

Abstract
Palaeoclimatic oscillations and different landscapes frequently result in complex population-level structure or the evolution of cryptic species. Elucidating the potential mechanisms is vital to understanding speciation events. However, such complex evolutionary patterns have rarely been reported in bats. In China, the Rhinolophus macrotis complex contains a large form and a small form, suggesting the existence of a cryptic bat species. Our field surveys found these two sibling species have a continuous and widespread distribution with partial sympatry. However, their evolutionary history has received little attention. Here, we used extensive sampling, morphological and acoustic data, as well as different genetic markers to investigate their evolutionary history. Genetic analyses revealed discordance between the mitochondrial and nuclear data. Mitochondrial data identified three reciprocally monophyletic lineages: one representing all small forms from Southwest China, and the other two containing all large forms from Central and Southeast China, respectively. The large form showed paraphyly with respect to the small form. However, clustering analyses of microsatellite and Chd1 gene sequences support two divergent clusters separating the large form and the small form. Moreover, morphological and acoustic analyses were consistent with nuclear data. This unusual pattern in the R. macrotis complex might be accounted for by palaeoclimatic oscillations, shared ancestral polymorphism and/or interspecific hybridization.
Species are not only the basic taxonomic unit, but also one of the most important units of interaction between ecology and evolution. Species delimitations and their distributions have traditionally been through morphological traits1. However, not all evolutionary changes show plainly as observable phenotypic traits2. Complex geographic landscapes, palaeoclimatic oscillations, as well as biological and ecological traits of organisms often lead to complex population structure and speciation processes (e.g. refs 3, 4 and 5). Unravelling those evolutionary mechanisms and processes becomes increasingly important because it is crucial to understanding speciation, from the earliest stages of incipient speciation through complete reproductive isolation (e.g. refs 6 and 7).
In some taxa with complex evolutionary histories, unexpectedly widespread species-level paraphyly and polyphyly in mitochondrial (mt) DNA analyses have been reported6,7,8,9, suggesting conflicting geographic patterns between mtDNA and nuclear (nc) DNA or phenotype. Except for cases of imperfect taxonomy8, these prevalent instances of paraphyly uncovered by mtDNA in animals are primarily attributed to incomplete lineage sorting and/or introgressive hybridization8,9,10. Incomplete lineage sorting produces a failure of mtDNA haplotype fixation from ancestral polymorphisms when speciation events are short and ancestral effective population sizes are large. Alternatively, hybridization can lead to mitochondrial capture, such that the mitochondrial and nuclear genomes yield different histories. Natural hybridization among closely related animal species has been increasingly uncovered in recent years using genotypic data11, making this an important alternative hypothesis to consider.
Bats represent over 20% of mammal species12. However, documented cases of complex evolutionary histories involving paraphyly in this group are very rare13,14,15,16,17. Complex evolutionary histories are relatively common in birds (e.g. refs 8 and 10), and since bats have similar vagility to birds, the limited number of observations in bats may be due to limited studies. Rhinolophus is one of the most diverse and complex bat genera. We recently explored the taxonomy of big-eared horseshoe bats (Rhinolophus macrotis) based on sampling in one province. Since the large and small form are sympatric, and have significant differences in both morphology (size) and echolocation frequencies (~16 kHz) as well as a relatively high mitochondrial cytochrome b (Cytb) divergence (3.16–3.25%), this is a species complex with cryptic species18. This species complex is widely distributed in Southeast Asia, and their morphology and echolocation frequency are extensively variable19. However, based on our recent field surveys in China, we observed the species complex had a continuously and widespread distribution throughout Central and South China, and they frequently roost in colonies including both small and large forms (Fig. 1), which suggests that complex evolutionary processes might be occurring in the R. macrotis complex.
Figure 1. Mitochondrial gene tree and sample sites in this study.

(a) Phylogenetic tree based on the analysis of the combined mitochondrial Cytb and control region. Numbers above the branches are bootstrap support from in Maximum likelihood followed by posterior probabilities from Bayesian analyses. (b) Sampling sites for the mt lineages included in the study. Circle sizes are proportional to the number of individuals captured. Abbreviations of samples populations are indicated in Table S1. The map was made with QGIS 2.8 (http://www.qgis.org) and Natural Earth public domain map data (http://www.naturalearthdata.com/about/terms-of-use/), and modified in Illustrator.
In this study, in order to examine their evolutionary history and speciation, we used ten unlinked genetic markers, including mtDNA (Cytb and control region) and ncDNA (eight microsatellite loci and Chd1 gene) to reconstruct the phylogeny of the big-eared horseshoe bat species complex from China. We combined this with morphological data (i.e. external measurements) and acoustic data (i.e. echolocation frequency). Our study aimed to (i) determine genetic lineages in this species complex; (ii) determine whether the mtDNA variation is congruent or conflicts with the nuclear and phenotypic data (i.e. morphological and acoustic measurements); (iii) if conflicting, evaluate potential mechanisms leading to patterns within this species complex; (iv) and, evaluate the effect of palaeoclimatic changes on this species complex.
Results
Sequence characteristics
For 79 individuals in the R. macrotis complex sampled from 14 localities across the entire Chinese range of the species, we found much variation in Cytb (1,140 bp) and control region (464 bp). A total of 29 haplotypes of Cytb were defined based on 103 polymorphic sites (71 of which were parsimony informative). No insertions or deletions were found. Fifty haplotypes of the control region were defined based on 115 polymorphic sites (90 of which were parsimony informative). The alignment of the combined Cytb and control region (1,604 bp) resulted in 53 haplotypes. For the autosomal Chd1 gene (742 bp), a total of 20 haplotypes were defined from 71 individuals based on 24 polymorphic sites (6 of which were parsimony informative).
Mitochondrial trees and genetic divergence
Maximum likelihood and Bayesian phylogenetic reconstruction based on the combined mtDNA data (Fig. 1a) and on the separate mtDNA genes (Figs S1 and S2, Supporting Information) resulted in similar tree topologies in which three well-supported clades were recovered. Clade 1 contained all small forms of R. macrotis from Southwest China, including those small forms identified previously18. Clades 2 and 3 contained all large forms of R. macrotis from Central and Southeast China, respectively. The large forms identified in Sun et al.18 were all contained in Clade 3. No haplotype was shared among the three clades.
The ranges of the three clades were partially sympatric (Fig. 1b). Both SXi and CQ populations contained individuals from Clades 2 and 3 (all large forms), both the HuN and JX1 populations contained individuals from Clades 1 and 3, and the GD1 population contained individuals from Clades 1 and 2.
In general, intraspecific genetic divergence levels among bats are typically less than 2% within Cytb20. Thus we used Cytb to calculate the intra-lineage and inter-lineage genetic differences. In this species complex, the maximum level of uncorrected intra-clade divergence was ≤2% (Table 1). Among the three clades, the minimum uncorrected divergence was over 2% (Table 1), though never exceeded 4%.
Table 1. Estimates of uncorrected genetic divergences (minimum-maximum values) for Cytb gene within and between mt lineages (expressed as percent divergent).
| mt Clade 1 | mt Clade 2 | mt Clade 3 | |
|---|---|---|---|
| mt Clade 1 | 0.1–2.0 | ||
| mt Clade 2 | 2.4–3.2 | 0.1–1.6 | |
| mt Clade 3 | 3.1–3.9 | 2.4–3.2 | 0.1–0.8 |
Since the clades were distinct genetic pools, population genetics analyses were performed within each lineage. Genetic diversity was highest in Clade 1, followed by Clades 3 and 2 for both the Cytb and control region (Table 2).
Table 2. Diversity estimates for different mt lineages of each form, based on the combined Cytb and control region, control region and microsatellites.
| mt lineage | Small form | Large form |
|||
|---|---|---|---|---|---|
| Clade 1 | Clade 2 | Clade 3 | |||
| mtDNA | Cyt-b | Nind/Nh | 36/11 | 22/8 | 21/10 |
| h | 0.83 | 0.85 | 0.91 | ||
| π(%) | 1 | 0.83 | 0.37 | ||
| control region | Nind/Nh | 36/25 | 22/11 | 21/14 | |
| h | 0.97 | 0.87 | 0.94 | ||
| π(%) | 4.24 | 4.09 | 2.83 | ||
| ncDNA | microsatellites | Nind/A | 32/12.25 | 46/14.875 | |
| AR | 12.25 | 13.66 | |||
| Ho | 0.71 | 0.76 | |||
| He | 0.85 | 0.87 | |||
| Chd1 | Nind/Nh | 35/9 | 36/12 | ||
| π(%) | 0.15 | 0.16 | |||
| Ho | 0.05 | 0.08 | |||
| He | 0.09 | 0.10 | |||
Number of individuals (Nind), number of haplotypes (Nh), haplotype diversity (h), nucleotide diversity (π), mean allele number (A), allelic richness (AR), expected heterozygosity (Ho) and observed heterozygosity (He).
Demographic inference
We estimated divergence times among clades using Cytb (Table 3). The inferred TMRCA for all R. macrotis complex was 1.51 Ma (Million years ago), with similar divergence times mt Clades 1 and 2 (0.64 and 0.66 Ma, respectively). In contrast, mt Clade 3 originated much later. Tajima’s D values and Fu’s Fs tests indicated that none of the three mt lineages deviated significantly from neutrality (Table 4). Unlike in mt Clades 2 and 3, analysis of mt Clade 1 failed to reject the model of population expansion (Table 4), with an estimated timing of expansion occurring 78.8 Ka (Thousand years ago) (95% CI: 38.1–96.1 Ka).
Table 3. Mean estimates of time to most recent common ancestor (TMRCA) and 95% credible intervals for each mt lineage based on Cytb gene.
| Node | TMRCA (Ma) | CI (95%) |
|---|---|---|
| mt Clade 1 (small forms) | 0.64 | 0.44–0.85 |
| mt Clade 2 (large forms) | 0.66 | 0.43–0.89 |
| mt Clade 3 (large forms) | 0.33 | 0.19–0.49 |
| mt Clades 1 + 2 | 1.16 | 0.87–1.47 |
| mt Clades 1 + 2 + 3 (all) | 1.51 | 1.14–1.88 |
Analyses were performed using a molecular clock rate of 1.3% per million years.
Table 4. Results of Fu’s Fs test, Tajima’s D test, mismatch distribution analysis and estimation of the time of population expansion for Rhinolophus macrotis complex based on control region.
| Fu’s Fs | Tajima’s D | SSD | r | Tao (95% CI) | TMD(95% CI) ka | |
|---|---|---|---|---|---|---|
| Clade 1 | −1.643 | 0.371 | 0.008 | 0.008 | 29.207 (15.191–36.018) | 78.7 (40.9–97.0) |
| Clade 2 | 3.949 | 0.760 | 0.043** | 0.077** | — | — |
| Clade 3 | −0.482 | 0.599 | 0.026** | 0.077** | — | — |
Statistically significant results are indicated by asterisks: *P < 0.05, **P < 0.01.
Nuclear genetic analyses
Based on microsatellites and the Chd1 gene, the large form populations generally showed higher genetic diversity than the small form (Table 3, Table S2, Supporting information). All eight microsatellite loci were polymorphic within each form, and only 3.4% of 120 population-locus combinations significantly deviated from HWE. No sign of linkage disequilibrium was detected. Micro-checker identified a potential of 3.8% null alleles in population-locus tests. However, null alleles and deviations from HWE were not associated more frequently with any particular locus. Therefore we retained all loci for subsequent analyses.
Using the Evanno criterion with structure output suggested that K = 2 was most likely, with the large form and small form largely identified as distinct clusters. The exception was one large form population (GD1; Fig. 2a) in which only one individual was correctly identified as the large form, while the other five were assigned to the small form. For K = 3 or 4, no new clusters were detected in the large form, but some cryptic genetic structure was found in the small form. Similar results were obtained from the NJ tree, which demonstrated that the small forms made two clades, rather than a single clade (Fig. 2b), while the large form individuals from mt Clade 2 and mt Clade 3 did not form independent monophyletic groups and were instead scattered throughout the tree. In the FCA (Fig. 2c), all small forms were clearly identified as a separate genetic group, while large forms from mt Clades 2 and 3 were intermixed. Moreover, the specimens from GD1 population of large form were located between the small and large forms in this analysis. Results from GENELAND (Fig. S3, Supporting Information), which can detect population structure in relation to geographic and genetic information, also supported the existence of two main genetic clusters associated with the small form and the large form, except for GD1 and HuN populations, where the two forms are sympatric. In three independent runs, GD1 and HuN populations were inferred to cluster with the small form and the large form (Fig. S3a,b), respectively, while the opposite result occurred in the other two runs (Fig. S3c,d).
Figure 2. Individual clustering based on the analysis of microsatellite data.

(a) Bayesian cluster analyses with Structure (K = 2, 3, 4) of all samples based on eight microsatellites. Each vertical bar represents one individual and its probability of being assigned to a cluster. (b) Unrooted neighbor-joining tree reconstructed from Nei et al.63 DA distances based on microsatellite genotypes of all individuals. Colours indicate the membership of each mt lineage. (c) Factorial corresponding analysis based on the microsatellite data. All the asterisks represent the individuals from GD1 population in the large form.
Consistent with the microsatellite results, the large form individuals from mt Clade 2 and mt Clade 3 did not form independent Chd1 haplogroups and instead shared five haplotypes (Fig. 3). However, the large form and the small form had their own distinct haplotypes with one exception (Fig. 3).
Figure 3. Haplotype network of Rhinolophus macrotis species complex samples based on nuclear Chd1 gene sequences.
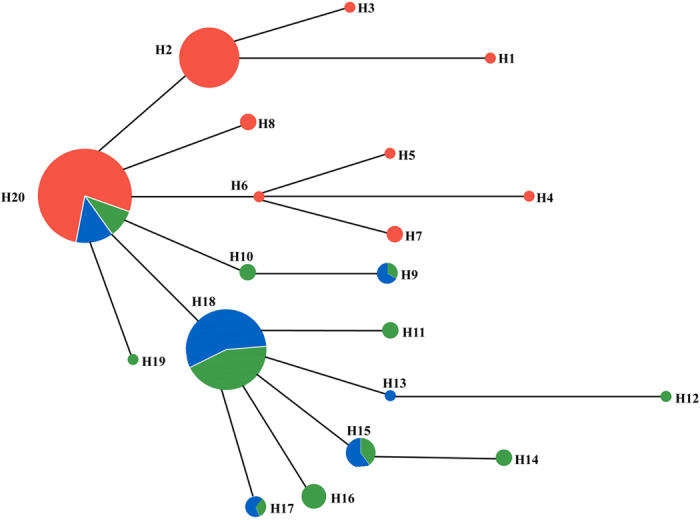
Colours indicate the membership of each mt lineage, including Clade 1 (red), Clade 2 (green) and Clade 3 (blue).
We focused on the geographic pattern of nuclear genetic variation within mt Clades 2 and 3, as the above analyses failed to detect their genetic differentiation in individual clustering (Figs 2 and 3). No significant IBD was found within the large forms (P > 0.05). However, when excluding the GD1 population, the test of IBD suggested that the two lineages had not evolved separately but were connected because smooth clinal variation rather than a geographic break was observed in the nuclear genetic variation (Fig. 4).
Figure 4. Regression of genetic distance based on microsatellite data versus geographic distances for pairwise population comparisons in large forms including mt Clades 2 and 3.
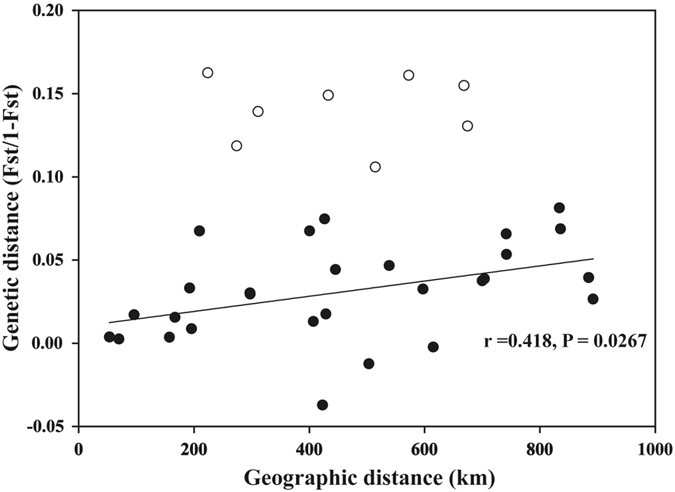
White circles represent the distances between GD1 population in large forms and other populations.
Phenotypic differences
Phenotypic analyses distinguished the large and small forms using either forearm length or resting frequency of echolocation calls (Fig. 5), including in sympatric populations.
Figure 5. The variation of forearm length and resting frequency in echolocation calls among the large form and the small form of R. macrotis species complex.

Circles represent the mean values. Colours indicate the membership of each mt lineage, including Clade 1 (red), Clade 2 (green) and Clade 3 (blue). The asterisks represent the individuals from GD1 population in large forms.
Within the large form, morphological differentiation was not significant between mt Clade 2 and mt Clade 3. Forearm length was significantly explained by site, but not by sex and mt lineage, or by interactions among variables (Table 5). Resting frequency was significantly explained by sex and mt lineage, as well as by site. No significant interaction was found between effects (Table 5).
Table 5. Morphological and acoustic differentiation of large forms in Rhinolophus macrotis complex (i.e. mt Clade 2 + mt Clade 3) according to mt lineage, sex and site, analyzed by general linear model (GLM).
| Factor | Forearm length |
Resting frequency |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| d.f. | Sum Sq | Mean Sq | F value | P | d.f. | Sum Sq | Mean Sq | F value | P | |
| Sex | 1 | 0.038 | 0.038 | 0.008 | 0.931 | 1 | 36.345 | 36.345 | 11.953 | 0.001* |
| mt lineage | 1 | 11.326 | 11.326 | 2.253 | 0.139 | 1 | 153.082 | 153.082 | 50.347 | 0.000* |
| Sex × mt lineage | 1 | 4.990 | 4.990 | 0.992 | 0.324 | 1 | 3.907 | 3.907 | 1.285 | 0.262 |
| Error | 53 | 266.486 | 5.028 | 53 | 161.147 | 3.041 | ||||
| Factor | d.f. | Sum Sq | Mean Sq | F value | P | d.f. | Sum Sq | Mean Sq | F value | P |
| Site | 8 | 75.831 | 9.479 | 2.255 | 0.040* | 8 | 172.740 | 21.593 | 33.240 | 0.000* |
| mt lineage | 1 | 2.582 | 2.582 | 0.614 | 0.437 | 1 | 0.805 | 0.805 | 1.239 | 0.271 |
| Site × mt lineage | 1 | 1.698 | 1.698 | 0.404 | 0.528 | 1 | 0.013 | 0.013 | 0.020 | 0.888 |
| Error | 46 | 193.347 | 4.203 | 46 | 29.882 | 0.650 | ||||
We focused on the geographic pattern of acoustic variation within mt Clade 2/Clade 3 of the large form as we found no significant morphological differences between these two clades. We detected positive significant associations between resting frequency and both longitude (r = 0.39, P = 0.0011) and latitude (r = 0.284, P = 0.0199). When excluding GD1, clinal variation was much more significant (longitude: r = 0.48, P < 0.0001; RF vs latitude: r = 0.74, P < 0.0001).
Discussion
This study highlights the complex evolutionary history of big-eared horseshoe bats in China, and adds to the limited literature documenting complex evolutionary histories in bats. We document here the incongruence between mt and nuclear genetic structures. Three mt lineages were identified: Clade 1 (small form) and Clade 2 (large form), which are closely related and separated by short genetic distances, and the more distantly related Clade 3 (large form). In contrast, only two genetic entities were identified on the basis of nuclear data: small form (mt Clade 1) and large form (combined mt Clades 2 and 3). The morphological and acoustic data were consistent with the nuclear genetic data, as they also did not separate the mt Clade 2 and mt Clade 3 lineages. These results suggest the occurrence of complex processes during the evolution of the big-eared horseshoe bats in the study area, such as palaeoclimatic changes, allopatric speciation, incomplete lineage sorting and/or genetic introgression.
The effect of palaeoclimatic changes and allopatric speciation of the small form
Pleistocene climatic fluctuations and accompanying ecological changes in China have presented severe challenges to the survival of bat species, greatly affecting their population differentiation and contemporary distribution3,21,22. In this study, the TMRCA of the entire R. macrotis complex populations dated to 1.51 Ma. During this period, China was experiencing glacial-interglacial cycles, including the Poyang glacial stage (1.8 Ma) and the Dagu glacial stage (1.1 Ma)23. Climatic changes and temperature decline may have forced the big-eared horseshoe bats to diverge from other bat species and evolve separately.
The TMRCAs of the combined mt Clade 1 (small form) and mt Clade 2 (large form) dated to 1.16 Ma, suggesting that the two mt lineages differentiated from each other between 1.16–0.64 Ma. This time was consistent with two major cold glacial stages, Xixiabangma glaciations (1.17–0.8 Ma)24 and the Naynayxungla glaciations (0.71–0.59 Ma)25. The former stage is one of the coldest recent climatic periods based on a high mass accumulation rate for Chinese loess26. At that time, the populations might have become isolated into different refugia and occupied different habitats and climate zones either due to, or eventually leading to, local ecological adaptation, and two distinct lineages distributed in Central and Southwest China.
Despite this recent divergence, there is strong evidence to suggest that the small form is genetically isolated from the large form (mt Clades 2 and 3). The small forms clearly formed a distinct genetic pool on the basis of nuclear genetic analyses (Figs 2 and 3). Moreover, both the morphological and acoustic data showed an obvious differentiation between small and large forms, even for sympatric populations (Fig. 5), consistent with those found in Sun et al.18. Given their current geographic distribution (Fig. 1), it seems possible that small forms diverged from the widely distributed large forms during glaciations, which would represent a case of allopatric or peripatric speciation. However, for peripatric speciation, an expectation might be that estimates of the genetic diversities of the small form would be lower than in the large forms due to a founder effect. Since all estimates of the small form (haplotype and nucleotide diversities for mtDNA, allelic richness for microsatellites, and genetic diversity for the Chd1) were similar between the small and large forms (Table 2), allopatric speciation is more likely.
Deep mitochondrial divergence in the large form
The mitochondrial genetic analyses indicated that large forms diverged into two distinct lineages, Clade 2 and Clade 3 with a level of divergence similar to that between the small and large forms. Additionally, these two mt clades show distinct geographic distribution, with Clade 2 primarily occurring Central China, whereas Clade 3 is primarily in Southeast China (Fig. 1), although there is sympatry in two localities (CQ and SXi). These results suggest a cryptic mitochondrial lineage in the large form in China.
However, it was not possible to differentiate between the specimens of the two mt lineages on the basis of nuclear microsatellite and Chd1 data, morphological measurements or acoustic signals. When considering sites, we detected variation in forearm length and call resting frequency, though there was no significant relationship for mt lineage. Additionally, in this study, we found no distinction in call frequency in the sympatric populations, CQ and SXi. For instance, the mean values of resting frequency were 50.05 kHz in mt Clade 2 and 49.32 kHz in mt Clade 3 within CQ, and 52.82 kHz in mt Clade 2 and 52.26 kHz in mt Clade 3 within SXi (Table S3, Supporting information). This phenomenon is also found in Pipistrellus kuhlii27, which has two divergent mitochondrial lineages, but similar nuclear genotypes in Western Europe.
We did detect a clinal central-southeast variation in call frequency (with frequency gradually decreasing with longitude, but increasing with latitude). The observed pattern of geographic variation could be due, at least in part, to cultural drift. In general, cultural transmission and copying errors are major drivers of stochastic divergence in learned vocal signals28,29, including in some bat species, i.e. Rhinolophus ferrumequinum30, Hipposideros armiger31, Rhinolophus pumilus32. However, environmental factors in call clinal variation could not be ruled out. The smooth cline rather than a geographic break in acoustic variation suggests the occurrence of regular gene flow between the two mt lineages in large forms.
Large form mitochondrial gene-tree paraphyly relative to small form
An increasing number of studies have reported mitochondrial species-level paraphyly (e.g. refs 6, 7 and 8). In our study, phylogenetic analyses revealed that mt Clade 1 (small forms) was nested within the mt Clades 2 + 3 group (large forms), rendering large forms paraphyletic with respect to small forms. Two evolutionary processes may account for this mitochondrial paraphyly: (i) incomplete lineage sorting and the persistence of highly divergent mt lineages, and (ii) mitochondrial introgression.
The persistence of highly divergent mt lineages within a species may reflect maintenance of two lineages in a panmictic population with a large effective population size33. Since genetic drift is weak in very large populations, this leads to increasing coalescence times and the probability of observing the persistence of highly divergent lineages. Moreover, the greater spatial homogenization of the nuclear genome relative to the mt genome (such as we observed) can also occur after two mt lineages come into contact. Since most bat species have female philopatry and male-biased dispersal34, highly divergent mt lineages may spread more slowly than the nuclear genome, as occurs in the big brown bat (Eptesicus fuscus)35. We determined that the current clinal north-south distribution of mt Clades 2 and 3 was likely a consequence of isolation in different Pleistocene refugia, followed by range expansion and the establishment of a secondary contact zone. Thus, retention of unique mt lineages that arose in isolation, followed by homogenization of the nuclear genome, is possible in the R. macrotis complex.
Alternatively, high mtDNA divergence could also be caused by hybridization with a closely related species, leading to introgression of mtDNA9,36. If this is the case, then we have presumably sampled the nuclear genome of a single species, together with its mt genome plus an introgressed mt genome. This scenario implies that another unsampled species with a different nuclear genome and one of the mt lineages identified in our data set exists elsewhere in China. One of individual from JX1 population in mt Clade 3 had 0.79–1.76% mitochondrial Cytb divergence with an unidentified Rhinolophus species, Rhinolophus sp1.37, suggesting that introgression of the mt genome may have occurred. We reconstructed trees using all of our data with the published Cytb sequence of R. sp1. (Fig. S4, Supporting Information), which showed that R. sp1. clustered within mt Clade 3. This result suggested that mt Clade 3 may have an introgressed mt genome from R. sp1. Certainly, sampling efforts should focus on population JX1 to sample more R. sp1. in the future. Alternatively, the ‘unsampled species’ may have gone extinct after ancient hybridization events. If so, its mt lineage now only survives in its new host, and a process of competitive replacement of mtDNA has occurred, with no nuclear introgression or morphological changes, such as has been suggested in some other animals, i.e., hare38 and lizard39.
Secondary contact between small form and large form
In our previous study18, the large and small forms were identified from sympatric large-eared horseshoe bats in China, suggesting the existence of a cryptic species. In this study, three sympatric populations (GD1, HuN and JX1) were detected between small and large forms (Fig. 1), suggesting secondary contact after putative speciation.
Based on our analyses of population demographic history, the small form might have undergone post-glacial range expansion after the Baiyu (the Last) Glaciation24, following secondary contact with the large forms. In the mitochondrial trees, all individuals from the three sympatric areas diverged recently (see the top of mt Clade 1; Figs S1 and S2). Moreover, the number of small forms is less than large forms in three sympatric areas (Fig. 1), suggesting the small form may be expanding into areas occupied by the large form.
For the HuN and JX1 populations, the individuals were assigned to the correct forms based on microsatellite NJ tree, FCA and Bayesian clustering, suggesting a recent secondary contact between two forms or the maintenance of reproductive isolating barriers in two populations. However, for the GD1 population, only one individual was correctly assigned to the large form group, but five individuals were genetically assigned to the small form group (Fig. 2), though their morphological and acoustic data were obviously different from those of the small form (Fig. 5). Furthermore, the different GENELAND assignments for the GD1 and HuN populations during five independent runs (Fig. S3, Supporting Information) emphasizes the complexity of these two sympatric areas. Such discordance between ncDNA and phenotypic patterns in GD1 might suggest hybridization followed by backcrossing to the small form at this site, such that the number of microsatellite loci we sampled was insufficient to separate large and small forms40. If hybridization between the small and large forms has occurred, it is expected that hybrids may have intermediate phenotypes, at least for several generations. In our study, the acoustic data of GD1 may support this hypothesis. When excluding GD1, call frequency was more significantly correlated with longitude in large forms (Fig. 5), suggesting that the call frequency of GD1 obviously deviated from the clinal variation of other large forms (Fig. 5d). Additionally, when excluding GD1, the microsatellite genetic distance is significantly related with geographic distance (Fig. 4), suggesting different genetics between GD1 and other large forms.
Conclusion
In summary, the evolutionary history of the R. macrotis complex appears to be complex. This complexity has led to strong incongruence between mt genetic structure, and that from nuclear loci, morphology and acoustic patterns in this species complex. The divergence between the small and large form likely occurred in allopatry due to isolation of populations during Pleistocene glaciation events, and current areas of sympatry appear to be due to more recent secondary contact (likely driven by range expansion of the small form). Current hybridization may be occurring in one sympatric population, though there appears to be reproductive isolation between the small and large forms in other sympatric populations. The occurrence of two highly divergent mt lineages in large forms conflicts with the nuclear plus phenotypic data (i.e. morphological and acoustic measurements), which might be due to the persistence of highly divergent mt lineages, geographically structured during Pleistocene glaciations, and/or genetic introgression from a species yet to be identified. While such complex patterns have been observed frequently in birds, they have rarely been reported in bats, which are also quite vagile. Our observations add to several other studies on bats, and suggest that the limited studies in bats (relative to birds) may be due to the paucity of studies on bats rather than a fundamental difference in their evolutionary histories. Overall, our results suggest that additional studies in bats may help highlight other cases of complex evolutionary histories.
Materials and Methods
Taxa
In China, 79 individuals of big-eared horseshoe bat complex were sampled during 2006–2012 from 14 localities (Fig. 1 and Table S1, Supporting Information), including the 12 specimens of the 7 small form and 5 large form bats published previously18. All bats were identified following Csorba et al.19.
Sampling sizes were different for morphological characters, echolocation calls, mtDNA, microsatellite DNA and the Chd1 gene. For morphological characters (forearm length), 133 independent samples were measured. For echolocation calls, 109 independent samples were recorded. From those individuals with morphological and echolocation data, 79 samples were sequenced for mtDNA, 78 and 71 were examined using microsatellite DNA and Chd1 analyses, respectively. For each sampling locality, we determined the latitude and longitude using GPS (eTrex Vista) and then calculated the geographic distance matrixes among localities.
Genetics
DNA extraction and sequencing
We used a UNIQ-10 column animal genomic DNA isolation kit (Sangon, China) to extract total genomic DNA from collected bat wing membranes which had been preserved in 99% ethanol.
We sequenced two mtDNA fragments: the complete Cytb gene (1,140 bp) and 464 bp of the control region. The complete Cytb fragment for 67 individuals was amplified using primers L14724 and H1591541,42, which were combined with 12 sequences reported by Sun et al.18 to obtain the final data set (Table S1, Supporting Information). PCR and sequencing protocols were the same as those described in Sun et al.18. The fragment of mitochondrial control region for all 79 individuals was amplified using primers P and E, and the PCR methods described in Wilkinson and Chapman43. Amplified products were purified and sequenced by Shanghai Sangon Biotechnology Co., Ltd. All Cytb and control region haplotypes of R. macrotis complex were deposited in GenBank with accession numbers KX261888-KX261916 and KX261917- KX261966, respectively. All sequences were aligned using GENEIOUS PRO (v. 5.6.6)44 and revised manually.
Eight microsatellite loci, Reffer 15, 22, 24, 27, H3, PD3, A4 and PH69A, were amplified using fluorescently labeled primers for 78 individuals. Primer sequences and PCR conditions followed those of Rossiter et al.45 and Hua et al.46. Amplified PCR products were analyzed using the ABI 3730 automated DNA sequencer. The resulting sequences were analyzed using GeneMapper ID 3.2 (Applied Biosystems). All loci were screened for null alleles and large allele dropouts using Micro-Checker v2.2.347. Tests for deviation from Hardy-Weinberg equilibrium and linkage disequilibrium were performed for each population in Genepop v4.048.
The autosomal Chd1 gene was amplified using two sets of primers (EX26F and EX27R; EMB26F and EMB27R)49 because some individuals failed to amplify with one set, and so we tried a second set. For the first set of primers, the PCR conditions were an initial denaturation at 95 °C for 5 min, followed by 36 cycles of denaturation at 94 °C for 30 s, annealing at 63 °C for 30 s, and extension at 70 °C for 2 min 30 s, and a final extension at 72 °C for 10 min. For the second set, the PCR conditions were 35 cycles of denaturation at 94 °C for 45 s, annealing at 59 °C for 45 s, and extension at 72 °C for 1 min. Heterozygous sites in Chd1 sequences were resolved to two haplotypes per individual using PHASE 2.150 implemented in DnaSP v551.
Genetic analyses
Haplotype diversity (h) and nucleotide diversity (π) were calculated for mitochondrial Cytb and control region for each population and mt lineage. The uncorrected Cytb genetic distances within and among mt lineages were calculated in MEGA 5.0552. For microsatellites, expected heterozygosity (He) and observed herterozygosity (Ho) were calculated using Genepop, and mean allele number and allelic richness for per locus and per taxon was assessed in Fstat 2.9.353. For the Chd1 gene, π, He and Ho for each taxon was calculated using Arlequin v3.554.
To examine the demographic history of each mt lineage, we only analyzed the control region due to its greater variability at this level. We used Tajima’s D55 and Fu’s Fs tests as implemented in Arlequin to test for neutrality. The goodness-of-fit of distributions was tested and its significant difference from a model of sudden expansion was assessed using the sum of squared deviations (SSD) and the raggedness index (r) with 1,000 parametric bootstrap replicates. Generally, low and non-significant SSD and r values indicate population expansion. For the expanding mt lineages, we estimated the time of expansion (t) from τ = 2ut, where τ is calculated as the time to expansion in mutational units and u is the mutation rate per generation for the entire sequence. The u is equal to μgk, where μ is the mutation rate per nucleotide, g is the generation time and k is the sequence length. We used a mutation rate of 20% ⁄ Myr for the current control region based on previous studies on bats17,56,57 and a generation time of 2 years based on data from a congeneric species21.
We calculated nuclear genetic distances between localities using Slatkin’s linearized FST (given by FST/(1 − FST)). An isolation by distance (IBD) model was estimated from the large forms based on microsatellites.
Phylogenetic analyses
For the mitochondrial analyses, two closely related species, Rhinolophus mashalli and R. pusillus were selected as outgroups (GenBank accession EU434938, EF217391, EU053160 and DQ642898). The nuclear Chd1 gene was not included in this analysis as part of the goal was to explore the different signals between the mitochondrion and the nuclear genome. Phylogenetic reconstruction using maximum likelihood (ML) in PhyML-aBayes58,59 and Bayesian Inference49 in MrBayes 3.1.260 was undertaken for the Cytb and control region separately as well as combined.
We used jModelTest61 and the Akaike information criterion to select the best model of evolution for ML and BI analyses. The best-fit models were as follows: (1) TrN + G [G = 0.072] for Cytb, (2) TIM3 + I + G [I = 0.478; G = 0.507] for control region, TrN + I + G [I = 0.651; G = 0.714] for the combined mtDNA data. For ML analysis, the starting tree was obtained with BIONJ62 and we evaluated support of the resulting topologies using 1,000 nonparametric bootstraps. For BI analysis, we used the molecular evolution model parameters estimated for each data set and two simultaneous runs of Markov chain Monte Carlo (MCMC) analysis, each comprising four chains and 106 generations. Trees and parameters were sampled every 10 generations. When the run terminated, the deviation of split frequencies reached a value <0.01. The ln-likelihoods of trees reached an asymptote. The first 25% of the sampled trees were discarded as a burn-in.
For the microsatellite analyses, we used a phylogenetic tree based on Nei et al.63 DA distance between individuals and reconstructed using a NJ algorithm using Populations v1.2.30 64. Second, we used a Bayesian approach using structure v2.3.165 on the microsatellite data. The admixture model without a priori designation for populations was used and the number of tested clusters (K) was based on the mtDNA results. For K = 1 to 5, 10 runs were performed with 500,000 iterations after 100,000 iterations were discarded as “burn-in”. The most likely number of populations was determined using the ΔK method described by Evanno et al.66. The graphical display of the genetic structure was produced by DISTRUCT67. Third, we used GENETIX v4.05.3 68 for factorial correspondence analysis (FCA) on the microsatellite data to further examine the degree of population substructuring among R. macrotis species complex. Fourth, we used GENELAND69 to determine the genetic boundaries between bat clusters based on K = 2 which is most likely according to STRUCTURE and FCA outputs. Five independent runs were performed for K = 2, using 107 iterations with a burn-in of 30,000 iterations and 250 thinning. As the correlated model seems to be more prone to algorithm instabilities and more sensitive to departure from model assumptions, such as the presence of isolation by distance, we used the uncorrelated model of allele frequencies for microsatellite loci70. Consistency of the resulting inference was checked by comparing parameter estimates from 5 independent runs of GENELAND. For the Chd1 analysis, we used Network71 to construct a haplotype network for comparison of genealogical relationships among the haplotypes.
Time of divergence
To estimate the time of divergence among clades, we only used Cytb sequences because the molecular evolutionary rate of this gene is well-known for inter-clade and interspecific comparisons. The time to the most recent common ancestor (TMRCA) among lineages was estimated in BEAST v1.7.472. A strict molecular clock was applied with a fixed mean substitution rate of 1.30 × 10−8 subs/site/year [calculated based on Nabholz et al.73; Nabholz, pers. com.; n = 223 Chiropteran species]. The TrN + I substitution model was used, as determined by AIC implemented in jModelTest. No outgroup was specified and the Yule process was used as a tree prior. UPGMA was used to construct the starting tree. The program was run for 5,000,000 generations (10% discarded as burn-in) and sampled every 1,000, which were then combined in Tracer version 1.474. ESS values exceeded 2,000 for all parameters. All other initial parameters settings were the default provided by BEAST.
Echolocation call recording and analysis
Echolocation calls were recorded with a D980 Pettersson bat detector (Pettersson Electronik AB, Uppsala, Sweden; frequency response 8 and 160 kHz ± 3.5 dB). This detector was positioned approximately 30 cm in front of hand-held bats. The recordings were transferred to a computer with a time expansion of ×10 using BatSound Pro 3.10 (sampling frequency of 441 kHz, 16 bit precision). The resting frequency that the bats emitted under these conditions is the stable constant frequency portion of the echolocation pulses emitted by the motionless bat. We selected a portion of a recording in which resting frequency calls were continuously uttered. Using criteria proposed by Sun et al.30, 5 high-quality calls were measured for the constant frequency component in the dominant second harmonic from power spectra of a call from each individual, and the mean value was used in the analysis.
Phenotypic analyses
Geographic variation for phenotypic characters was explored via box plots, with longitude and latitude on the abscissa. The relationship of resting frequency to either longitude or latitude was estimated by linear regression analysis, with averaging of frequency by site.
To estimate the phenotypic differentiation between mt Clade 2 and mt Clade 3 in the large form, we used a general linear model with type III sums of squares to test for an effect of sex, site and mt lineage on forearm length and echolocation frequency. All variables were treated as fixed effects in the model.
Additional Information
Accession Codes: All sequence data has been uploaded to GenBank (KX261888-KX261966, KX826057-KX826076). Microsatellite genotypes, sequence alignments by markers, phylogenetic trees and morphological and acoustic data files are uploaded on the Dryad Digital Repository (doi:10.5061/dryad.b1c88).
How to cite this article: Sun, K. et al. The complex evolutionary history of big-eared horseshoe bats (Rhinolophus macrotis complex): insights from genetic, morphological and acoustic data. Sci. Rep. 6, 35417; doi: 10.1038/srep35417 (2016).
Supplementary Material
Acknowledgments
We thank Lei Wang, Yuyan You, Sen Liu, Shi Li, Guanjun Lu, Limin Shi and Xu Zhu for field support. This work was financed by the National Natural Science Foundation of China (Grant Nos. 31370399, 31270414 and 31570390), the Specialized Research Fund for the Doctoral Program of Higher Education (No. 20120043130002), the Fundamental Research Funds for the Central Universities (No. 2412016KJ045) and the Program for Introducing Talents to Universities (B16011).
Footnotes
Author Contributions K.S. designed the study, performed much of the sampling, implemented most of genetic methodologies and analyses, and wrote the manuscript. R.T.K. assisted with interpretation of data and gave important comments on multiple versions of the manuscript. T.L. and X.W. contributed part of genetic methodologies. L.J., T.J. and A.L. assisted with sample collection. J.F. assisted with study design, provided laboratory space, reagents, and some funding. All authors reviewed the manuscript.
References
- Dayrat B. Towards integrative taxonomy. Biol. J. Lin. Soc. 85, 407–415 (2005). [Google Scholar]
- Avise J. C. A role for molecular genetics in the recognition and conservation of endangered species. Trends Ecol. Evol. 4, 279–281 (1989). [DOI] [PubMed] [Google Scholar]
- Flanders J., Wei L., Rossiter S. J. & Zhang S. Y. Identifying the effects of the Pleistocene on the greater horseshoe bat, Rhinolophus ferrumequinum, in East Asia using ecological niche modelling and phylogenetic analyses. J. Biogeogr. 38, 439–452 (2011). [Google Scholar]
- Hewitt G. M. [Ice ages: their impact on species distributions and evolution]. Evolution on Planet Earth [Rothschild L. J. & Lister A. M. (eds.)] [339–361] (Academic Press, New York, 2003). [Google Scholar]
- Lin A. Q. et al. Phylogeography of Hipposideros armiger (Chiroptera: Hipposideridae) in the Oriental Region: the contribution of multiple Pleistocene glacial refugia and intrinsic factors to contemporary population genetic structure. J. Biogeogr. 41, 317–327 (2014). [Google Scholar]
- Dong F. et al. Testing hypotheses of mitochondrial gene-tree paraphyly: unravelling mitochondrial capture of the Streak-breasted Scimitar Babbler (Pomatorhinus ruficollis) by the Taiwan Scimitar Babbler (Pomatorhinus musicus). Mol. Ecol. 23, 5855–5867 (2014). [DOI] [PubMed] [Google Scholar]
- Pages M. et al. Cytonuclear discordance among Southeast Asian black rats (Rattus rattus complex). Mol. Ecol. 22, 1019–1034 (2013). [DOI] [PubMed] [Google Scholar]
- Funk D. J. & Omland K. E. Species-level paraphyly and polyphyly: frequency, causes, and consequences, with insights from animal mitochondrial DNA. Annu. Rev. Ecol. Systemat. 34, 397–423 (2003). [Google Scholar]
- Toews D. P. & Brelsford A. The biogeography of mitochondrial and nuclear discordance in animals. Mol. Ecol. 21, 3907–3930 (2012). [DOI] [PubMed] [Google Scholar]
- McKay B. D. & Zink R. M. The causes of mitochondrial DNA gene tree paraphyly in birds. Mol. Phylogenet. Evol. 54, 647–650 (2010). [DOI] [PubMed] [Google Scholar]
- Freeland J. R., Kirk H. & Petersen S. Molecular Ecology Second Edition (Wiley-Blackwell, 2011). [Google Scholar]
- Simmons N. B. Order Chiroptera (Johns Hopkins University Press, 2005). [Google Scholar]
- Berthier P., Excoffier L. & Ruedi M. Recurrent replacement of mtDNA and cryptic hybridization between two sibling bat species Myotis myotis and Myotis blythii. Proc. R. Soc. Lond. B. Biol. Sci. 273, 3101–3109 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann F. G., Owen J. G. & Baker R. J. mtDNA perspective of chromosomal diversification and hybridization in Peters’ tent-making bat (Uroderma bilobatum: Phyllostomidae). Mol. Ecol. 12, 2981–2993 (2003). [DOI] [PubMed] [Google Scholar]
- Khan F. A., Phillips C. D. & Baker R. J. Timeframes of speciation, reticulation, and hybridization in the bulldog bat explained through phylogenetic analyses of all genetic transmission elements. Syst. Biol. 63, 96–110 (2014). [DOI] [PubMed] [Google Scholar]
- Mao X. G. et al. Historical introgression and the persistence of ghost alleles in the intermediate horseshoe bat (Rhinolophus affinis). Mol. Ecol. 22, 1035–1050 (2013). [DOI] [PubMed] [Google Scholar]
- Mao X. G., Zhang J. P., Zhang S. Y. & Rossiter S. J. Historical male-mediated introgression in horseshoe bats revealed by multilocus DNA sequence data. Mol. Ecol. 19, 1352–1366 (2010). [DOI] [PubMed] [Google Scholar]
- Sun K. P. et al. A new crptic species of Rhinolophus macrotis (Chiroptera: Rhinolophidae) from Jiangxi Province, China. Acta Chiropterol. 10, 1–10 (2008). [Google Scholar]
- Csorba G., Ujhelyi P. & Thomas N. Horseshoe bats of the world (Chiroptera: Rhinolophidae) (Alana Books, Shropshire, 2003). [Google Scholar]
- Bradley R. D. & Baker R. J. A test of the genetic species concept: Cytochrome-b sequences and mammals. J. Mammal. 82, 960–973 (2001). [Google Scholar]
- Flanders J. O. N. et al. Phylogeography of the greater horseshoe bat, Rhinolophus ferrumequinum: contrasting results from mitochondrial and microsatellite data. Mol. Ecol. 18, 306–318 (2009). [DOI] [PubMed] [Google Scholar]
- You Y. Y. et al. Pleistocene glacial cycle effects on the phylogeography of the Chinese endemic bat species, Myotis davidii. BMC Evol. Biol. 10, 208 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing C. R. & Liu H. P. On the glacial and interglacial stages in Quaternary of China. J. Cheng Du Univ. Tech. 26, 97–100 (1999). [Google Scholar]
- Zheng B. X., Xu Q. Q. & Shen Y. P. The relationship between climate change and Quaternary glacial cycles on the Qinghai-Tibetan Plateau: review and speculation. Quatern. Int. 97-8, 93–101 (2002). [Google Scholar]
- Yi C., Cui Z. & Xiong H. Numerical periods of Quaternary glaciations in China. Quatern. Sci. 25, 609–619 (2005). [Google Scholar]
- Zhan X. J., Zheng Y. F., Wei F. W., Bruford M. W. & Jia C. X. Molecular evidence for Pleistocene refugia at the eastern edge of the Tibetan Plateau. Mol. Ecol. 20, 3014–3026 (2011). [DOI] [PubMed] [Google Scholar]
- Andriollo T., Naciri Y. & Ruedi M. Two mitochodnrial barcodes for one biological species: the case of European Kuhl’s pipostrelles (Chiroptera). PLoS One 10, e0134881 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch A. [The population memetics of birdsong]. Ecology and evolution of acoustic communication in birds [Kroodsma D. E. & Miller E. H. (eds.)] (Cornell University Press, New York, 1996). [Google Scholar]
- Rendell L. & Whitehead H. Culture in whales and dolphins. Behav. Brain. Sci. 24, 309–324 (2001). [DOI] [PubMed] [Google Scholar]
- Sun K. et al. Geographic variation in the acoustic traits of greater horseshoe bats: testing the importance of drift and ecological selection in evolutionary processes. PLoS One 8, e70368 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A. Q. et al. Geographical variation in echolocation vocalizations of the Himalayan leaf-nosed bat: contribution of morphological variation and cultural drift. Oikos 124, 364–371 (2015). [Google Scholar]
- Yoshino H., Armstrong K. N., Izawa M., Yokoyama J. & Kawata M. Genetic and acoustic population structuring in the Okinawa least horseshoe bat: are intercolony acoustic differences maintained by vertical maternal transmission? Mol. Ecol. 17, 4978–4991 (2008). [DOI] [PubMed] [Google Scholar]
- Webb W. C., Marzluff J. M. & Omland K. E. Random interbreeding between cryptic lineages of the Common Raven: evidence for speciation in reverse. Mol. Ecol. 20, 2390–2402 (2011). [DOI] [PubMed] [Google Scholar]
- Greenwood P. J. Mating systems, philopatry and dispersal in birds and mammals. Anim. Behav. 28, 1140–1162 (1980). [Google Scholar]
- Turmelle A. S., Kunz T. H. & Sorenson M. D. A tale of two genomes: contrasting patterns of phylogeographic structure in a widely distributed bat. Mol. Ecol. 20, 357–375 (2011). [DOI] [PubMed] [Google Scholar]
- Coyne J. A. & Orr A. H. Speciation (Sinauser Associates, Sunderland, 2004). [Google Scholar]
- Sun K. P., Feng J., Zhang Z. Z., Xu L. J. & Liu Y. Cryptic diversity in Chinese rhinolophids and hipposiderids (Chiroptera: Rhinolophidae and Hipposideridae). Mammalia 73, 135–141 (2009). [Google Scholar]
- Melo-Ferreira J. et al. The rise and fall of the mountain hare (Lepus timidus) during Pleistocene glaciations: expansion and retreat with hybridization in the Iberian Peninsula. Mol. Ecol. 16, 605–618 (2007). [DOI] [PubMed] [Google Scholar]
- Renoult J. P., Geniez P., Bacquet P., Benoit L. & Crochet P. A. Morphology and nuclear markers reveal extensive mitochondrial introgressions in the Iberian Wall Lizard species complex. Mol. Ecol. 18, 4298–4315 (2009). [DOI] [PubMed] [Google Scholar]
- Liu T. et al. Extensive hybridization and introgression between Melastoma candidum and M. sanguineum. PLoS One 9, e96680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin D. M., Kocher T. D. & Wilson A. C. Evolution of the cytochrome-B gene of mammals. J. Mol. Evol. 32, 128–144 (1991). [DOI] [PubMed] [Google Scholar]
- Kocher T. D. et al. Dynamics of mitochondrial-DNA evolution in animals - amplification and sequencing with conserved primers. Proc. Natl. Acad. Sci. USA 86, 6196–6200 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson G. S. & Chapman A. M. Length and sequence variation in evening bat D-loop mtDNA. Genetics 128, 607–617 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A. J. et al. Geneious v5.5, http://www.geneious.com 7 (2011).
- Rossiter S. J., Burland T. M., Jones G. & Barratt E. M. Characterization of microsatellite loci in the greater horseshoe bat Rhinolophus ferrumequinum. Mol. Ecol. 8, 1959–1960 (1999). [DOI] [PubMed] [Google Scholar]
- Hua P. Y., Guo T. T., Liu W. C., Zhang S. Y. & Rossiter S. Isolation and characterization of 13 microsatellite loci in Rhinolophus pusillus (least horseshoe bat) with cross-amplification in five related species. Conserv. Genet. 10, 597–600 (2009). [Google Scholar]
- van Oosterhout C., Hutchinson W. F., Wills D. P. M. & Shipley P. Micro-Checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 4, 535–538 (2004). [Google Scholar]
- Rousset F. genepop'007: a complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 8, 103–106 (2008). [DOI] [PubMed] [Google Scholar]
- Lim B. K., Engstrom M. D., Bickham J. W. & Patton J. C. Molecular phylogeny of New World sheath-tailed bats (Emballonuridae: Diclidurini) based on loci from the four genetic transmission systems in mammals. Biol. J. Linn. Soc. 93, 189–209 (2008). [Google Scholar]
- Stephens M., Smith N. J. & Donnelly P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 68, 978–989 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado P. & Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009). [DOI] [PubMed] [Google Scholar]
- Tamura K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudet J. FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). Updated from Goudet (1995). Available at: http://www2.unil.ch/popgen/softwares/fstat.htm (2001).
- Excoffier L. & Lischer H. E. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010). [DOI] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S. F., Rossiter S. J., Faulkes C. G. & Jones G. Population genetic structure and demographic history of the endemic Formosan lesser horseshoe bat (Rhinolophus monoceros). Mol. Ecol. 15, 1643–1656 (2006). [DOI] [PubMed] [Google Scholar]
- Petit E. & Mayer F. Male dispersal in the noctule bat (Nyctalus noctula): where are the limits? Proc. R. Soc. Lond. B. Biol. Sci. 266, 1717–1722 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S. Bayesian estimation of divergence times from large sequence alignments. Mol. Biol. Evol. 27, 1768–1781 (2010). [DOI] [PubMed] [Google Scholar]
- Guindon S. & Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 (2003). [DOI] [PubMed] [Google Scholar]
- Ronquist F. & Huelsenbeck J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003). [DOI] [PubMed] [Google Scholar]
- Posada D. jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256 (2008). [DOI] [PubMed] [Google Scholar]
- Gascuel O. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 14, 685–695 (1997). [DOI] [PubMed] [Google Scholar]
- Nei M., Tajima F. & Tateno Y. Accuracy of estimated phylogenetic trees from molecular data. J. Mol. Evol. 19, 153–170 (1983). [DOI] [PubMed] [Google Scholar]
- Langella O. Populations 1.2.30: Population genetic software (individuals or population distances, phylogenetic trees) (2007).
- Pritchard J. K., Stephens M. & Donnelly P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evanno G., Regnaut S. & Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620 (2005). [DOI] [PubMed] [Google Scholar]
- Rosenberg N. A. DISTRUCT: a program for the graphical display of population structure. Mol. Ecol. Notes 4, 137–138 (2004). [Google Scholar]
- Belkhir K., Borsa P., Chikhi L., Raufaste N. & Bonhomme F. GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations. Laboratoire Génome, Populations, Interactions, CNRS UMR 5171, Université de Montpellier II, Montpellier (1996–2004).
- Guillot G., Estoup A., Mortier F. & Cosson J. F. A spatial statistical model for landscape genetics. Genetics 170, 1261–1280 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillot G., Santos F. & Estoup A. Analysing georeferenced population genetics data with Geneland: a new algorithm to deal with null alleles and a friendly graphical user interface. Bioinformatics 24, 1406–1407 (2008). [DOI] [PubMed] [Google Scholar]
- Bandelt H. J., Forster P. & Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48 (1999). [DOI] [PubMed] [Google Scholar]
- Drummond A. J. & Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabholz B., Glemin S. & Galtier N. Strong variations of mitochondrial mutation rate across mammals–the longevity hypothesis. Mol. Biol. Evol. 25, 120–130 (2008). [DOI] [PubMed] [Google Scholar]
- Rambaut A. & Drummond A. J. Tracer, version 1.4. Avaliable from: http://tree.bio.ed.ac.uk/software/tracer (2007).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.