk of DVT was higher across all categories of gene variants and cancer with the hazard ratios increasing to 23.9-fold (95% CI 10.4–55.3) in subjects with one risk allele and to 29.7-fold (95% CI 13.3–66.6) in those with two risk alleles. In subjects with a previous cancer, the risk of a VTE was 5-fold (95% CI 2.7–9.3) higher in F5 rs4524 heterozygous subjects and 4.3-fold (95% CI 2.3–7.9) higher in homozygous subjects. The risk estimates were 4.4-fold (95% CI 1.4–13.9) and 3.5-fold (95% CI 0.8–14.5) higher for VTE and DVT, respectively, in homozygous subjects with active cancer when non-carriers with active cancer were used as the reference group (Table 3). When adjusting for FVL, the risk estimates remained essentially similar (data not shown). We observed a supra-additive interaction (RERI; 15 95% CI 7.14–22.81) between active cancer and the single nucleotide polymorphism in F5 rs4524, demonstrating a strong synergistic effect, which was supported by an AP value of 0.71 (Table 4). Interaction on a multiplicative scale was not significant (HR 2.0, 95% CI 0.6–7.1).
Based on the entire Tromsø Study cohort (our source population for the case-cohort), we estimated that 4.2% of the subjects with cancer developed a VTE during the active cancer period. The cumulative incidence of VTE during the active cancer period in subjects without and with risk alleles is shown in Figure 3. There was a notable increase in the cumulative incidence of VTE during the initial 6 months following a cancer diagnosis, with a substantially steeper slope in the incidence curve for carriers compared to that of non-carriers of the risk alleles (Figure 3 panels A and B). The majority of the VTE events occurred during the first 6 months after a cancer diagnosis. The cumulative incidence of VTE was 10.3% in the period 0–6 months before the cancer diagnosis, and increased to 29.4% at 6 months after the cancer diagnosis for those heterozygous for FVL. Likewise, the cumulative incidence of VTE increased from 6.42% in the period 0–6 months before the cancer diagnosis to 20.2% at 6 months after the cancer diagnosis for those homozygous for F5 rs4524. The cumulative incidence of VTE at the end of the active cancer period was notably higher for individuals with one FVL allele and individuals with one or two F5 rs4524 alleles compared to those without a mutation.
Finally, we tested the impact of competing risk by death on our risk estimates for VTE by applying the Fine-Gray model. The hazard ratios and sub-distribution hazard ratios (SHR), as estimates of the relative risk of overall VTE, were nearly identical when comparing the presence of risk alleles in subjects with active cancer to that in subjects with the wild-type allele and active cancer: HR 1.9 (95% CI 1.1–3.3) versus SHR 1.9 (95% CI 0.9–3.9) for FVL, and HR 3.5 (95% CI 0.8–11.9) versus SHR 3.9 (95% CI 1.1–13.2) for F5 rs4524.
Figure 4.
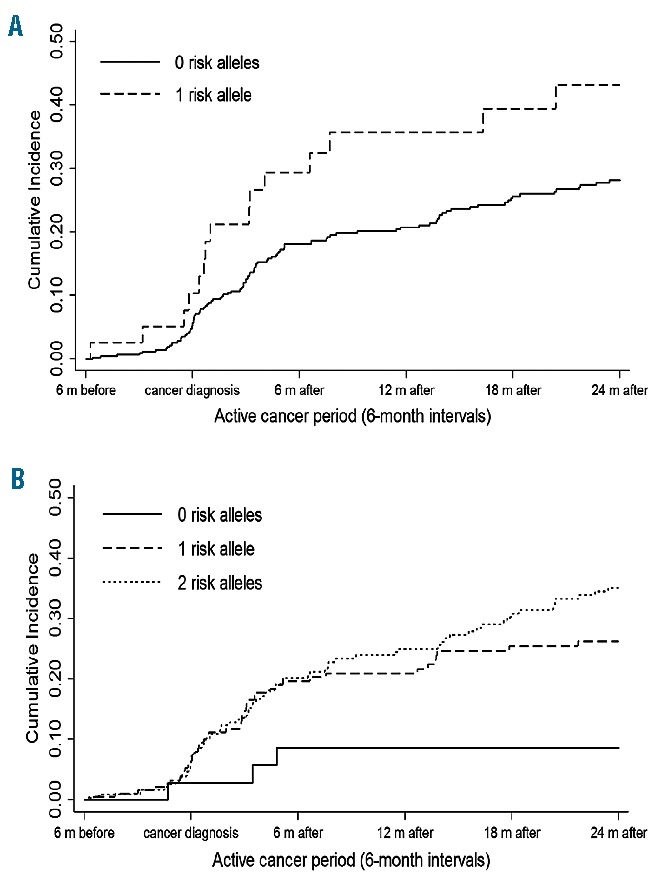
Cumulative incidence of VTE in the presence of F5 rs6025 (FVL) and F5 rs4524 risk alleles during the active cancer period. (A) Kaplan-Meier estimates of cumulative incidence in the presence of F5 rs6025 (FVL) risk alleles during the active cancer period. (B) Kaplan-Meier estimates of cumulative incidence in the presences of F5 rs4524 risk alleles during the active cancer period.
Discussion
We found that both F5 variants (rs4524 and FVL) were associated with a higher risk of VTE, and substantially so in individuals with active cancer. On an additive scale, there was a synergistic effect of active cancer and the F5 rs4524 and FVL variants on the risk of VTE. The risk of VTE increased for each additional risk allele. The incidence of VTE was highest during the first 6 months following the cancer diagnosis, which is likely due to cancer treatment-related factors, such as surgery, chemotherapy, acute infections, and central venous catheters.
Previous studies determined that the presence of F5 rs4524 risk alleles is associated with a moderately higher risk (26–67%) of DVT in middle-aged populations,36 of VTE in post-menopausal women,37 and of VTE in the antenatal period.38 The risk of VTE increased weakly in an allele-dependent manner and risk estimates were higher in post-menopausal women with additional risk factors.37 Accordingly, we found that F5 rs4524 in individuals without cancer was associated with a moderate allele-dependent increased risk of VTE. To date, there have been, to the best of our knowledge, no other studies exploring the impact of F5 rs4524 on cancer-related VTE. In our study, active cancer had a synergistic effect with F5 rs4524 on the risk of VTE. The combined effect was stronger for DVT than for PE. The genetic risk difference between DVT and PE appears to follow the same pattern as has been previously described for FVL.39 We observed that the presence of F5 rs4524 had a particularly strong impact on the incidence of VTE during the first 6 months after the diagnosis of cancer.
Active cancer displayed synergism, on an additive scale, with FVL on the VTE risk, an effect mainly driven by the impact of the interaction on DVT risk. Previously, few studies had investigated the role of FVL in cancer-related VTE. Initial results from small cancer cohorts, including 75–353 patients, reported either no40,41 or a 2- to 4.4-fold higher risk24,42 of VTE in cancer patients with FVL compared to those without the FVL mutation. In a large population-based VTE case-control study with cancer diagnoses registered during 5 years prior to the inclusion date, Blom and colleagues found a 2.4-fold and 12-fold higher VTE risk in individuals with FVL and cancer compared to those with cancer without the mutation, and those without cancer and the mutation, respectively.25 The latter findings support our observation of a joint effect of cancer and FVL on VTE risk. In a larger cohort of 982 cancer patients, Pabinger and colleagues reported a 2.0-fold (95% CI 1.0–3.97) higher risk of cancer-related VTE among subjects with FVL.43 In agreement with our findings, they described that the presence of the FVL had a particular strong impact on the incidence of VTE during the first 6 months after the cancer diagnosis.
Interactions can be statistical or biological. A statistical interaction refers to the departure from the underlying form of a statistical model, and for regression analysis, it is normally assessed by entering a product term into the regression model. A biological interaction means that two causes are both required to precipitate disease; i.e. that the two causes are component causes in the same causal model (the effect of one is biologically dependent on the presence of another). A biological interaction is not dependent on the underlying statistical model as it always refers to departure from additivity.44,45 The underlying mechanisms for the synergistic effect of cancer and mutations at sites rs6025 and rs4524 in the F5 gene on VTE risk may involve factors related to APC resistance. Both FVL and F5 rs4524 have been reported to exhibit their prothrombotic actions by attenuated down-regulation of activated FV by APC.19 Previous studies have also found an increased prevalence of acquired APC resistance in patients with cancer.46–48 It is licit to assume that two sources of APC resistance (i.e. acquired and inherited) in cancer patients would greatly increase the risk of a VTE, similarly to the synergistic effect of oral contraceptive use in individuals with FVL, as both result in a poor response to APC.49,50
Our findings of a pronounced increased risk of VTE during the first 6 months after cancer diagnosis in subjects with risk alleles fit well with the thrombosis potential model.51 This model illustrates how individual risk factors alone may be insufficient to trigger VTE. Inherited risk factors, such as FVL and rs4524, alone only mildly increased the VTE risk. Cancer itself is a strong provoking factor for VTE, and in the presence of inherited risk factors the thrombosis potential is further increased. Additional treatment-related provoking factors such as surgery, chemotherapy, and central venous catheters along with treatment-related complications including acute infections and immobilization, further elevate the risk of VTE. Accordingly, VTE occurs when the thrombosis potential exceeds the thrombosis threshold as a consequence of additive or synergistic effects of accumulated risk factors. These aggregated conditions may explain why, in individuals with these risk alleles, there is such a considerable increase in the number of VTE events in the initial months following a cancer diagnosis.
According to a recent Cochrane review, thromboprophylaxis with low molecular weight heparin given to ambulatory cancer patients receiving chemotherapy lowered the incidence of symptomatic VTE by 47% at the expense of a non-significant increase in major bleeding (30%).52 Even though cancer patients are at a high risk of VTE, and cancer patients with VTE have shortened life expectancy,8,10 current international guidelines do not recommend medical thromboprophylaxis for ambulatory cancer patients without additional risk factors due to an uncertain benefit to harm (i.e. bleeding risk) ratio.53,54 It is, therefore, very important to recognize individuals at high risk of cancer-related VTE in order to identify those patients who would benefit from prophylaxis. Current risk prediction models,55,56 are based on cancer localization and laboratory parameters influenced by several factors (i.e. infection/inflammation, surgery, various medications, and dehydration). A validation study revealed that these risk prediction models may have limited clinical utility because of their low potential to identify patients in the high-risk category (12% of the entire cancer cohort study population) and inadequate capacity to predict VTE in the high-risk subjects, as only 7% of the high-risk subjects developed a VTE over 2.5 months.55 As risk alleles for VTE in the F5 gene are common, exhibit synergistic effects with active cancer on VTE risk, and particularly discriminate patients at risk during the first 6 months after cancer diagnosis, these may be attractive candidates to pursue in future research on prediction models of VTE risk in cancer patients.
A major strength of our study is its prospective design with subjects recruited from a general population. The high participation rate in the Tromsø study and the broad age range formed a cohort that is representative of the general population and also eliminated selection bias in the sub-cohort. The long duration of follow-up enabled us to capture a large quantity of cancer events in the study population. Furthermore, all VTE and cancer events were objectively confirmed and systematically validated. The main limitation of our study was insufficient power for subgroup analysis of VTE (i.e. PE) in individuals with active cancer, demonstrated by the wide confidence intervals for our risk estimates in these categories. Secondly, information regarding cancer treatment modalities was unfortunately not available. Such data could have provided additional insight into the relationship between genes and treatment-related risk factors.
In conclusion, there is a synergistic effect of the FVL and rs4524 single nucleotide polymorphisms and active cancer on the risk of VTE. Future studies should address whether information on the presence of these polymorphisms can improve prediction of VTE in cancer patients.
Acknowledgments
The K.G. Jebsen Thrombosis Research and Expertise Center is supported by an independent grant from the K.G. Jebsen foundation.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/9/1046
References
- 1.Heit JA. Venous thromboembolism: disease burden, outcomes and risk factors. J Thromb Haemost. 2005;3(8):1611–1617. [DOI] [PubMed] [Google Scholar]
- 2.Naess IA, Christiansen SC, Romundstad P, Cannegieter SC, Rosendaal FR, Hammerstrom J. Incidence and mortality of venous thrombosis: a population-based study. J Thromb Haemost. 2007;5(4):692–699. [DOI] [PubMed] [Google Scholar]
- 3.Cushman M, Tsai AW, White RH, et al. Deep vein thrombosis and pulmonary embolism in two cohorts: the longitudinal investigation of thromboembolism etiology. Am J Med. 2004;117(1):19–25. [DOI] [PubMed] [Google Scholar]
- 4.Bouillaud S. De l’obliteration des veines et de son influence sur la formation des hydropisies partielles: considerations sur l’hydropisie passive et generale. Arch Gen Med. 1823;1:188–204. [Google Scholar]
- 5.Trousseau A. Lectures on clinical medicine: Delivered at the Hotel-Dieu, Paris. The New Syndenham Society 1872. [Google Scholar]
- 6.Agnelli G, Verso M. Thrombosis and cancer: clinical relevance of a dangerous liaison. Haematologica. 2005;90(2):154–156. [PubMed] [Google Scholar]
- 7.Heit JA, Silverstein MD, Mohr DN, et al. Risk factors for deep vein thrombosis and pulmonary embolism: a population-based case-control study. Arch Intern Med. 2000;160(6):809–815. [DOI] [PubMed] [Google Scholar]
- 8.Timp JF, Braekkan SK, Versteeg HH, Cannegieter SC. Epidemiology of cancer-associated venous thrombosis. Blood. 2013;122(10):1712–1723. [DOI] [PubMed] [Google Scholar]
- 9.Sorensen HT, Mellemkjaer L, Olsen JH, Baron JA. Prognosis of cancers associated with venous thromboembolism. N Engl J Med. 2000;343(25):1846–1850. [DOI] [PubMed] [Google Scholar]
- 10.Khorana AA, Francis CW, Culakova E, Kuderer NM, Lyman GH. Thromboembolism is a leading cause of death in cancer patients receiving outpatient chemotherapy. J Thromb Haemost. 2007;5(3):632–634. [DOI] [PubMed] [Google Scholar]
- 11.Monreal M, Trujillo-Santos J. Lessons from VTE registries: the RIETE experience. Best Pract Res Clin Haematol. 2009;22(1):25–33. [DOI] [PubMed] [Google Scholar]
- 12.Larsen TB, Sorensen HT, Skytthe A, Johnsen SP, Vaupel JW, Christensen K. Major genetic susceptibility for venous thromboembolism in men: a study of Danish twins. Epidemiology. 2003;14(3): 328–332. [PubMed] [Google Scholar]
- 13.Souto JC, Almasy L, Borrell M, et al. Genetic susceptibility to thrombosis and its relationship to physiological risk factors: the GAIT study. Genetic Analysis of Idiopathic Thrombophilia. Am J Hum Genet. 2000;67(6):1452–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morange PE, Tregouet DA. Current knowledge on the genetics of incident venous thrombosis. J Thromb Haemost. 2013;11(Suppl 1):111–121. [DOI] [PubMed] [Google Scholar]
- 15.Heit JA, Cunningham JM, Petterson TM, Armasu SM, Rider DN, de Andrade M. Genetic variation within the anticoagulant, procoagulant, fibrinolytic and innate immunity pathways as risk factors for venous thromboembolism. J Thromb Haemost. 2011;9(6):1133–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mann KG, Kalafatis M. Factor V: a combination of Dr Jekyll and Mr Hyde. Blood. 2003;101(1):20–30. [DOI] [PubMed] [Google Scholar]
- 17.Nicolaes GA, Dahlback B. Factor V and thrombotic disease: description of a Janus-faced protein. Arterioscler Thromb Vasc Biol. 2002;22(4):530–538. [DOI] [PubMed] [Google Scholar]
- 18.Vos HL. Inherited defects of coagulation factor V: the thrombotic side. J Thromb Haemost. 2006;4(1):35–40. [DOI] [PubMed] [Google Scholar]
- 19.Kostka H, Siegert G, Schwarz T, et al. Frequency of polymorphisms in the B-domain of factor V gene in APC-resistant patients. Thromb Res. 2000;99(6):539–547. [DOI] [PubMed] [Google Scholar]
- 20.Severinsen MT, Overvad K, Johnsen SP, et al. Genetic susceptibility, smoking, obesity and risk of venous thromboembolism. Br J Haematol. 2010;149(2):273–279. [DOI] [PubMed] [Google Scholar]
- 21.Kujovich JL. Factor V Leiden thrombophilia. Genet Med. 2011;13(1):1–16. [DOI] [PubMed] [Google Scholar]
- 22.Sode BF, Allin KH, Dahl M, Gyntelberg F, Nordestgaard BG. Risk of venous thromboembolism and myocardial infarction associated with factor V Leiden and prothrombin mutations and blood type. CMAJ. 2013;185(5):E229–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rees DC. The population genetics of factor V Leiden (Arg506Gln). Br J Haematol. 1996;95(4):579–586. [DOI] [PubMed] [Google Scholar]
- 24.Pihusch R, Danzl G, Scholz M, et al. Impact of thrombophilic gene mutations on thrombosis risk in patients with gastrointestinal carcinoma. Cancer. 2002;94(12): 3120–3126. [DOI] [PubMed] [Google Scholar]
- 25.Blom JW, Doggen CM, Osanto S, Rosendaal FR. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA. 2005;293(6):715–722. [DOI] [PubMed] [Google Scholar]
- 26.Kennedy M, Andreescu ACM, Greenblatt MS, et al. Factor V Leiden, prothrombin 20210A and the risk of venous thrombosis among cancer patients. Br J Haematol. 2005;128(3):386–388. [DOI] [PubMed] [Google Scholar]
- 27.Eroglu A, Ulu A, Cam R, Kurtman C, Akar N. Prevalence of factor V 1691 G-A (Leiden) and prothrombin G20210A polymorphisms and the risk of venous thrombosis among cancer patients. J Thromb Thrombolysis. 2007;23(1):31–34. [DOI] [PubMed] [Google Scholar]
- 28.Jacobsen BK, Eggen AE, Mathiesen EB, Wilsgaard T, Njølstad I. Cohort profile: the Tromsø study. Int J Epidemiol. 2011;41(4): 961–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.White RH, Chew HK, Zhou H, et al. INcidence of venous thromboembolism in the year before the diagnosis of cancer in 528 693 adults. Arch Intern Med. 2005;165(15):1782–1787. [DOI] [PubMed] [Google Scholar]
- 30.Walker AJ, Card TR, West J, Crooks C, Grainge MJ. Incidence of venous thromboembolism in patients with cancer - a cohort study using linked United Kingdom databases. Eur J Cancer. 2013;49(6):1404–1413. [DOI] [PubMed] [Google Scholar]
- 31.Blix K, Severinsen M, Braekkan S, et al. Cancer-related venous thromboembolism in the general population: results from the Scandinavian Thrombosis and Cancer (STAC) study. J Thromb Haemost. 2015; 13(Suppl 2):549. [Google Scholar]
- 32.Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc. 1999;94(446):496–509. [Google Scholar]
- 33.Morange PE, Tregouet DA. Lessons from genome-wide association studies in venous thrombosis. J Thromb Haemost. 2011;9(Suppl 1):258–264. [DOI] [PubMed] [Google Scholar]
- 34.Germain M, Chasman Daniel I, de Haan H, et al. Meta-analysis of 65,734 Individuals Identifies TSPAN15 and SLC44A2 as two susceptibility loci for venous thromboembolism. Am J Hum Genet. 2015;96(4):532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mozsik G, Rumi G, Domotor A, et al. Involvement of serum retinoids and Leiden mutation in patients with esophageal, gastric, liver, pancreatic, and colorectal cancers in Hungary. World J Gastroenterol. 2005;11(48):7646–7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bezemer ID, Bare LA, Doggen CJ, et al. Gene variants associated with deep vein thrombosis. JAMA. 2008;299(11):1306–1314. [DOI] [PubMed] [Google Scholar]
- 37.Smith NL, Hindorff LA, Heckbert SR, et al. Association of genetic variations with nonfatal venous thrombosis in postmenopausal women. JAMA. 2007;297(5):489–498. [DOI] [PubMed] [Google Scholar]
- 38.Dahm AE, Tiscia G, Holmgren A, et al. Genetic variations in the annexin A5 gene and the risk of pregnancy-related venous thrombosis. J Thromb Haemost. 2015;13(3):409–413. [DOI] [PubMed] [Google Scholar]
- 39.Corral J, Roldán V, Vicente V. Deep venous thrombosis or pulmonary embolism and factor V Leiden: enigma or paradox. Haematologica. 2010;95(6):863–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ravin AJ, Edwards RP, A Krohn M, Kelley JR, Christopherson WA, Roberts JM. The factor V Leiden mutation and the risk of venous thromboembolism in gynecologic oncology patients. Obstet Gynecol. 2002;100(6):1285–1289. [DOI] [PubMed] [Google Scholar]
- 41.Ramacciotti E, Wolosker N, Puech-Leao P, et al. Prevalence of factor V Leiden, FII G20210A, FXIII Val34Leu and MTHFR C677T polymorphisms in cancer patients with and without venous thrombosis. Thromb Res. 2003;109(4):171–174. [DOI] [PubMed] [Google Scholar]
- 42.Otterson GA, Monahan BP, Harold N, Steinberg SM, Frame JN, Kaye FJ. Clinical significance of the FV:Q506 mutation in unselected oncology patients. Am J Med. 1996;101(4):406–412. [DOI] [PubMed] [Google Scholar]
- 43.Pabinger I, Ay C, Dunkler D, et al. Factor V Leiden mutation increases the risk for venous thromboembolism in cancer patients – results from the Vienna Cancer And Thrombosis Study (CATS). J Thromb Haemost. 2014;13(1):17–22. [DOI] [PubMed] [Google Scholar]
- 44.Rothman KJ, Greenland S, Walker AM. Concepts of Interaction. Am J Epidemiol. 1980;112(4):467–470. [DOI] [PubMed] [Google Scholar]
- 45.Knol MJ, van der Tweel I, Grobbee DE, Numans ME, Geerlings MI. Estimating interaction on an additive scale between continuous determinants in a logistic regression model. Int J Epidemiol. 2007;36(5): 1111–1118. [DOI] [PubMed] [Google Scholar]
- 46.Haim N, Lanir N, Hoffman R, Haim A, Tsalik M, Brenner B. Acquired activated protein C resistance is common in cancer patients and is associated with venous thromboembolism. Am J Med. 2001;110(2):91–96. [DOI] [PubMed] [Google Scholar]
- 47.Green D, Maliekel K, Sushko E, Akhtar R, Soff GA. Activated-protein-C resistance in cancer patients. Haemostasis. 1997;27(3):112–118. [DOI] [PubMed] [Google Scholar]
- 48.De Lucia D, De Vita F, Orditura M, et al. Hypercoagulable state in patients with advanced gastrointestinal cancer: evidence for an acquired resistance to activated protein C. Tumori. 1997;83(6):948–952. [DOI] [PubMed] [Google Scholar]
- 49.Vandenbroucke JP, Koster T, Rosendaal FR, Briët E, Reitsma PH, Bertina RM. Increased risk of venous thrombosis in oral-contraceptive users who are carriers of factor V Leiden mutation. Lancet. 1994;344(8935): 1453–1457. [DOI] [PubMed] [Google Scholar]
- 50.Hellgren M, Svensson PJ, Dahlback B. Resistance to activated protein C as a basis for venous thromboembolism associated with pregnancy and oral contraceptives. Am J Obstet Gynecol. 1995;173(1):210–213. [DOI] [PubMed] [Google Scholar]
- 51.Rosendaal FR. Venous thrombosis: a multicausal disease. Lancet. 1999;353(9159): 1167–1173. [DOI] [PubMed] [Google Scholar]
- 52.Di Nisio M, Porreca E, Otten HM, Rutjes AW. Primary prophylaxis for venous thromboembolism in ambulatory cancer patients receiving chemotherapy. Cochrane Database Syst Rev. 2014;(8):CD008500. [DOI] [PubMed] [Google Scholar]
- 53.Kahn SR, Lim W, Dunn AS, et al. Prevention of VTE in nonsurgical patients: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(Suppl 2):e195S–226S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lyman GH, Khorana AA, Kuderer NM, et al. Venous thromboembolism prophylaxis and treatment in patients with cancer: American Society of Clinical Oncology clinical practice guideline update. J Clin Oncol. 2013; 31(17):2189–2204. [DOI] [PubMed] [Google Scholar]
- 55.Khorana AA, Kuderer NM, Culakova E, Lyman GH, Francis CW. Development and validation of a predictive model for chemotherapy-associated thrombosis. Blood. 2008;111(10):4902–4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ay C, Dunkler D, Marosi C, et al. Prediction of venous thromboembolism in cancer patients. Blood. 2010;116(24):5377–5382. [DOI] [PubMed] [Google Scholar]