
Figure 1.
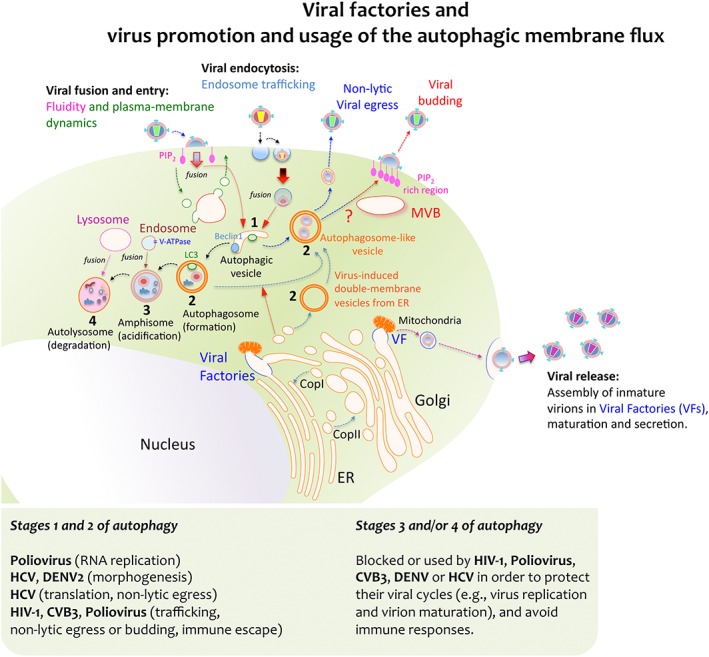
Viral factories and virus‐triggered autophagic membrane flux for replication and egression. Some viruses achieve replication by exploiting the cell's membrane transport pathways, thereby generating membrane organelles named Viral Factories (VFs). These VFs are organised by different viral proteins, and they represent specialized compartments for viral‐gene replication, morphogenesis, export, maturation and release. Moreover, these compartments also serve to override or evade the immune responses directed against viral genomes. Viral proteins can enter secretory pathways by co‐translational translocation into the ER in order for them to be further transported to the Golgi complex, either in vesicles or in a coatomer protein complex (COP) II‐dependent manner. Viral complexes formed inside the VFs communicate with vesicles, mitochondria, Golgi cisternae and ER membranes. This interaction allows viral complexes to be transported through the Golgi network to the plasma membrane and it promotes their final release as viral particles. Alternatively, some viruses take advantage of the host's autophagic machinery for their own replication and pathogenesis. Viruses first initiate the formation of vesicles that bear key autophagic proteins, such as Beclin‐1 and LC3, capturing portions of membranes from the ER and other cytoplasmic elements. This assembly evolves toward an immature double‐membrane vesicle (DMV) that serves as an aggresome compartment to recruit viruses or newly formed viral replication complexes. Several RNA viruses induce the formation of these autophagosome‐like vesicles (also referred to as DMVs) to enhance viral replication and non‐lytic egression, such as poliovirus and CVB3, HIV‐1 and HCV. How these viruses trigger the accumulation of autophagosome‐like vesicles and DMVs remains unclear. Some theories involve blocking the fusion of nascent autophagosomes with late endosomes and lysosomes, as in the case of HIV‐1 Nef, which appears to cause autophagosome accumulation by inhibiting their progression towards more mature stages. Indeed, autophagosome‐like vesicles may represent a trafficking pathway for these viruses, connecting to multivesicular bodies (MVBs), and assuring virus assembly and budding at the cell surface while protecting them from intrinsic antiviral factors and immune responses. The morphogenesis and release of mature and infectious HBV particles also require Tsg101 and depend on the ESCRT‐MVB system. Under standard conditions the lumen of autophagosomes acidifies after fusion with endosomes that carry vacuolar (H+)‐ATPase (V‐ATPase) to form amphisomes. The autophagic membrane flux progresses by fusing with lysosomes in order to form the autolysosome that contains the former's proteinases. Poliovirus inhibition of autophagosome formation attenuates viral replication while inhibiting autolysosome formation, and thus, catalytic activity does not affect the virus. However, degradation of cellular triglycerides by autophagy benefits DENV replication and autolysosome degradation dampens IFN activation following HCV infection