

Abstract
We report the synthesis of M2NH3 which is a tetracationic analogue of our prototypical acyclic CB[n]-type molecular container M2. Both M1NH3 and M2NH3 possess excellent solubility in D2O and do not undergo intermolecular self-association processes that would impinge on their molecular recognition properties. Compounds M1NH3 and M2NH3 do, however, undergo an intramolecular self-complexation process driven by ion-dipole interactions between the ureidyl C=O portals and the OCH2CH2NH3 arms along with inclusion of one aromatic wall in its own hydrophobic cavity. The Ka values for M1NH3 and M2NH3 toward seven nucleotides were determined by 1H NMR titration and found to be quite modest (Ka in the 102 – 103 M−1 range) although M2NH3 is slightly more potent. The more highly charged guests (e.g. ATP) form stronger complexes with M1NH3 and M2NH3 than the less highly charged guest (e.g. ADP, AMP). The work highlights the dominant influence of the ureidyl C=O portals on the molecular recognition behavior of acyclic CB[n]-type receptors and suggests routes (e.g. more highly charged arms) to enhance their recognition behavior toward anions.
Introduction
Since the pioneering work of Pedersen, Lehn, and Cram more than 40 years ago the development of the field of supramolecular chemistry has witnessed the creation ever more complex receptor molecules whose structures are tailored toward specific molecular targets.1 For example, macrocyclic molecular container compounds (e.g. crown ethers, cyclodextrins, calixarenes, cyclophanes, pillararenes)2 have served as core structures of more complex receptors prepared by covalent reactions or by self-assembly processes. These exquisitely designed hosts have been used for a variety of applications including chemical sensing,3 drug delivery,4 to promote supramolecular polymerization,5 supramolecular approaches to catalysis,6 to stabilize otherwise unstable compounds and conformations of molecules,7 to promote the assembly and/or disassembly of protein-protein interactions,8 and for various imaging applications.9
Over the past 15 years, the synthetic and supramolecular chemistry of the cucurbit[n]uril (CB[n], n = 5, 6, 7, 8, 10, 14) family of molecular containers has been unfolding rapidly.10,11 CB[n] compounds are prepared by condensation of glycoluril and formaldehyde under aqueous acidic conditions and feature n glycoluril units connected by 2n CH2-bridges.12 CB[n] compounds possess a hydrophobic cavity that is guarded by two symmetry equivalent ureidyl carbonyl lined portals. The molecular recognition properties of CB[n] containers are unrivaled among synthetic molecular containers in that they display remarkable binding affinity (Ka up to 1017 M−1) and very high selectivity toward their guests (hydrophobic cations and even neutral molecules) in aqueous solution.13,14 CB[n] host-guest complexes form the basis for complex multistate supramolecular systems because they undergo changes in constitution and conformation in response to appropriate stimuli (e.g. pH, chemical, electrochemical, photochemical).15 For example, unfunctionalized CB[n] containers have featured prominently in a variety of applications including drug delivery,16 chemical sensing ensembles,17 supramolecular catalysis, peptide and protein recognition,18 supramolecular materials,11,19 and affinity capture materials.20 In order to further augment the recognition properties of the CB[n] systems, the development of methods for the selective (mono)functionalization of CB[n] have been pursued. As early as 2003, Kim’s group developed the perhydroxylation of CB[n] to generate (HO)2nCB[n] and has used these compounds as starting materials to prepare supramolecular systems capable of affinity capture, targeted drug delivery, and tissue engineering.14,21 In 2011 and 2012, the Isaacs group utilized their mechanistic knowledge of the CB[n] forming reaction to devise the first routes to CB[6] and CB[7] derivatives containing a single reactive functional group.22 Subsequently, the perhydroxylation reaction was tamed which allowed the isolation of monohydroxylated CB[n] compounds by the groups of Scherman, Kim, and Bardelang.23 The Isaacs group along with the Sindelar group has also been pursuing a different approach to the preparation of functionalized CB[n]-type receptors for advanced applications in the form of acyclic CB[n]-type receptors whose structures are typified by that of M1 (Figure 1).24,25-27 M1 features a central C-shaped glycoluril tetramer which endows it with the ability to bind to hydrophobic cations, two aromatic walls which form π−π interactions with aromatic target compounds, and four sodium sulfonate solubilizing groups. M1 and its relatives have been used in a variety of applications including the solubilization of insoluble pharmaceutical agents and carbon nanotubes, the creation of sensing ensembles, and the in vivo reversal of neuromuscular block.24,25,26 We have studied the influence of several structural variables (e.g. aromatic sidewall identity, solubilizing group identity (NH3+ versus OH versus SO3Na), and length of central glycoluril oligomer (monomer – tetramer)) on their function as solubilizing excipients for insoluble drugs.24,25,26 Interestingly, in our study of the recognition properties of the cationic analogue of M1 (M1NH3, Figure 1) we found that the container is not a good host for cationic compounds due to self-complexation of the NH3+ arms at the ureidyl carbonyl portals, but displayed modest affinity toward adamantanecarboxylate (Ka = 678 M−1) in 20 mM sodium phosphate buffer at pH 7.4.26 Accordingly, it appeared that the cationic arms turned M1NH3 into an anion receptor. Based on this observation, we recently prepared cationic acyclic cucurbit[n]uril dendrimers and demonstrated their ability to bind to and compact plasmid DNA although, unfortunately, gene transfection ability was quite modest.28 Previous supramolecular researchers have shown that polycationic receptors are capable of recognizing nucleotide anions in aqueous solution due to electrostatic and other non-covalent interactions.29 Within the CB[n] field, bambusurils and related compounds are known to be potent receptors for anions in water.30 Given the generally high affinity of CB[n] compounds toward their guests, we decided to investigate the binding properties of tetracationic hosts M1NH3 and M2NH3 toward nucleotides because of the confluence of aromatic moieties which are known to bind nicely to acyclic CB[n] and the anionic phosphates which should interact with the ammonium ion arms.
Figure 1.

Chemical structures of CB[n] and prototypical acyclic CB[n]-type molecular containers.
Results and Discussion
This results and discussion section is organized as follows. First, we describe the synthesis of M2NH3 and determination of its solubility and self-association in water. Next, we describe the structural features of this class of host molecules. Then, we measure the binding constants of M1NH3 and M2NH3 toward seven nucleotide guests by 1H NMR spectroscopic titration. Finally, we discuss the trends in binding affinity as a function of guest structure and as a function of ionic strength of the aqueous binding media.
Synthesis of M2NH3
Scheme 1 shows the synthesis of tetracationic acyclic CB[n]-type molecular container M2NH3. As starting materials, we selected methylene bridged glycoluril tetramer bis (cyclic ether) 131 and 1,4-bis(2-chloroethoxy)naphthalene 2.32 Upon heating in a 1:1 mixture of TFA and Ac2O, 1 and 2 undergo double electrophilic aromatic substitution reactions to install the two naphthalene walls yielding 3 in 36% yield. Tetrachloro acyclic CB[n] derivative 3 could be transformed into the correspond tetraazide functionalized acyclic CB[n]-type molecular container 4 by treatment with sodium azide in hot DMSO (80 °C) by simple SN2 chemistry. Finally, 4 could be transformed into the target tetracationic acyclic CB[n]-type molecular container M2NH3 by treatment with triphenylphospine in aqueous DMSO followed by treatment with HCl. Compound M2NH3 was characterized by all of the usual spectroscopic techniques including 1H NMR, 13C NMR, infrared spectroscopy, and electrospray ionization mass spectrometry (ES-MS). The high resolution ES-MS spectrum showed a molecular ion that was consistent with the depicted molecular formula of the singly protonated form of the free base of M2NH3 (C58H69N20O12) at m/z = 1237.5409. The 1H NMR and 13C NMR spectra recorded for 3 and 4 (in DMSO) and M2NH3 in D2O are relatively simple which reflects their overall C2v-symmetry. For example, the 13C NMR spectra (Supporting Information) display only two resonances for the ureidyl C=O groups, five resonances for the C-atoms of the aromatic ring, two CH3 signals, two resonances for the OCH2CH2X arms, along with three CH2 and four signals for the equatorial glycoluril C-atoms. Similarly, the 1H NMR spectrum of M2NH3 displays the typical AA’BB’ pattern for aromatic protons Ha and Hb, two singlets for the CH3-groups, two resonances for the equatorial glycoluril CH groups (Hk and Hl) and three pairs of doublets for the diastereotopic CH2-groups connecting two glycoluril units or a glycoluril unit with an aromatic sidewall. As expected based on theory, the resonances for the OCH2CH2NH3 arms display patterns that reflect the fact that the protons on each CH2-group are diastereotopic. Interestingly, the chemical shift of Ha and Hb of the naphthalene sidewalls of M2NH3 measured in D2O as solvent are significantly upfield shifted relative to the related chemical shifts measured for 4 in DMSO-d6 (Figure 2). This observation suggests that M2NH3, similar to M1NH3,26 assumes a self-folded conformation in which the naphthalene rings undergo π−π interactions.
Figure 2.
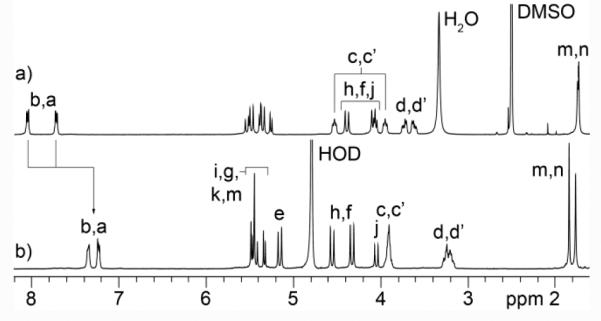
NMR spectra recorded for: a) 4 (DMSO-d6, 400 MHz, RT), b) M2NH3 (D2O, 400 MHz, RT).
X-ray Crystal Structure of 4
We were unable to obtain single crystals of M2NH3, but were lucky enough to obtain crystals and solve the x-ray crystal structure for compound 4 (CCDC-1454834). Figure 3 shows a cross-eyed stereoview of one molecule of 4 in the crystal. As seen previously for M2, compound 4 assumes a helical conformation in which its two terminal aromatic rings are splayed out of plane with respect to the glycoluril tetramer backbone. A molecule of formic acid fills the cavity of 4 in the crystal and forms one H(C=O)O-H•••O=C H-bond (O•••O distance: 2.77 Å) to the ureidyl C=O portal of 4. Reciprocally, two molecules of H2O form a H-bonded chain from the included formic acid terminated at the opposite ureidyl C=O portal. The individual molecules of 4 exhibit an interesting three dimensional packing in the crystal whereby molecules of one handedness form zig-zag chains along the a-axis which repeat along the b-axis to give sheets of a single handedness in the ab-plane (Supporting Information). These sheets pack along the z-axis with alternating senses of chirality driven in part by interactions between naphthalene sidewalls in adjacent slabs.
Figure 3.
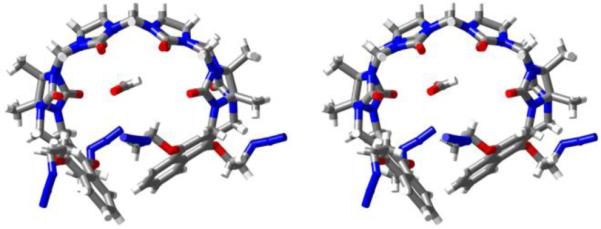
Cross-eyed stereoview of the x-ray crystal structure of one molecule of 4 in the crystal. Color code: C, grey; H, white; N, blue; O, red; H-bonds, red-yellow stripped.
Solubility Properties of M1NH3 and M2NH3
The ability to use a given molecular container compound in relevant applications requires sufficient solubility in appropriate solvents. Unfunctionalized CB[n] (n = 6, 8, 10) exhibit low solubility in water (< 100 μM) whereas CB[n] derivatives and acyclic CB[n] can exhibit dramatically enhanced solubility. For this reason we decided to determine the inherent solubility of M1NH3 and M2NH3 before proceeding to perform molecular recognition studies. Experimentally, we added M1NH3 or M2NH3 to water until a heterogenous mixture was formed and then excess insoluble host was filtered off and the concentration of M1NH3 and M2NH3 in solution was measured by 1H NMR spectroscopy relative to trimesic acid as an internal standard of known concentration. In this manner we measured the high inherent solubility of M1NH3 (250 mM) and M2NH3 (≥ 207 mM).
Container M2NH3 Does Not Undergo Significant Self-Association
Before proceeding to study any new host molecule – particularly for studies in aqueous solution – it is important to determine whether the host undergoes self-association processes that would impinge upon the expected host-guest recognition processes. Previously, we have investigated the use of M1NH3 as a solubilizing excipient for insoluble drugs and studied its self-association by 1H NMR dilution experiments in 20 mM sodium phosphate buffered D2O and did not observe any changes in chemical shift that would be indicative of self-association processes.26 Accordingly, we performed an analogous dilution experiment for M2NH3 (20 mM to 0.125 mM) and looked for changes in 1H NMR chemical shift as a function of concentration (Figure 4). As can be readily seen the host protons – most significantly those of the aromatic sidewalls – do not undergo any significant changes in chemical shift which would be indicative of self-association processes over the experimentally accessible concentration range. Accordingly, we conclude that M2NH3 exists in its monomeric form under the conditions used for Ka determination by 1H NMR titrations decribed below.
Figure 4.

1H NMR spectra recorded (D2O, 400 MHz, 20 mM sodium phosphate, pD = 7.4, RT) during the dilution experiment for M2NH3: a) 20 mM, b) 15 mM, c) 10 mM, d) 5 mM, e) 2.5 mM, f) 1 mM, g) 0.5 mM, h) 0.25 mM, i) 0.125 mM.
Structures of M1NH3 and M2NH3
Previously, we have determined the x-ray crystal structures of M1 and M2 whose aromatic sidewalls are roughly orthogonal and help define a hydrophobic box whereas the sulfonate solubilizing groups do not interact with the ureidyl C=O groups of the host. In sharp contrast, the x-ray crystal structure of M1NH3 26 (Figure 5) displays a self-folding phenomenon whereby the two o-xylylene walls undergo π−π interactions with each other which reduces the available cavity volume. In addition, one of the CH2CH2NH3 sidearms of M1NH3 folds back to the ureidyl C=O portal to form ion-dipole interactions which helps explain the fact that M1NH3 is not a good receptor for cations. Unfortunately, we have not been able to obtain x-ray quality crystals of M2NH3. However, on the basis of the x-ray crystal structure of M1NH3, we infer that M2NH3 may undergo a related self-folding phenomenon although the presence of the longer naphthalene walls of M2NH3 may induced some strain within a self-folded geometry. As described above, the observation of upfield shifting of Ha and Hb for M2NH3 in water relative to 4 as model compound strongly suggests a self-folded geometry for M2NH3 driven by C=O•••NH3+ ion-dipole interactions and resulting in intramolecular π−π interactions between the naphthalene walls of M2NH3.
Figure 5.
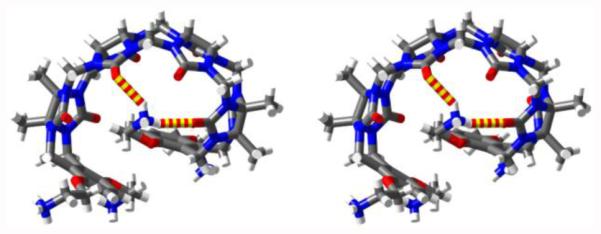
Cross-eyed stereoview of the X-ray crystal structure of M1NH3. Color code: C, grey; H, white; N, blue; O, red; H-bonds, red-yellow striped.
Binding Studies of M1NH3 and M2NH3 Toward the Nucleotide Guests
Next, we decided to measure the binding constants for the interaction of M1NH3 and M2NH3 with the various nucleotide guests. For this purpose, we decided to use 1H NMR spectroscopy and monitor the change in chemical shift of the protons on the nucleotide upon titration with the acyclic CB[n]-type molecular containers M1NH3 and M2NH3 with the expectation that the direction and magnitude of the induced changes in chemical shift (Δδ) would deliver information about the structure of the host-guest complex. Figure 7 shows the 1H NMR recorded for the titration of a fixed concentration of GTP (1 mM) with M1NH3 (0 – 8 mM) in 20 mM sodium phosphate buffered D2O at pD 7.4. The 1H NMR chemical shifts of the aromatic proton of GTP (Hp) shifts slightly upfield upon complex formation with M1NH3. In addition, the proton attached to the anomeric center (Hq) and the sugar ring (Hs) also experience small upfield shifts upon complex formation. We performed a simultaneous global fitting of the plot of change in chemical shift data versus [M1NH3] (Figure 6f) to extract the binding constant for the M1NH3•GTP complex (Ka = 234 M−1). Analogous 1H NMR titrations were performed for the M1NH3 and M2NH3 with the other nucleotides and the binding constants are presented in Table 1.
Figure 7.

a) 1H NMR spectra (400 MHz, 20 mM sodium phosphate, pD = 7.4, RT) recorded during the titration of GTP (1 mM) with M1NH3: a) 0 mM, b) 0.5 mM, c) 2 mM, d) 4 mM, e) 6 mM. Global fitting of the Δδ versus [M1NH3] data to a 1:1 binding model to determine Ka = 234 ± 20 M−1.
Figure 6.

Structures and selected proton labeling of guests used in this study.
Table 1.
Binding constants (Ka, M−1) measured for the complexes between M1NH3 and M2NH3 and the nucleotide guests.
| M1NH3 | M2NH3 | |
|---|---|---|
| ATP | 937 ± 75 | (1.47 ± 0.16) × 103 |
| UTP | 461 ± 40 | (1.10 ± 0.12) × 103 |
| GTP | 234 ± 20 | 648 ± 114 |
| CTP | 564 ± 49 | 556 ± 37 |
| ADP | 507 ± 80 | (1.25 ± 0.13) × 103 |
| AMP | 199 ± 20 | 853 ± 173 |
| cAMP | 198 ± 68 | 82 ± 15 |
The complexes between M1NH3 or M2NH3 with the seven nucleotides are all relatively weak with binding constants in the 102 – 103 M−1 range. However, there are useful trends in the binding affinity data that can be discerned. First, the binding constants of M2NH3 toward a given nucleotide are generally larger than that of M1NH3. Previously, we have observed similar trends in the binding affinity of M1 and M2 toward insoluble drugs and attributed this effect to the fact that M2 is shaped by two naphthalene rings which results in a larger hydrophobic cavity and one that offers a larger amount of π-surface area to its guests.25 We surmise that the superior binding affinity of M2NH3 relative to M1NH3 may be due to similar effects; confirmation of this interpretation awaits an x-ray crystal structure of M2NH3. Second, across the series of guests ATP, ADP, AMP, cAMP that differ only in the number of negative charges we observe a decrease in Ka toward M1NH3 and M2NH3 as guest charge decreases. The 5-fold (18-fold) decrease observed between the complexes of M1NH3•ATP versus M1NH3•cAMP (M2NH3•ATP versus M2NH3•cAMP) suggest that a significant portion of the binding free energy of these complexes are due to electrostatic interactions between the negatively charged phosphate groups and the positively charged arms of M1NH3 and M2NH3. Third, among the purines, we find that ATP forms tighter complexes than GTP toward both M1NH3 and M2NH3. We believe that this trend is due to the fact that the nucleobase of GTP is more hydrophilic than that of ATP because of the additional oxo-group. Accordingly, binding GTP in the hydrophobic cavity of M1NH3 or M2NH3 would require more extensive desolvation of the guest. Consistently, we do not observe a similar trend in the binding constants of UTP and CTP toward M1NH3 and M2NH3 because of the more comparable substituents on the bases.
To obtain structural information about the complexes between M1NH3 or M2NH3 and the seven nucleotides we obtained the maximal complexation induced changes in chemical shift (Δδ) from the simultaneous global fitting of the titration data which are summarized in Table 2. Table 2 presents Δδ values for the protons on the aromatic rings (Ho and Hp), the anomeric center (Hq), and for GTP proton (Hs) on the sugar residue. Although we were unable to monitor the other protons across the full titration because of spectral complexity or the obscuring effect of the D2O solvent we do observe that most resonances tend to shift slightly upfield upon complex formation which is consistent with the nucleotides residing in the hydrophobic cavity of M1NH3 and M2NH3. Figure 8 presents one MMFF minimized geometry of the M2NH3•ATP complex although given the relatively low Ka values we suspect a range of similar geometries are plausible. As can be seen, the aromatic base and the sugar ring are bound within the anisotropic shielding region of the acyclic CB[n]-type cavity whereas the ammonium ion arms extend toward the cavity to complement the anionic triphosphate. We believe that analogous complex geometries are likely for the complexes of M1NH3 and M2NH3 with the other nucleotides. 31P NMR measured with ATP in presence of M1NH3 or M2NH3 show a slight downshift of phosphate units compare to ATP only (for M1NH3 Δδ: Pα 0.21; Pβ 0.33; Pγ 0.39 ppm; for M2NH3 Δδ: Pα 0.02; Pβ 0.17; Pγ 0.32 ppm; Supporting Information). This observation is consistent with electrostatic / H-bonding interactions between the phosphate chain and the ammonium ion arms. To assess the importance of electrostatic interactions on the strength of the M2NH3•ATP complex we decided to perform 1H NMR titrations as a function of the concentration of the sodium phosphate buffer. Figure 9 shows a plot of Ka as a function of sodium phosphate concentration. As expected the observed Ka values decrease as sodium phosphate concentration increases presumably due to the competition / screening effect of the added salt.
Table 2.
Complexation induced changes in chemical shift (A5) for the various protons of the nucleotides for their complexes with M1NH3 and M2NH3.
| M1NH3 | M2NH3 | |||||
|---|---|---|---|---|---|---|
| Hp | Ho | Hq | Hp | Ho | Hq | |
| ATP | −0.17 | −0.05 | −0.18 | −0.26 | −0.18 | −0.26 |
| UTP | −0.18 | −0.14 | −0.14 | −0.20 | −0.17 | −0.18 |
| GTP | −0.03 | −0.07 | −0.14 | −0.04 | −0.04 (Hs) | −0.09 |
| CTP | −0.19 | −0.17 | −0.20 | −0.31 | −0.29 | −0.31 |
| ADP | −0.15 | −0.05 | −0.15 | −0.21 | −0.13 | −0.20 |
| AMP | −0.13 | −0.03 | −0.15 | −0.07 | −0.04 | −0.07 |
| cAMP | −0.01 | −0.02 | −0.04 | −0.16 | −0.19 | −0.20 |
Figure 8.
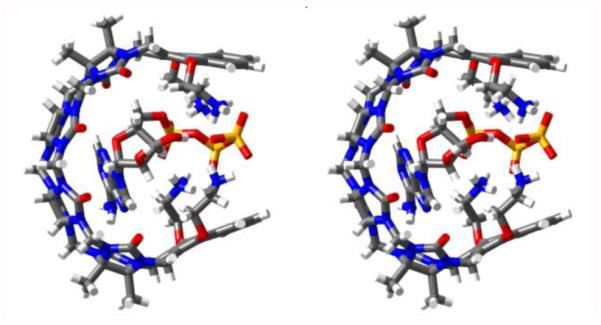
Cross-eyed stereoview of an MMFF minimized geometry for M2NH3•ATP. Color code: C, gray; H, white; N, blue; O, red; P, orange.
Figure 9.
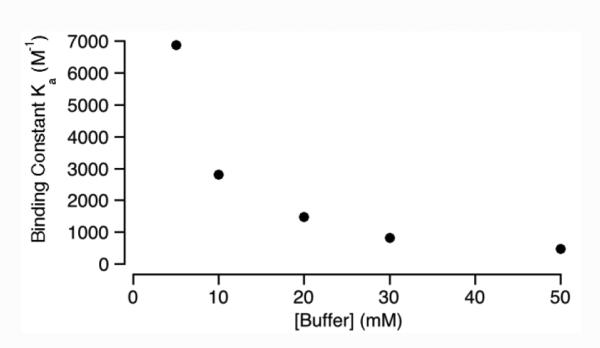
Plot of Ka versus [Buffer] for the M2NH3•ATP complex. Conditions: sodium phosphate (5 – 50 mM) buffered D2O, pD = 7.4, RT.
Conclusion
In summary, we have reported the synthesis of the tetracationic analogue of M2 which we refer to as M2NH3. We find that both M1NH3 (250 mM) and M2NH3 (≥ 207 mM) possess outstanding solubility in D2O. The results of 1H NMR dilution experiments establish that neither M1NH3 nor M2NH3 undergo significant self-association which enables their ability to act as hosts. By virtue of their electrostatically negative ureidyl C=O portals and their cationic (CH2)2NH3+ arms both M1NH3 and M2NH3 form self-complexed structures due to ion-dipole interactions at the portals and π−π interactions of one sidewall folding into its own cavity. The results of 1H NMR titrations establish that both M1NH3 and M2NH3 are relatively weak hosts toward nucleotides with Ka in the 102 – 103 M−1 range although M2NH3 is somewhat more potent than M1NH3 toward a given guest. We attribute this relatively modest affinity to the electrostatically negative ureidyl C=O portals which interact with the NH3+ arms and thereby decrease electrostatic interactions toward the negatively charged guests. As expected, along the series from ATP, ADP, AMP, to cAMP we observe that the Ka values toward M1NH3 or M2NH3 decreases as the charge on the guest decreases. In conclusion, the work highlights the powerful influence of the ureidyl C=O portals on acyclic cucurbituril recognition processes and suggests that future designs of acyclic CB[n] containers for anion recognition requires the use of arms that are more preorganized, more highly charged, and coordinate less well with the C=O portals to enhance their anion recognition abilities.
Experimental Details
Starting materials were purchased from commercial suppliers and were used without further purification. Compounds 1, 2, and M1NH3 were synthesized following the literature procedures.26,31,32 Melting points were measured on a Meltemp apparatus in open capillary tubes and are uncorrected. IR spectra were recorded on a JASCO FT/IR 4100 spectrometer and are reported in cm−1. 1H and 13C NMR spectra were measured on 400, 500, or 600 MHz instruments. Mass spectrometry was performed using a JEOL AccuTOF electrospray instrument (ESI).
Compound 3
A solution of 1 (1.498 g, 1.95 mmol) in TFA/Ac2O (1:1, 15 mL) was treated with 1,4-bis(2-chloroethoxy)naphthalene (2) (1.944 g, 6.81 mmol) and the mixture was stirred at 70 °C for 3 h under nitrogen. The mixture was poured into MeOH (100 mL) and the precipitate was isolated by filtration. The crude solid was washed with water (100 mL) and filtered. The solid was resuspended in acetone (100 mL) and filtered. Finally, the crude solid was dissolved in formic acid (10 mL), precipitated by addition of water (7 mL) and centrifuged, three times. Compound 3 was dried under high vacuum and obtained as a white solid (932 mg, 36%). M.p. > 300 °C (dec.). IR (ATR, cm−1): 2927w, 1723s, 1462s, 1310s, 1221s, 1183m, 1081m, 796s. 1H NMR (500 MHz, DMSO-d6): 8.11-8.09 (m, 4H), 7.70-7.68 (m, 4H), 5.55 (d, J = 14.6 Hz, 2H), 5.48 (d, J = 15.0 Hz, 4H), 5.38 (d, J = 9.0 Hz, 2H), 5.37 (d, J = 16.1 Hz, 4H), 5.26 (d, J = 9.0 Hz, 2H), 4.66-4.61 (m, 4H), 4.37 (d, J = 16.1 Hz, 4H), 4.10-3.93 (m, 18H), 1.74 (s, 6H), 1.72 (s, 6H). 13C NMR (125 MHz, DMSO-d6): 155.3, 154.2, 147.7, 127.8, 127.5, 126.7, 122.6, 77.5, 76.1, 74.0, 70.8, 70.4, 52.9, 48.4, 44.7, 35.9, 16.0, 15.6. MS (ESI): m/z 726 ([M+p-xylenediamine+2H]2+, calculated for C66H74Cl4N18O122+: 726).
Compound 4
A mixture of tetrachloride 3 (673 mg, 0.51 mmol) and NaN3 (667 mg, 10.26 mmol) was stirred in DMSO (10 mL) at 80 °C for 12 h. The reaction mixture was cooled to RT and water (40 mL) was added. The mixture was filtered and the solid was washed with water (40 mL) and then filtered again. Subsequently, the solid was suspended in MeOH (40 mL) and filtered, twice. Compound 4 was dried under high vacuum and obtained as a white solid (606 mg, 88%). M.p. > 300 °C (dec.). IR (ATR, cm−1): 2930w, 2102m, 1721s, 1461s, 1307s, 1220s, 1182s, 1081m, 796s. 1H NMR (400 MHz, DMSO-d6): 8.07-8.05 (m, 4H), 7.74-7.72 (m, 4H), 5.54 (d, J = 14.5 Hz, 2H), 5.49 (d, J = 15.1 Hz, 4H), 5.39 (d, J = 9.0 Hz, 2H), 5.36 (d, J = 16.0 Hz, 4H), 5.26 (d, J = 9.0 Hz, 2H), 4.55-4.51 (m, 4H), 4.39 (d, J = 16.0 Hz, 4H), 4.09 (d, J = 15.1 Hz, 4H), 4.07 (d, J = 14.5 Hz, 2H), 3.98-3.93 (m, 4H), 3.76-3.59 (m, 8H), 1.74 (s, 6H), 1.73 (s, 6H). 13C NMR (125 MHz, DMSO-d6): 155.3, 154.2, 147.8, 127.7 127.3, 126.7, 122.6, 77.5, 76.1, 73.3 70.8, 70.4, 52.9, 50.9, 48.4, 35.9, 16.1, 15.6. MS (ESI): m/z 739 ([M+p-xylenediamine+2H]2+, calculated for C66H74N30O122+: 739). X-ray crystal structure (CCDC-1454834).
Compound M2NH3
A mixture of tetraazide 4 (212 mg, 0.16 mmol) and PPh3 (314 mg, 1.20 mmol) were stirred in DMSO/H2O (5:1, 6 mL) at 80 °C for 4 h. The mixture was cooled to RT and acidified to pH 1 with HCl (6 M). Acetone (40 mL) was added and the precipitate was isolated by centrifugation. The solid was dissolved in water (2 mL) and precipitated with acetone (40 mL), four times. The solid was dried under high vacuum and obtained as a yellow solid (178 mg, 81%). M.p. > 300°C (dec.). IR (ATR, cm−1): 3359w, 2975w, 1722s, 1692s, 1461s, 1311s, 1227s, 1151s, 1064s, 1007m, 977m, 821m, 797s. 1H NMR (400 MHz, D2O): 7.36-7.34 (m, 4H), 7.24-7.22 (m, 4H), 5.49-5.41 (m, 8H), 5.33 (d, J = 8.6 Hz, 2H), 5.15 (d, J = 16.0 Hz, 4H), 4.56 (d, J = 16.2 Hz, 4H), 4.33 (d, J = 15.8 Hz, 4H), 4.05 (d, J = 15.5 Hz, 2H), 3.95-3.86 (m, 8H), 3.32-3.14 (m, 8H), 1.84 (s, 6H), 1.76 (s, 6H). 13C NMR (125 MHz, D2O, dioxane as reference): 156.9, 148.9, 128.2, 127.9, 126.5, 122.9, 80.0, 78.8, 72.2, 71.6, 71.2, 53.6, 48.9, 40.4, 37.0, 16.1, 15.4 (only 17 of the 18 expected resonances were observed. HR-MS (ESI): m/z 1237.5409 ([M+H-4HCl]1+, calculated for C58H69N20O12+: 1237.5404), 413.1853 ([M+3H-4HCl]3+, calculated for C58H71N20O123+: 413.1854).
Supplementary Material
Scheme 1.

Synthesis of cationic acyclic CB[n]-type container M2NH3. Conditions: a) CF3CO2H / Ac2O, 70 °C, 3 h, 36%, b) DMSO, NaN3, 80 °C, 12 h, 88%, c) DMSO, H2O, PPh3, 80 °C, 4h then HCl, 81%.
Acknowledgement
We thank the National Cancer Institute of the National Institutes of Health (CA168365 to L. I.) for financial support.
References
- 1.Cram DJ. Angew. Chem., Int. Ed. Engl. 1988;27:1009. [Google Scholar]; Lehn J-M. Angew. Chem., Int. Ed. Engl. 1988;27:89. [Google Scholar]; Pedersen CJ. Angew. Chem. Int. Ed. Engl. 1988;27:1021. [Google Scholar]; Gutsche CD. Acc. Chem. Res. 1983;16:161. [Google Scholar]; Rebek J. Acc. Chem. Res. 2009;42:1660. doi: 10.1021/ar9001203. [DOI] [PubMed] [Google Scholar]; Pluth MD. [Google Scholar]; Raymond KN. Chem. Soc. Rev. 2007;36:161. doi: 10.1039/b603168b. [DOI] [PubMed] [Google Scholar]; Yoshizawa M. [Google Scholar]; Klosterman J. [Google Scholar]; Fujita M. Angew. Chem., Int. Ed. 2009;48:3418. doi: 10.1002/anie.200805340. [DOI] [PubMed] [Google Scholar]; Kumari H. [Google Scholar]; Deakyne CA. [Google Scholar]; Atwood JL. Acc. Chem. Res. 2014;47:3080. doi: 10.1021/ar500222w. [DOI] [PubMed] [Google Scholar]; Kay ER. [Google Scholar]; Leigh DA. Angew. Chem., Int. Ed. 2015;54:10080. doi: 10.1002/anie.201503375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Diederich F. Angew. Chem., Intl. Ed. Engl. 1988;27:362. [Google Scholar]; Boehmer V. Angew. Chem., Int. Ed. Engl. 1995;34:713. [Google Scholar]; Rekharsky MV, Inoue Y. Chem. Rev. 1998;98:1875. doi: 10.1021/cr970015o. [DOI] [PubMed] [Google Scholar]; Ogoshi T, Kanai S, Fujinami S, Yamagishi T-A, Nakamoto Y. J. Am. Chem. Soc. 2008;130:5022. doi: 10.1021/ja711260m. [DOI] [PubMed] [Google Scholar]; Xue M, Yang Y, Chi X, Zhang Z, Huang F. Acc. Chem. Res. 2012;45:1294. doi: 10.1021/ar2003418. [DOI] [PubMed] [Google Scholar]
- 3.You L, Zha D, Anslyn EV. Chem. Rev. 2015;115:7840. doi: 10.1021/cr5005524. [DOI] [PubMed] [Google Scholar]
- 4.Ma X, Zhao Y. Chem. Rev. 2015;115:7794. doi: 10.1021/cr500392w. [DOI] [PubMed] [Google Scholar]
- 5.Harada A, Takashima Y, Nakahata M. Acc. Chem. Res. 2014;47:2128. doi: 10.1021/ar500109h. [DOI] [PubMed] [Google Scholar]
- 6.Garcia-Simon C, Gramage-Doria R, Raoufmoghaddam S, Parella T, Costas M, Ribas X, Reek Joost NH. J. Am. Chem. Soc. 2015;137:2680. doi: 10.1021/ja512637k. [DOI] [PubMed] [Google Scholar]; Ramamurthy V. Acc. Chem. Res. 2015;48:2904. doi: 10.1021/acs.accounts.5b00360. [DOI] [PubMed] [Google Scholar]
- 7.Warmuth R. Angew. Chem. Int. Ed. 1997;36:1347. [Google Scholar]; Mal P, Breiner B, Rissanen K, Nitschke JR. Science. 2009;324:1697. doi: 10.1126/science.1175313. [DOI] [PubMed] [Google Scholar]; Cram DJ, Tanner ME, Thomas R. Angew. Chem., Int. Ed. 1991;30:1024. [Google Scholar]
- 8.Cummings CG, Hamilton AD. Curr. Opin. Chem. Biol. 2010;14:341. doi: 10.1016/j.cbpa.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Gassensmith JJ, Baumes JM, Smith BD. Chem. Commun. 2009:6329. doi: 10.1039/b911064j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee JW, Samal S, Selvapalam N, Kim H-J, Kim K. Acc. Chem. Res. 2003;36:621. doi: 10.1021/ar020254k. [DOI] [PubMed] [Google Scholar]; Lagona J, Mukhopadhyay P, Chakrabarti S, Isaacs L. Angew. Chem., Int. Ed. 2005;44:4844. doi: 10.1002/anie.200460675. [DOI] [PubMed] [Google Scholar]; Nau WM, Florea M, Assaf KI. Isr. J. Chem. 2011;51:559. [Google Scholar]; Assaf KI, Nau WM. Chem. Soc. Rev. 2015;44:394. doi: 10.1039/c4cs00273c. [DOI] [PubMed] [Google Scholar]; Masson E, Ling X, Joseph R, Kyeremeh-Mensah L, Lu X. RSC Adv. 2012;2:1213. [Google Scholar]
- 11.Barrow SJ, Kasera S, Rowland MJ, del Barrio J, Scherman OA. Chem. Rev. 2015;115:12320. doi: 10.1021/acs.chemrev.5b00341. [DOI] [PubMed] [Google Scholar]
- 12.Freeman WA, Mock WL, Shih N-Y. J. Am. Chem. Soc. 1981;103:7367. [Google Scholar]; Kim J, Jung I-S, Kim S-Y, Lee E, Kang J-K, Sakamoto S, Yamaguchi K, Kim K. J. Am. Chem. Soc. 2000;122:540. [Google Scholar]; Day AI, Arnold AP, Blanch RJ, Snushall B. J. Org. Chem. 2001;66:8094. doi: 10.1021/jo015897c. [DOI] [PubMed] [Google Scholar]; Day AI, Blanch RJ, Arnold AP, Lorenzo S, Lewis GR, Dance I. Angew. Chem., Int. Ed. 2002;41:275. doi: 10.1002/1521-3773(20020118)41:2<275::aid-anie275>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]; Liu S, Zavalij PY, Isaacs L. J. Am. Chem. Soc. 2005;127:16798. doi: 10.1021/ja056287n. [DOI] [PMC free article] [PubMed] [Google Scholar]; Cheng X-J, Liang L-L, Chen K, Ji N-N, Xiao X, Zhang J-X, Zhang Y-Q, Xue S-F, Zhu Q-J, Ni X-L, Tao Z. Angew. Chem., Int. Ed. 2013;52:7252. doi: 10.1002/anie.201210267. [DOI] [PubMed] [Google Scholar]
- 13.Mock WL, Shih N-Y. J. Org. Chem. 1986;51:4440. [Google Scholar]; Liu S, Ruspic C, Mukhopadhyay P, Chakrabarti S, Zavalij PY, Isaacs L. J. Am. Chem. Soc. 2005;127:15959. doi: 10.1021/ja055013x. [DOI] [PubMed] [Google Scholar]; Rekharsky MV, Mori T, Yang C, Ko YH, Selvapalam N, Kim H, Sobransingh D, Kaifer AE, Liu S, Isaacs L, Chen W, Moghaddam S, Gilson MK, Kim K, Inoue Y. Proc. Natl. Acad. Sci. U. S. A. 2007;104:20737. doi: 10.1073/pnas.0706407105. [DOI] [PMC free article] [PubMed] [Google Scholar]; Cao L, Šekutor M, Zavalij PY, Mlinarić-Majerski K, Glaser R, Isaacs L. Angew. Chem., Int. Ed. 2014;53:988. doi: 10.1002/anie.201309635. [DOI] [PubMed] [Google Scholar]; Biedermann F, Uzunova VD, Scherman OA, Nau WM. [Google Scholar]; De Simone A. J. Am. Chem. Soc. 2012;134:15318. doi: 10.1021/ja303309e. [DOI] [PubMed] [Google Scholar]; Moghaddam S, Yang C, Rekharsky M, Ko YH, Kim K, Inoue Y, Gilson MK. J. Am. Chem. Soc. 2011;133:3570. doi: 10.1021/ja109904u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shetty D, Khedkar JK, Park KM, Kim K. Chem. Soc. Rev. 2015;44:8747. doi: 10.1039/c5cs00631g. [DOI] [PubMed] [Google Scholar]
- 15.Isaacs L. Acc. Chem. Res. 2014;47:2052. doi: 10.1021/ar500075g. [DOI] [PMC free article] [PubMed] [Google Scholar]; Del Barrio J, Horton P, Lairez D, Lloyd G, Toprakcioglu C, Scherman O. J. Am. Chem. Soc. 2013;135:11760. doi: 10.1021/ja406556h. [DOI] [PubMed] [Google Scholar]; Ko YH, Kim E, Hwang I, Kim K. Chem. Commun. 2007:1305. doi: 10.1039/b615103e. [DOI] [PubMed] [Google Scholar]; Loh XJ, del Barrio J, Toh PPC, Lee T-C, Jiao D, Rauwald U, Appel EA, Scherman OA. Biomacromolecules. 2012;13:84. doi: 10.1021/bm201588m. [DOI] [PubMed] [Google Scholar]
- 16.Vazquez J, Remon P, Dsouza RN, Lazar AI, Arteaga JF, Nau WM, Pischel U. Chem. - Eur. J. 2014;20:9897. doi: 10.1002/chem.201403405. [DOI] [PubMed] [Google Scholar]; Walker S, Oun R, McInnes FJ, Wheate NJ. Isr. J. Chem. 2011;51:616. [Google Scholar]; Dong N, Xue S-F, Zhu Q-J, Tao Z, Zhao Y, Yang L-X. Supramol. Chem. 2008;20:659. [Google Scholar]; Macartney DH. Isr. J. Chem. 2011;51:600. [Google Scholar]
- 17.Dsouza R, Hennig A, Nau W. Chem. Eur. J. 2012;18:3444. doi: 10.1002/chem.201103364. [DOI] [PubMed] [Google Scholar]; Ghale G, Lanctot AG, Kreissl HT, Jacob MH, Weingart H, Winterhalter M, Nau WM. Angew. Chem., Int. Ed. 2014;53:2762. doi: 10.1002/anie.201309583. [DOI] [PubMed] [Google Scholar]; Norouzy A, Azizi Z, Nau WM. Angew. Chem., Int. Ed. 2015;54:792. doi: 10.1002/anie.201407808. [DOI] [PubMed] [Google Scholar]
- 18.Dang D, Nguyen H, Merkx M, Brunsveld L. Angew. Chem., Int. Ed. 2013;52:2915. doi: 10.1002/anie.201208239. [DOI] [PubMed] [Google Scholar]; Lee HH, Choi TS, Lee SJC, Lee JW, Park J, Ko YH, Kim WJ, Kim K, Kim HI. Angew. Chem., Int. Ed. 2014;53:7461. doi: 10.1002/anie.201402496. [DOI] [PubMed] [Google Scholar]; Chinai JM, Taylor AB, Ryno LM, Hargreaves ND, Morris CA, Hart PJ, Urbach AR. J. Am. Chem. Soc. 2011;133:8810. doi: 10.1021/ja201581x. [DOI] [PMC free article] [PubMed] [Google Scholar]; Urbach AR, Ramalingam V. Isr. J. Chem. 2011;51:664. [Google Scholar]; Smith LC, Leach DG, Blaylock BE, Ali OA, Urbach AR. J. Am. Chem. Soc. 2015;137:3663. doi: 10.1021/jacs.5b00718. [DOI] [PubMed] [Google Scholar]; Rekharsky MV, Yamamura H, Ko YH, Selvapalam N, Kim K, Inoue Y. Chem. Commun. 2008:2236. doi: 10.1039/b719902c. [DOI] [PubMed] [Google Scholar]
- 19.Appel E, del Barrio J, Loh X, Scherman O. Chem. Soc. Rev. 2012;41:6195. doi: 10.1039/c2cs35264h. [DOI] [PubMed] [Google Scholar]
- 20.Lim S, Kim H, Selvapalam N, Kim K-J, Cho SJ, Seo G. [Google Scholar]; Kim K. Angew. Chem. Int. Ed. 2008;47:3352. doi: 10.1002/anie.200800772. [DOI] [PubMed] [Google Scholar]; Miyahara Y, Abe K, Inazu T. Angew. Chem., Int. Ed. 2002;41:3020. doi: 10.1002/1521-3773(20020816)41:16<3020::AID-ANIE3020>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; Tian J-A, Ma S-Q, Thallapally PK, Fowler D, McGrail BP, Atwood JL. Chem. Commun. 2011;47:7626. doi: 10.1039/c1cc12689j. [DOI] [PubMed] [Google Scholar]
- 21.Jon SY, Selvapalam N, Oh DH, Kang J-K, Kim S-Y, Jeon YJ, Lee JW, Kim K. J. Am. Chem. Soc. 2003;125:10186. doi: 10.1021/ja036536c. [DOI] [PubMed] [Google Scholar]; Kim E, Kim D, Jung H, Lee J, Paul S, Selvapalam N, Yang Y, Lim N, Park CG, Kim K. Angew. Chem., Int. Ed. 2010;49:4405. doi: 10.1002/anie.201000818. [DOI] [PubMed] [Google Scholar]; Jung H, Park KM, Yang J-A, Oh EJ, Lee D-W, Park K, Ryu SH, Hahn SK, Kim K. Biomaterials. 2011;32:7687. doi: 10.1016/j.biomaterials.2011.06.060. [DOI] [PubMed] [Google Scholar]; Lee D-W, Park K, Banerjee M, Ha S, Lee T, Suh K, Paul S, Jung H, Kim J, Selvapalam N, Ryu S, Kim K. Nat. Chem. 2011;3:154. doi: 10.1038/nchem.928. [DOI] [PubMed] [Google Scholar]; Jung H, Park JS, Yeom J, Selvapalam N, Park KM, Oh K, Yang J-A, Park KH, Hahn SK, Kim K. Biomacromolecules. 2014;15:707. doi: 10.1021/bm401123m. [DOI] [PubMed] [Google Scholar]; Jang M, Kim H, Lee S, Kim HW, Khedkar JK, Rhee YM, Hwang I, Kim K, Oh JH. Adv. Funct. Mater. 2015;25:4882. [Google Scholar]; Yeom J, Kim SJ, Jung H, Namkoong H, Yang J, Hwang BW, Oh K, Kim K, Sung YC, Hahn SK. Adv. Healthcare Mater. 2015;4:237. doi: 10.1002/adhm.201400304. [DOI] [PubMed] [Google Scholar]
- 22.Lucas D, Minami T, Iannuzzi G, Cao L, Wittenberg JB, Anzenbacher P, Isaacs L. J. Am. Chem. Soc. 2011;133:17966. doi: 10.1021/ja208229d. [DOI] [PubMed] [Google Scholar]; Cao L, Isaacs L. Org. Lett. 2012;14:3072. doi: 10.1021/ol3011425. [DOI] [PubMed] [Google Scholar]; Vinciguerra B, Cao L, Cannon JR, Zavalij PY, Fenselau C, Isaacs L. J. Am. Chem. Soc. 2012;134:13133. doi: 10.1021/ja3058502. [DOI] [PMC free article] [PubMed] [Google Scholar]; Cao L, Hettiarachchi G, Briken V, Isaacs L. Angew. Chem., Int. Ed. 2013;52:12033. doi: 10.1002/anie.201305061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao N, Lloyd G, Scherman O. Chem. Commun. 2012;48:3070. doi: 10.1039/c2cc17433b. [DOI] [PubMed] [Google Scholar]; Ahn Y, Jang Y, Selvapalam N, Yun G, Kim K. Angew. Chem., Int. Ed. 2013;52:3140. doi: 10.1002/anie.201209382. [DOI] [PubMed] [Google Scholar]; Ayhan MM, Karoui H, Hardy M, Rockenbauer A, Charles L, Rosas R, Udachin K, Tordo P, Bardelang D, Ouari O. J. Am. Chem. Soc. 2015;137:10238. doi: 10.1021/jacs.5b04553. [DOI] [PubMed] [Google Scholar]
- 24.Ma D, Hettiarachchi G, Nguyen D, Zhang B, Wittenberg JB, Zavalij PY, Briken V, Isaacs L. Nat. Chem. 2012;4:503. doi: 10.1038/nchem.1326. [DOI] [PubMed] [Google Scholar]; Ma D, Zhang B, Hoffmann U, Sundrup MG, Eikermann M, Isaacs L. Angew. Chem., Int. Ed. 2012;51:11358. doi: 10.1002/anie.201206031. [DOI] [PubMed] [Google Scholar]; Shen C, Ma D, Meany B, Isaacs L, Wang Y. J. Am. Chem. Soc. 2012;134:7254. doi: 10.1021/ja301462e. [DOI] [PubMed] [Google Scholar]; Gilberg L, Zhang B, Zavalij PY, Sindelar V, Isaacs L. Org. Biomol. Chem. 2015;13:4041. doi: 10.1039/c5ob00184f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang B, Isaacs L. J. Med. Chem. 2014;57:9554. doi: 10.1021/jm501276u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang B, Zavalij PY, Isaacs L. Org. Biomol. Chem. 2014;12:2413. doi: 10.1039/c3ob42603c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stancl M, Necas M, Taraba J, Sindelar V. J. Org. Chem. 2008;73:4671. doi: 10.1021/jo800699s. [DOI] [PubMed] [Google Scholar]; Stancl M, Hodan M, Sindelar V. Org. Lett. 2009;11:4184. doi: 10.1021/ol9017886. [DOI] [PubMed] [Google Scholar]; Stancl M, Gargulakova Z, Sindelar V. J. Org. Chem. 2012;77:10945. doi: 10.1021/jo302063j. [DOI] [PubMed] [Google Scholar]; Stancl M, Gilberg L, Ustrnul L, Necas M, Sindelar V. Supramol. Chem. 2014;26:168. [Google Scholar]
- 28.Sigwalt D, Ahlbrand S, Zhang M, Vinciguerra B, Briken V, Isaacs L. Org. Lett. 2015:5914. doi: 10.1021/acs.orglett.5b03145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuchelmeister HY. [Google Scholar]; Schmuck C. Chem. Eur. J. 2011;17:5311. doi: 10.1002/chem.201003393. [DOI] [PubMed] [Google Scholar]; Inclan M, Albelda MT, Carbonell E, Blasco S, Bauza A, Frontera A. doi: 10.1002/chem.201303861. [DOI] [PubMed] [Google Scholar]; Garcia-Espana E. Chem. Eur. J. 2014;20:3730. doi: 10.1002/chem.201303861. [DOI] [PubMed] [Google Scholar]; Hosseini MW, Blacker AJ, Lehn J-M. J. Am. Chem. Soc. 1990;112:3896. [Google Scholar]; Guo Y-H, Ge Q-C, Lin H, Lin H-K, Zhu S-R. Polyhedron. 2002;21:1005. [Google Scholar]; Delepine A-S, Tripier R, Baccon ML, Handel H. Eur. J. Org. Chem. 2010:5380. [Google Scholar]; Moreno-Corral R, Lara KO. Supramol. Chem. 2008;20:427. [Google Scholar]; Bazzicalupi C, Bencini A, Biagini S, Faggi E, Meini S, Giorgi C, Spepi A, Valtancoli B. J. Org. Chem. 2009;74:7349. doi: 10.1021/jo901423m. [DOI] [PubMed] [Google Scholar]; Arranz-Mascaros P, Bazzicalupi C, Bianchi A, Giorgi C, Goldino-Salido ML, Gutierrez-Valero MD, Lopez-Garzon R, Valtancoli B. New J. Chem. 2011;35:1883. [Google Scholar]; Abe H, Mawatari Y, Teraoka H, Fujimoto K, Inouye M. J. Org. Chem. 2004;69:495. doi: 10.1021/jo035188u. [DOI] [PubMed] [Google Scholar]; Bencini A, Biagini S, Giorgi C, Handel H, Baccon ML, Mariani P, Paoletti P, Paoli P, Rossi P, Tripier R, Valtancoli B. Eur. J. Org. Chem. 2009:5610. [Google Scholar]
- 30.Svec J, Necas M, Sindelar V. Angew. Chem., Int. Ed. 2010;49:2378. doi: 10.1002/anie.201000420. [DOI] [PubMed] [Google Scholar]; Svec J, Dusek M, Fejfarova K, Stacko P, Klan P, Kaifer AE, Li W, Hudeckova E, Sindelar V. Chem. - Eur. J. 2011;17:5605. doi: 10.1002/chem.201003683. [DOI] [PubMed] [Google Scholar]; Havel V, Sindelar V, Necas M, Kaifer AE. Chem. Commun. 2014;50:1372. doi: 10.1039/c3cc47828a. [DOI] [PubMed] [Google Scholar]; Yawer MA, Havel V, Sindelar V. Angew. Chem., Int. Ed. 2015;54:276. doi: 10.1002/anie.201409895. [DOI] [PubMed] [Google Scholar]; Prigorchenko E, Oeren M, Kaabel S, Fomitsenko M, Reile I, Jarving I, Tamm T, Topic F, Rissanen K, Aav R. Chem. Commun. 2015;51:10921. doi: 10.1039/c5cc04101e. [DOI] [PubMed] [Google Scholar]; Lisbjerg M, Jessen BM, Rasmussen B, Nielsen BE, Madsen AO, Pittelkow M. Chem. Sci. 2014;5:2647. [Google Scholar]; Lisbjerg M, Valkenier H, Jessen BM, Al-Kerdi H, Davis AP, Pittelkow M. J. Am. Chem. Soc. 2015;137:4948. doi: 10.1021/jacs.5b02306. [DOI] [PubMed] [Google Scholar]; Singh M, Solel E, Keinan E, Reany O. Chem. - Eur. J. 2015;21:536. doi: 10.1002/chem.201404210. [DOI] [PubMed] [Google Scholar]; Parvari G, Annamalai S, Borovoi I, Chechik H, Botoshansky M, Pappo D, Keinan E. Chem. Commun. 2014;50:2494. doi: 10.1039/c3cc48284g. [DOI] [PubMed] [Google Scholar]
- 31.Ma D, Zavalij PY, Isaacs L. J. Org. Chem. 2010;75:4786. doi: 10.1021/jo100760g. [DOI] [PubMed] [Google Scholar]
- 32.Holliday BJ, Jeon Y-M, Mirkin CA, Stern CL, Incarvito CD, Zakharov LN, Sommer RD, Rheingold AL. Organomettalics. 2002;21:5713. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.