
Fig. 2.
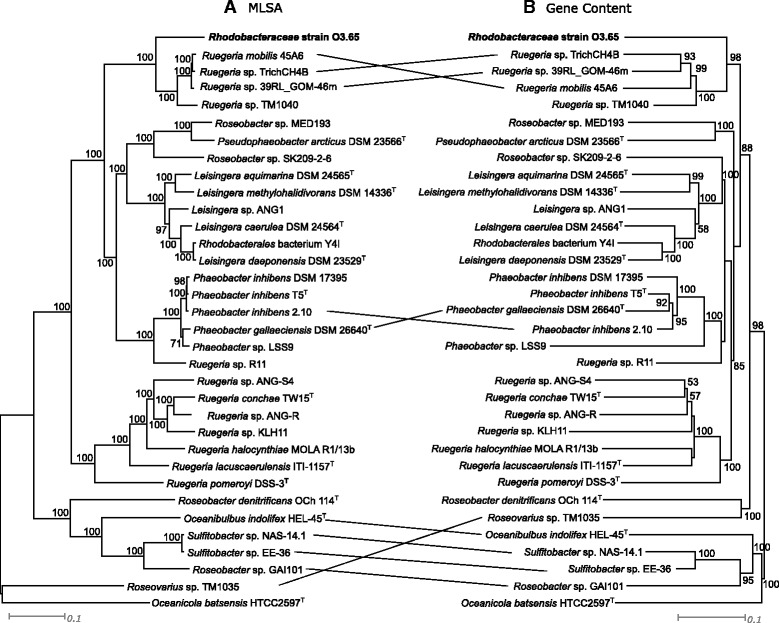
Tanglegram of genome based trees. a Maximum likelihood tree based on genomic data of organisms affiliated with the genera Phaeobacter, Pseudophaeobacter, Ruegeria, Leisingera and additional strains of the Roseobacter clade inferred with 500 bootstraps (BS) with RAxML after Stamatakis (2014) [100]. The alignment was created from 684 orthologous single-copy genes present in all genomes (Multilocus Sequence Analysis; MLSA) after total protein sequences of the genomes were extracted from the corresponding GenBank files and used for the downstream analysis with an in house pipeline at the Goettingen Genomics Laboratory (J. Vollmers, unpubl.). In brief, clusters of orthologs were generated using proteinortho version 5 [101], inparalogs were removed, the remaining sequences were aligned with MUSCLE [102] and poorly aligned positions automatically filtered from the alignments using Gblocks [103]. b Gene content tree including singletons of the same organisms as in A based on an orthologs-content matrix representing presence or absence of a gene in a certain genome, inferred with Neighbour Joining (1000 BS). Both scripts for this pipeline, PO_2_MLSA.py and PO_2_GENECONTENT.py, are available at github. Numbers at the nodes specify BS values ≥50 %. Scale bars represent 10 % sequence divergence. For Genbank accession numbers see Additional file 1: Table S1. For a clear view only lines were given linking the same species at different positions