

Abstract
Pathogenic strains of bacteria are known to cause various infectious diseases and there is a growing demand for molecular probes that can selectively recognize them. Here we report a special DNAzyme (catalytic DNA), RFD‐CD1, that shows exquisite specificity for a pathogenic strain of Clostridium difficile (C. difficile). RFD‐CD1 was derived by an in vitro selection approach where a random‐sequence DNA library was allowed to react with an unpurified molecular mixture derived from this strain of C. difficle, coupled with a subtractive selection strategy to eliminate cross‐reactivities to unintended C. difficile strains and other bacteria species. RFD‐CD1 is activated by a truncated version of TcdC, a transcription factor, that is unique to the targeted strain of C. difficle. Our study demonstrates for the first time that in vitro selection offers an effective approach for deriving functional nucleic acid probes that are capable of achieving strain‐specific recognition of bacterial pathogens.
Keywords: aptamers, bacterial detection, biosensors, DNAzymes, in vitro selection
Bacterial infections pose serious threats to public health and are responsible for many annual costly outbreaks.1 The issue has been further compounded by the emergence of hypervirulent and/or antibiotic‐resistant strains, which has now become a serious global problem.2 Early detection of specific pathogens has long been recognized as a vital strategy in the control of infectious diseases because it can lead to timely care of patients and prevent potential outbreaks. However, detection of specific bacteria represents a significant challenge because of the presence of many different species of bacteria in biological samples. Furthermore, for any given species of bacterium, only virulent strains are infectious while other strains of the same species may be harmless or even beneficial to human health. Therefore, there is a great need to develop molecular probes that are highly specific for pathogenic strains of bacteria.
DNAzymes refer to single‐stranded DNA molecules with catalytic capabilities.3 They have been widely explored as molecular tools for applications ranging from biosensing to gene regulation.4 DNAzymes can be generated de novo by in vitro selection, a simple technique that allows for the isolation of rare functional DNA sequences from a random DNA pool.5 We have previously developed an RNA‐cleaving fluorogenic DNAzyme (RFD) as an indicator for E. coli.6 RFD‐EC1 has since been used in a colorimetric assay for the field detection of E. coli and in a microfluidic‐based counting assay for the rapid detection of E. coli in blood.7, 8 Although RFD‐EC1 is highly specific for E. coli with minimal cross‐activity to other bacterial species,6 it non‐discriminatively recognizes all E. coli strains.8 The goal of this study was to investigate whether it was possible to derive strain‐specific RFDs by in vitro selection.
C. difficile, a gram‐positive bacterium that has been identified as the major cause of the diarrheic disease known as Clostridium difficile infection (CDI), was chosen for the current study. The incidence and mortality of CDI have increased dramatically over the past 15 years and CDI has become one of the most common healthcare‐associated infections in the Western hemisphere.9 These have been linked to the emergence of a hypervirulent clinical strain known as BI/027.10 In addition to being more virulent, this strain is also more resistant to antibiotics that are used to treat CDI.11 Therefore, developing a specific molecular probe for BI/027 has important clinical implications.
We used a locally isolated clinical strain of C. difficile for our investigation, which was confirmed to be a BI/027 strain by the typing experiment shown in Figure S1 in the Supporting Information. We refer this strain as BI/027‐H (H: Hamilton, Ontario). We employed two strategies to derive the desired DNAzymes. First, we used a complex mixture (rather than a defined target) derived from BI/027‐H for the selection—this strategy bypassed the challenging process of identifying a biomarker as the starting point. Second, in each round of selection, we incorporated a counter‐selection step where the DNA pool was selected against similar mixtures from control bacteria—this strategy was to ensure the desired recognition specificity. Three bacteria were chosen for this purpose, E. coli, B. subtilis (representing two different species of bacteria), and CD630 (representing a non‐BI/027 C. difficile strain).
RFD isolation was achieved using a DNA library (Figure 1, panel B) containing 40 random nucleotides and a selection scheme shown in Figure S2. Prior to the selection, a crude extracellular mixture (CEM) was prepared for BI/027‐H. This was done by culturing BI/027‐H to OD600 (optical density at 600 nm) of approximately 1, followed by cell removal using centrifugation. This CEM, named CEM‐CD, served as the positive selection target. A CEM was also prepared similarly for each control bacterium. These CEMs were combined and incubated with the DNA library for five hours. The uncleaved DNA molecules in this step were purified by 10 % denaturing polyacrylamide gel electrophoresis (dPAGE), and then incubated with CEM‐CD for 30 minutes. The cleaved DNA products in this step were purified by dPAGE, amplified by polymerase chain reaction (PCR), and used for the next round of selection (experimental details are described in the Supporting Information). In total, 19 iterations were conducted. Figure 1 A depicts the reactivity of representative DNA pools when incubated with CEM‐CD. By round 19, approximately 20 % cleavage was observed. The 19th DNA pool was cloned and sequenced. Five DNAzyme classes were discovered (Figure 1 B) and the DNAzyme with highest cleavage activity (class 2) was named RFD‐CD1 (Figure 1 C) and chosen for further investigation.
Figure 1.
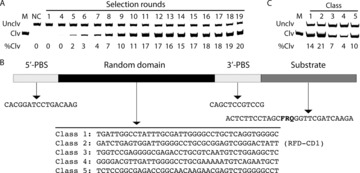
A) The activity of DNA pools. Each pool was incubated with CEM‐CD for 30 minutes, followed by dPAGE analysis. Unclv: uncleaved DNA pool; Clv: cleaved DNA pool; %Clv=(F Clv/6)/[(F Clv/6)+F Unclv)]. F Clv and F Unclv: fluorescence intensity of cleaved and uncleaved fractions of the DNA pool. Note that the cleavage leads to six‐fold fluorescence enhancement, which was taken into consideration for cleavage percentage calculation. M: marker for cleavage. NC: Negative control (pool 19 in the selection buffer). B) Five RFD classes. Each sequence is organized in the 5′‐3′ direction as 5′‐PBS (primer binding site), random domain, 3′‐PBS, and substrate. F: fluorescein‐dT; R: adenine ribonucleotide; Q: Dabcyl‐dT. C) Responses of top RFDs towards CEM‐CD. Reaction time: 30 minutes.
We examined the recognition specificity of RFD‐CD1. We first evaluated the reactivity of RFD‐CD1 towards the CEM of 13 bacterial species and found none of them was able to activate RFD‐CD1 (Figure 2 A). We then tested the reactivity of RFD‐CD1 to the CEM of 12 C. difficile strains. Only the CEM from BI/027 was able to activate RFD‐CD1 (lane 1, Figure 2 B). The results indicate that RFD‐CD1 is both species‐specific and strain‐selective.
Figure 2.
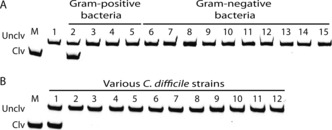
A) Responses of RFD‐CD1 to CEMs prepared from various bacteria. Lane 1: reaction buffer only; lanes 2–5: C. difficile, B. subtilis, Leuconostoc mesenteroides, and Pediococcus acidilactici. Lanes 6–15: Pseudomonas peli, Brevundimonas diminuta, Hafnia alvei, Yersinia ruckeri, Ochrobactrum grignonese, Achromobacter xylosoxidans, Moraxella osloensis, Acinetobacter lwoffi, Serratia fonticola, and E. coli. B) Responses of RFD‐CD1 to CEMs prepared from different C. difficile strains. Lane 1: BI/027‐H; Lanes 2–12: BAA‐1801, BAA‐1804, BAA‐1875, 43594, 43598, BAA‐1382 (also known as CD630), BAA‐1871, BAA‐1872, 43255, BAA‐1870, and BAA‐1814, respectively. Reaction time: 30 minutes.
Next we turned our attention to the identification of the target that activates RFD‐CD1. We first treated CEM‐CD with three proteases, trypsin (TP) and proteinase K (PK) and subtilisin (SL), to determine if the activator is a protein. We found that CEM‐CD treated with each protease was unable to activate RFD‐CD1 (Figure 3 A), signifying that the target is indeed a protein.
Figure 3.
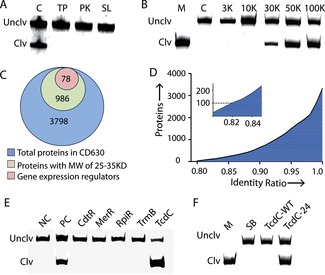
A) Responses of RFD‐CD1 to protease‐treated CEM‐CD. TP: trypsin; PK: proteinase K; SL: subtilisin. B) Estimation of the molecular weight of the protein target. CEM‐CD was passed through centrifugal filters with specified molecular weight cut‐offs, and the filtrates were then tested for reactivity with RFD‐CD1. C) The Venn diagram showing the number of proteins in CD630 in each listed category. D) Cumulative distribution of proteins in CD630 and CD196 having lowest to highest identity ratio. Insert shows the first 100 proteins with the lowest identity ratio. E) RFD‐CD1 treated with the CEM prepared from E. coli cells transformed with a plasmid expressing one of the five candidate targets: CdtR, MerR, RpiR, TrmB, and TcdC. NC: the negative control made of the CEM prepared from E. coli containing an empty plasmid. PC: positive control made of the CEM prepared from BI/027‐H. F) RFD‐CD1 treated with purified TcdC‐WT and TcdC‐24. SB: Selection buffer only. Reaction time in panels A, B, E and F: 30 minutes.
We evaluated the molecular weight of the target using molecular sizing. CEM‐CD was passed through centrifugal filters with molecular weight (MW) cut‐offs from 3–100 kilodaltons (KDa). The 3 KDa and 10 KDa filtrates did not induce the cleavage of RFD‐CD1 while the 30 KDa filtrate caused a small level of cleavage. In contrast, the 50 K and 100 K filtrates prompted strong cleavage (Figure 3 B). This experiment suggests that the protein target has a MW of about 30 000 Daltons.
C. difficile genome encodes nearly 4000 proteins.12 Given the estimated MW of the target, we tightened our search to 986 proteins with an MW between 25 000 and 35 000 Daltons (Figure 3 C). Based on the fact that the RFD‐CD1 is a DNA molecule, we hypothesized that the target might have an intrinsic ability to interact with DNA. Therefore, we decided to narrow down our search to gene expression regulators. After applying this filtration strategy using Procom software, the potential candidates were further reduced to 78 (Figure 3 C).
The strain specificity data presented in Figure 2 B indicates that RFD‐CD1 is only active with BI/027‐H but not with other strains including CD630. Previous studies have shown that there are numerous genetic differences between CD‐BI/027 and CD630.13 Taking these into account, we decided to compare the genomic difference between CD630 and CD196, a BI/027 strain whose genomic sequence has been reported.13
We calculated the sequence identity of each protein encoded by a given gene in these two strains by Blastx14 and the results are presented in Figure 3 D. Upon examination of the list of the top 100 proteins with lowest sequence identity with the list of the 78 candidates described earlier, we found that the following five proteins appeared on both: MerR, TcdC, RpiR, TrmB, and CdtR. Each candidate gene was then amplified from the genomic DNA of BI/027‐H and expressed in E. coli. The CEM from each cell line was then prepared and used to induce the cleavage of RFD‐CD1. Of the five cell lines, only the one harboring the tcdC gene produced a positive signal (Figure 3 E), indicating that TcdC, a transcription factor, is the target for RFD‐EC1.
To further verify that TcdC is the target for RFD‐CD1 and uncover the reason behind the strain specificity, we sequenced the tcdC genes in BI/027‐H and CD630 (Table 1). The tcdC gene from CD630 contains an open reading frame (ORF) of 696 nucleotides, translating into a TcdC protein that has a size of 232 amino acids, which is denoted TcdC‐WT.15 The nucleotide and protein sequences match those in the NCBI databank.16 The tcdC gene from BI/027‐H, however, has an internal truncation of 18 nucleotides located between 330–347. The same gene has been reported previously by others, which was named tcdC‐24 15 (or tcdC‐B 17). Thus, the TcdC protein from BI/027‐H, TcdC‐24, is 6‐amino acid shorter than TcdC‐WT.
Table 1.
Typing of tcdC genes of the C. difficile strains used in Figure 2 B for strain specificity test.
| Strain names | TcdC version | Size of TcdC |
|---|---|---|
| BI/027‐H | TcdC‐24 | 226 |
| BAA‐1382 (CD630) | TcdC‐WT | 232 |
| 43 255 | TcdC‐WT | 232 |
| 43 598 | TcdC‐WT | 232 |
| 43 594 | TcdC‐WT | 232 |
| BAA‐1804 | TcdC‐WT | 232 |
| BAA‐1814 | TcdC‐WT | 232 |
| BAA‐1871 | TcdC‐WT | 232 |
| BAA‐1872 | TcdC‐WT | 232 |
| BAA‐1870 | TcdC‐1 | 65 |
| BAA‐1875 | TcdC‐5 | 61 |
| BAA‐1801 | no TcdC | 0 |
We cloned and purified TcdC‐WT and TcdC‐24, which are predicted to have a molecular weight of 28 200 and 27 300 Daltons, respectively. These predictions are consistent with SDS‐PAGE analysis showing that TcdC‐24 has an increased gel mobility (Figure S3). Purified TcdC‐24 and TcdC‐WT were then used to induce the cleavage of RFD‐CD1. As expected, TcdC‐24 induced the cleavage of RFD‐CD1 while TcdC‐WT did not (Figure 3 F). These experiments confirm that TcdC is indeed the target for RFD‐CD1 and explain the specificity of RFD‐CD1 for BI/027‐H over CD630.
We next sequenced the tcdC genes from other strains used for the strain specificity test in Figure 2 B and the results are summarized in Table 1. BAA‐1801 is a non‐pathogenic strain and does not have a tcdC gene.18 BAA‐1870 and BAA‐1875 produce significantly truncated TcdC proteins containing only 65 and 61 amino acids, respectively, due to sequence mutations.15 All other C. difficile strains produce wild‐type TcdC. Taken together, the experiments above not only result in the discovery of TcdC as the target for RFD‐CD1, but also explain the origin of the strain specificity. That is: RFD‐CD1 has a high specificity for TcdC‐24 and recognizes neither full‐length TcdC nor the truncated TcdC variants.
In summary, using in vitro selection, we have obtained an RNA‐cleaving fluorogenic DNAzyme (RFD) that can recognize an infectious strain of C. difficle. This DNAzyme not only exhibits no cross‐reactivity to other bacterial species, but also is highly strain‐selective for C. difficile. To our knowledge, our work reports the first example of a functional nucleic acid that can recognize a specific strain of bacterium. Moreover, our approach also represents an unconventional strategy to molecular probe engineering as it does not begin with the identification of a specific biomarker for the probe development, which can be a lengthy and possibly unproductive process. Instead, our approach relies on a test‐tube selection process to come up with a solution. This is achieved through tandem selection steps where a complex biological mixture from the targeted bacterial strain is used as the positive‐selection target and similar mixtures from control bacteria are used as the subtractive selection target. Even though this approach does present the challenge of identifying the target upon the completion of in vitro selection, we can exploit simple bioinformatic tools and experimental techniques to progressively narrow down target candidates and ultimately identify the target. Taken together, our work here demonstrates that in vitro selection can be an effective solution to engineering functional DNA probes that are able to recognize specific strains of bacterial pathogens. This sets up the stage to exploit synthetic DNAzyme probes for infectious disease diagnosis.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Funding is provided by Natural Sciences and Engineering Research Council of Canada (NSERC) and the Canadian Institute of Health Research (CIHR) through a CHRP grant.
Z. Shen, Z. Wu, D. Chang, W. Zhang, K. Tram, C. Lee, P. Kim, B. J. Salena, Y. Li, Angew. Chem. Int. Ed. 2016, 55, 2431.
References
- 1.
- 1a. Klevens R. M., Edwards J. R., C. L. Richards, Jr. , Horan T. C., Gaynes R. P., Pollock D. A., Cardo D. M., Public Health Rep. 2007, 122, 160–166; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Mead P. S., Slutsker L., Dietz V., McCaig L. F., Bresee J. S., Shapiro C., Griffin P. M., Tauxe R. V., Emerging Infect. Dis. 1999, 5, 607–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antibiotic resistance threats in the United States, 2013 Centers for Disease Control and Prevention. It can be accessed at: http://www.cdc.gov/drugresistance/threat-report-2013/.
- 3.
- 3a. Breaker R. R., Nat. Biotechnol. 1997, 15, 427–431; [DOI] [PubMed] [Google Scholar]
- 3b. Schlosser K., Li Y., Chem. Biol. 2009, 16, 311–322. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Liu J., Cao Z., Lu Y., Chem. Rev. 2009, 109, 1948–1998; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Silverman S. K., Angew. Chem. Int. Ed. 2010, 49, 7180–7201; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 7336–7359. [Google Scholar]
- 5.
- 5a. Ellington A. D., Szostak J. W., Nature 1990, 346, 818–822; [DOI] [PubMed] [Google Scholar]
- 5b. Tuerk C., Gold L., Science 1990, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- 6. Ali M. M., Aguirre S. D., Lazim H., Li Y., Angew. Chem. Int. Ed. 2011, 50, 3751–3754; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 3835–3838. [Google Scholar]
- 7. Tram K., Kanda P., Salena B. J., Huan S., Li Y., Angew. Chem. Int. Ed. 2014, 53, 12799–12802; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13013–13016. [Google Scholar]
- 8. Kang D. K., Ali M. M., Zhang K., Huang S. S., Peterson E., Digman M. A., Gratton E., Zhao W., Nat. Commun. 2014, 5, 5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Bartlett J. G., Moon N., Chang T. W., Taylor N., Onderdonk A. B., Gastroenterology 1978, 75, 778–782; [PubMed] [Google Scholar]
- 9b. Larson H. E., Price A. B., Honour P., Borriello S. P., Lancet 1978, 311, 1063–1066; [DOI] [PubMed] [Google Scholar]
- 9c. Voelker R., JAMA J. Am. Med. Assoc. 2010, 303, 2017–2019. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Goorhuis A., Van der Kooi T., Vaessen N., Dekker F. W., Van den Berg R., Harmanus C., van den Hof S., Notermans D. W., Kuijper E. J., Clin. Infect. Dis. 2007, 45, 695–703; [DOI] [PubMed] [Google Scholar]
- 10b. Loo V. G., et al., N. Engl. J. Med. 2005, 353, 2242–2249; [Google Scholar]
- 10c. Redelings M. D., Sorvillo F., Mascola L., Emerging Infect. Dis. 2007, 13, 1417–1419; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10d. Killgore G., et al., J. Clin. Microbiol. 2008, 46, 431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cartman S. T., Heap J. T., Kuehne S. A., Cockayne A., Minton N. P., Int. J. Med. Microbiol. 2010, 300, 387–395. [DOI] [PubMed] [Google Scholar]
- 12. Sebaihia M., et al., Nat. Genet. 2006, 38, 779–786. [DOI] [PubMed] [Google Scholar]
- 13. Stabler R. A., et al., Genome Biol. 2009, 10, R102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J., J. Mol. Biol. 1990, 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 15. Dingle K. E., Griffiths D., Didelot X., Evans J., Vaughan A., PloS One 2011, 6, e19993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Samie A., Obi C. L., Franasiak J., Archbald-Pannone L., Bessong P. O., Alcantara-Warren C., Guerrant R. L., Am. J. Trop. Med. Hyg. 2008, 78, 577–585. [PubMed] [Google Scholar]
- 17. Spigaglia P., Mastrantonio P., J. Clin. Microbiol. 2002, 40, 3470–3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lemee L., Dhalluin A., Testelin S., Mattrat M. A., Maillard K., Lemeland J. F., Pons J. L., J. Clin. Microbiol. 2004, 42, 5710–5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary