

ABSTRACT
Previously we showed that THY-1 has a critical role in the initial stage of infection of certain cell types with human cytomegalovirus (HCMV) and that THY-1 is important for HCMV-mediated activation of phosphatidylinositol 3-kinase (PI3K)/Akt during virus entry. THY-1 is known to interact with integrins and is a major cargo protein of clathrin-independent endocytic vesicles. Since macropinocytosis involves integrin signaling, is PI3K/Akt dependent, and is a clathrin-independent endocytic process, we determined whether THY-1 has a role in HCMV entry by macropinocytosis. Using electron microscopy in two cell lines that support HCMV infection in a THY-1-dependent manner, we found that HCMV enters these cells by a macropinocytosis-like process. THY-1 associated with HCMV virions on the cell surface and colocalized with virus inside macropinosomes. 5-(N-Ethyl-N-isopropyl)amiloride (EIPA) and soluble THY-1 blocked HCMV infection in the cell lines by ≥80% and 60%, respectively. HCMV entry into the cells triggered increased influx of extracellular fluid, a marker of macropinocytosis, and this increased fluid uptake was inhibited by EIPA and by soluble THY-1. Blocking actin depolymerization, Na+/H+ exchange, PI3K, and Pak1 kinase, which are critical for macropinocytosis, impaired HCMV infection. Neither internalized HCMV virions nor THY-1 in virus-infected cells colocalized with transferrin as determined by confocal microscopy, indicating that clathrin-mediated endocytosis was not involved in THY-1-associated virus entry. These results suggest that HCMV has adapted to utilize THY-1, a cargo protein of clathrin-independent endocytotic vesicles, to facilitate efficient entry into certain cell types by a macropinocytosis-like process.
IMPORTANCE Human cytomegalovirus (HCMV) infects over half of the population and is the most common infectious cause of birth defects. The virus is the most important infection occurring in transplant recipients. The mechanism of how HCMV enters cells is controversial. In this study, we show that THY-1, a cell surface protein that is critical for the early stage of entry of HCMV into certain cell types, contributes to virus entry by macropinocytosis. Our findings suggest that HCMV has adapted to utilize THY-1 to facilitate entry of HCMV into macropinosomes in certain cell types. Further knowledge about the mechanism of HCMV entry into cells may facilitate the development of novel inhibitors of virus infection.
INTRODUCTION
Human cytomegalovirus (HCMV) infects over half of the general population. While the infection is often asymptomatic when occurring in young children, it poses a serious health threat in transplant recipients and in immunocompromised individuals. HCMV is the most common infectious cause of birth defects. The virus infects a variety of host cells in vivo, including epithelial, endothelial, and muscle cells as well as fibroblasts. HCMV encodes multiple glycoproteins, some of which are not shared with other human herpesviruses, such as UL128, UL130, and UL131A. Other glycoproteins are conserved, including glycoprotein gH, its chaperon protein gL, and gB. This broad array of glycoproteins contributes to diverse mechanisms for the virus to enter into host cells. Previous studies have shown that low-passage-number clinical isolates of HCMV encode a pentameric complex of gH/gL and UL128-UL131A, which functions as a determinant for infection of epithelial and endothelial cells and monocytes, and a gH/gL/gO trimer (1–6), which facilities HCMV infection in all cell types (7). However, during prolonged propagation of virus in cell culture, portions of the UL128-UL131A component of the pentamer are often lost from the viral genome, resulting in virus that is limited to infection of fibroblasts. The pentamer and trimer glycoprotein complexes interact with specific cellular receptors in a cell type-dependent manner (8), which has been postulated to lead to the recruitment of gB and to trigger membrane fusion (9). However, a recent study showed that HCMV gH/gL and gB form stable gB-gH/gL complexes that are incorporated into virions prior to receptor binding or membrane fusion and that only small amounts of gH/gL/gO and gH/gL/UL128-131A complexes are associated with gB (10).
Entry of viruses into cells involves a repertoire of multiple cellular proteins and diverse entry pathways (11). While herpes simplex virus (HSV), Epstein-Barr virus (EBV), and HCMV can penetrate directly through the plasma membrane by pH-independent fusion into the cytosol of certain cell types (12–14), the major route of entry for these viruses is through endocytic pathways, including clathrin-mediated endocytosis (CME), macropinocytosis, caveolar/lipid raft-mediated endocytosis, and phagocytosis (11, 12, 15–20). Engagement of specific cellular receptors and/or entry mediators by viral ligands plays a key role in sorting virions into different endocytic pathways (11).
Several host molecules have been postulated to have roles in facilitating HCMV entry, including platelet-derived growth factor receptor alpha (PDGFR-α) (18, 21), epidermal growth factor receptor (EGFR) (22), DC-SIGN (23), αVβ3 and β1 integrins (24, 25), and paxillin (26). The use of these molecules to facilitate HCMV entry is often cell type dependent (18, 27, 28). Previously, we showed that expression of cell surface antigen THY-1 (CD90) highly correlates with HCMV infectivity (29). Loss-of-function experiments using soluble THY-1, specific antibody, and small interfering RNAs (siRNAs) demonstrated that THY-1 contributes to HCMV infection at an early stage of entry; inhibition of THY-1 blocked HCMV-triggered activation of phosphatidylinositol 3-kinase (PI3K)/Akt signaling, which has been reported to be required for entry (6, 21, 30). Expression of exogenous THY-1 in a cell line that expresses little endogenous THY-1 and is refractory to HCMV infection enhanced HCMV infectivity within the first 60 min postinfection. Therefore, THY-1 likely functions as an HCMV entry mediator in certain cell types (29).
THY-1 is a 27-kDa glycosylphosphatidylinositol (GPI)-linked cell surface glycoprotein expressed on numerous cell types. It participates in several signaling pathways and affects numerous biological processes, including cellular adhesion, neurite outgrowth, tumor growth, migration, and cell death (31). THY-1 interacts with integrins and recruits paxillin (32). GPI anchors target proteins to specific lipid raft microdomains or detergent-resistant membranes that provide critical platforms for signaling and membrane trafficking (33, 34) and are important for herpesvirus infection (30, 35–38). THY-1 is a major cargo protein of clathrin-independent endocytotic carriers (39). Therefore, we investigated whether THY-1 contributes to HCMV infection by facilitating entry of the virus through endocytic pathways. Previous studies showed that HCMV enters dendritic cells through a macropinocytosis-like pathway in a pH-independent and cholesterol-dependent manner (40) and enters primary fibroblasts through macropinocytosis that is dynamin dependent (41). Since macropinocytosis occurs constitutively in dendritic cells and macrophages but not in epithelial cells, such as HEK293, HEp2, and A431 cells, that require growth factor stimulation (42–46), it is unclear whether HCMV utilizes the same pathway in other cells of different origins. Here we show that THY-1 mediates HCMV entry in three different cell lines through an endocytic pathway that has many of the major features of macropinocytosis in a low-pH- and dynamin-dependent manner, which provides additional evidence supporting the role of THY-1 in HCMV entry and the role of macropinocytosis in virus entry.
MATERIALS AND METHODS
Cells, viruses, and reagents.
Human diploid fibroblasts (MRC-5 cells) were obtained from the American Type Culture Collection (ATCC) (Manassas, VA) and maintained in minimum essential medium or F12 medium with 10% fetal bovine serum (FBS). HS-578T adenocarcinoma and SNB-19 glioblastoma cells were obtained from Charles River Laboratories (Frederick, MD) and grown in RPMI 1640 medium with 10% FBS.
HCMV Towne-GFP and AD169 were propagated in MRC-5 cells. Cell culture supernatants from virus-infected cells were centrifuged at 2,000 × g for 30 min at 4°C, and the clarified supernatants were used as virus stocks or further partially purified by centrifugation through a 20% sucrose or sorbitol cushion at 35,000 × g at 4°C for 60 min and resuspended in RPMI 1640 medium with 10% FBS.
Anti-HCMV pp65 monoclonal antibody (MAb) was purchased from Virusys (Taneytown, MD). THY-1 monoclonal antibody 5E10 and IgG1 isotype control antibody were purchased from BioLegend (San Diego, CA). Polyclonal goat anti-THY-1 was obtained from Novus (Littleton, CO). Transferrin-conjugated Alexa 488- and AlexaFluo-conjugated secondary antibodies were purchased from Invitrogen (Grand Island, NY). IPA-3 (EMD, Chicago, IL), dynasore monohydrate, and filipin III (Santa Cruz, Santa Cruz, CA), jasplakinolide (Calbiochem, San Diego, CA), 5-(N-ethyl-N-isopropyl)amiloride (EIPA), bafilomycin A1, and monensin (Sigma-Aldrich, St. Louis, MO), and LY294002 (Cell Signaling Technology, Boston, MA) were used to inhibit virus entry.
Scanning electron microscopy.
Cells were grown and infected on silicon chips, fixed with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, and then postfixed with 1.0% osmium tetroxide in 0.1 M sodium cacodylate buffer. Specimens were dehydrated with a graded ethanol series, critical point dried under CO2 in a Bal-Tec model cpd 030 drier (Balzers, Liechtenstein), mounted on aluminum studs, and sputter coated with 50 Å of iridium in a model IBSe ion beam sputter coater (South Bay Technologies, San Clemente, CA) prior to viewing at 5 kV in a Hitachi SU-8000 field emission scanning electron microscope (Hitachi, Tokyo, Japan).
Transmission electron microscopy.
Specimens were fixed overnight at 4°C with 2.5% glutaraldehyde–4% paraformaldehyde in 0.1 M sodium cacodylate buffer, pH 7.4. Samples were postfixed for 1 h with 0.5% osmium tetroxide–0.8% potassium ferricyanide, stained overnight with 1% uranyl acetate at 4°C, dehydrated with a graded ethanol series, and embedded in Spurr's resin. Thin sections were cut with an RMC MT-7000 ultramicrotome (Ventana, Tucson, AZ) and stained with 1% uranyl acetate and Reynold's lead citrate prior to viewing at 80 kV on an FEI BT Tecnai transmission electron microscope (FEI, Hillsboro, OR). Digital images were acquired with an AMT digital camera system (AMT, Chazy, NY).
Immune labeling and cryo-immunoelectron microscopy.
Cells in suspension were fixed with 0.1% glutaraldehyde–2% paraformaldehyde in 0.1 M phosphate buffer overnight. Cells were washed with phosphate-buffered saline (PBS), resuspended in 10% liquid gelatin, centrifuged into a pellet, and then allowed to solidify at 4°C for 2 h. The gelatin pellet was cut into small cubes and immersed in 2.3 M sucrose overnight. Sections of 70 nm were cut at −120°C, picked up with a mixture of methylcellulose and sucrose, and placed on freshly glow-discharged 200-mesh nickel grids with Formvar-carbon coating. The grids were incubated on 20-μl droplets of 0.1% glycine in phosphate buffer for 5 min and then blocking reagent (1% bovine serum albumin [BSA]–Tris) for 10 min. Isotype control IgG1 and monoclonal THY-1 antibodies were diluted 1:10 in 1% BSA–Tris, and grids were incubated on 10-μl droplets for 30 min. After four 2-min washes with blocking buffer, the grids were incubated with 5-nm BBI colloidal gold-conjugated anti-mouse secondary antibody (Ted Pella, Redding, CA) diluted 1:25. Specimens were imaged by transmission electron microscopy as described above.
Confocal microscopy.
HCMV AD169 and transferrin-Alexa 488 conjugates were incubated with cells on ice for 60 min to allow binding. The temperature was then raised to 37°C for 45 min to internalize virus. HCMV and transferrin reagent that still remained outside the cells after internalization were inactivated and quenched, respectively, with low-pH citrate buffer (40 mM sodium citrate, 10 mM potassium chloride, 135 mM sodium chloride, pH 3.2) at room temperature for 3 min. The cells were then fixed with 2% paraformaldehyde and 0.25% glutaraldehyde in PBS and mounted with DAPI (4′,6′-diamidino-2-phenylindole)–Fluoromount-G (Southern Biotech, Birmingham, AL). Confocal imaging was performed with a Leica SP5 X-WLL microscope.
Inhibition of HCMV infection with chemical inhibitors and RT-qPCR to monitor infectivity.
Chemical inhibitors were dissolved in buffer or solvent as specified by the manufacturer and further diluted with cell culture medium to reduce the amount of solvent used. HS-578T cells were pretreated with inhibitors at 37°C for 60 to 90 min. HCMV was then added in the presence of the inhibitors on ice and left for 45 min, and the temperature was raised to 37°C for 5.5 h to initiate infection before the cells were lysed and total RNA was extracted using an RNeasy minikit (Qiagen, Valencia, CA) following the manufacturer's instructions. To eliminate DNA contamination, RNA was treated with DNase I (Roche Applied Science, Indianapolis, IN) and purified a second time with an RNeasy minikit. Quantitative real-time reverse transcription-PCR (RT-qPCR) was performed using One-step RT-PCR Master mix reagent (Promega, Madison, WI) with a 7500 real-time PCR machine. Primers and probes for detection of HCMV immediate early gene UL123 and late gene UL55 were described previously (108), and the following oligonucleotides were used: UL123, 5′-TCCCGCTTATCCTCAGGTACA, 5′-TGAGCCTTTCGAGGAGATGAA, and 5′-FAM-TCTCATACATGCTCTGCATAGTTAGCCCAATACA; UL55, 5′-TGGGCGAGGACAACGAA, 5′-TGAGGCTGGGAAGCTGACAT, and 5′-FAM-TGGGCAACCACCGCACTGAGG. Primers and Vic-labeled probe for GAPDH (glyceraldehyde-3-phosphate dehydrogenase) were purchased from Applied Biosystems (Carlsbad, CA). Serial dilutions of HCMV Bac DNA or human GAPDH plasmid were used to generate standard curves, and copy numbers of HCMV RNAs were normalized to copy numbers of human GAPDH amplified from the same wells. DNA contamination was monitored by performing PCR amplification without reverse transcriptase.
Fluid-phase marker uptake.
Cells were serum starved in OPTI-MEM medium (Invitrogen, Grand Island, NY) for 5 h. HCMV (Towne) was then added to cells and bound on ice for 60 min. Fluorophore-conjugated dextran (Invitrogen, Grand Island, NY) was added at 0.5 mg/ml. The temperature was shifted to 37°C for 60 min. The cells were then treated with low-pH citrate buffer (40 mM sodium citrate, 10 mM potassium chloride, 135 mM sodium chloride, pH 3.2) for 3 min to quench the remaining extracellular dextran-fluorescein isothiocyanate (dextran-FITC) and virus (29) and washed extensively with PBS. The uptake of dextran was quantified by fluorescence-activated cell sorter (FACS) analysis.
RESULTS
HCMV virions enter HS-578T and SNB-19 cells via macropinocytosis but not by clathrin-coated endocytosis.
Previously we showed that HS-578T adenocarcinoma cells support productive HCMV infection and produce progeny virus and that inhibiting THY-1 expression or blocking its activity reduced HCMV infection within the first hour of infection (29). We also found that blocking THY-1 inhibited virus-induced activation of PI3K/Akt. THY-1 is known to interact with integrins (47–50) and is a major protein present in clathrin-independent endocytic vesicles (39). Since macropinocytosis is PI3K/Akt dependent, involves integrin signaling, and is a clathrin-independent endocytic process, we determined whether THY-1 has a role in HCMV entry by macropinocytosis. Macropinosome formation is a result of organized movement of the plasma membrane and actin cytoskeleton, a multistep process including membrane ruffling and cup-shaped invagination of the plasma membrane (ruffle cup closure) (42). Virus-induced membrane ruffling can include different forms, including lamellopodial ruffles and filopodial protrusions and blebs. The variation in morphology is determined by the properties of the virus, the cell type infected, the receptor used, and the signaling pathways that are engaged (51).
To visualize HCMV entry we bound virus onto cells at 4°C and then raised the temperature to 37°C for internalization. The cells were then fixed and studied by electron microscopy. Scanning electron microscopy showed that HCMV induced cell surface membrane ruffling and blebbing in HS-578T cells, and virus particles were observed to associate with lamellipodium-like protrusions and ruffles (Fig. 1). A similar association of virions with membrane ruffles was also observed by transmission electron microscopy and showed features consistent with macropinocytosis (42, 52). Large vesicles (diameter of >200 nm) harboring HCMV virions were readily observed just under the plasma membrane (Fig. 2). The morphology of HCMV-containing macropinosomes was closely analogous to what has been observed with other viruses that enter cells by macropinocytosis (40, 52, 53). Like that of HS-578T cells, HCMV infection of SNB-19, a glioblastoma cell line, is THY-1 dependent at the entry step (29). Electron microscopy showed that most internalized HCMV particles were localized inside macropinosomes within the first 90 min of infection (Fig. 3), thus providing additional evidence for THY-1-mediated HCMV entry through macropinocytosis in an additional cell type.
FIG 1.
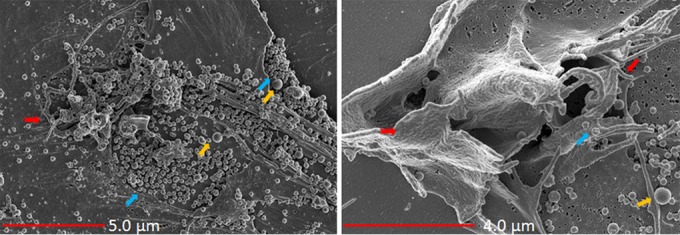
HCMV induces membrane ruffling and blebbing in HS-578T cells. HS-578T cells were incubated with HCMV on ice. The temperature was then raised to 37°C for 60 min to initiate internalization. Scanning electron microscopy was then performed as described in Materials and Methods. HCMV (blue arrows) was observed in close contact with lamellipodium-like protrusions and ruffles (red arrows) and membrane blebs (orange arrows).
FIG 2.

HCMV virions enter HS-578T cells via macropinocytosis. HCMV was bound to HS-578T cells and internalized for 90 min as described in the legend to Fig. 1. Transmission electron microscopy of HCMV-infected cells was performed, and virions were observed undergoing macropinocytosis with ruffling of membranes and ruffle closure over virions (panels A and B). Virions are seen inside macropinosomes (C to F). In 32 electron microscopy fields with virus-infected HS-278T cells, we observed that 81% (74/91) of HCMV particles were inside macropinosomes, while 19% (17/91) of particles were free in the cytoplasm within 90 min of infection.
FIG 3.
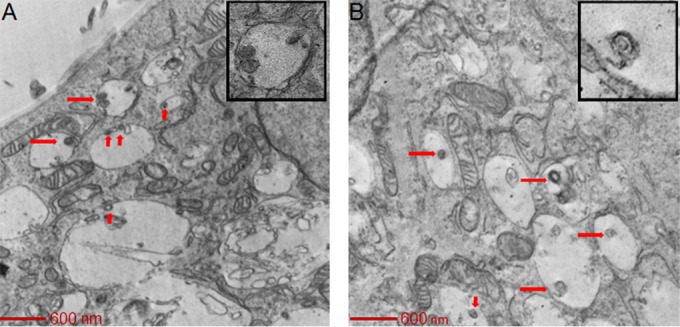
HCMV virions enter SNB-19 cells via macropinocytosis. HCMV was added to SNB-19 cells on ice for 60 min and then internalized for 90 min and observed under a transmission electron microscope as described in the legend to Fig. 2. In 20 electron microscopy fields with virus-infected SNB-19 cells, 87% (33/38) of HCMV particles were inside macropinosomes, while 13% (5/38) of HCMV particles were free in the cytoplasm.
To rule out the involvement of clathrin-dependent endocytosis in HCMV entry, we performed confocal microscopy using an endosomal marker, transferrin, in HS-578T cells (54). Purified HCMV AD169 virus and Alexa 488-conjugated transferrin were bound to the cell surface on ice, and entry was initiated at 37°C for 45 min. HCMV was identified by using a monoclonal antibody against virus-encoded tegument protein pp65. In the absence of transferrin, internalized HCMV was distributed evenly in the cytoplasm (Fig. 4A). Transferrin was also distributed evenly in the cytoplasm in the absence of HCMV (Fig. 4B). Entry of HCMV resulted in redistribution of transferrin from a diffuse to a more clumped pattern; however, HCMV did not colocalize with transferrin (Fig. 4C). Since transferrin has been shown to enter cells using clathrin-coated endosomes (54), these results indicate that HCMV does not use this pathway for entry in HS-578T cells. THY-1 did not colocalize with transferrin-Alexa 488 in either the absence (Fig. 4D) or presence (Fig. 4E) of virus, indicating that THY-1 was not present in clathrin-coated endosomes. These data are consistent with a previous report that THY-1 is excluded from clathrin-coated pits (55).
FIG 4.

HCMV internalization does not involve clathrin-coated endosomes. The results of a confocal microscopy study using a Leica SP5 microscope are shown. (A) HS-578T cells were exposed to HCMV on ice, followed by internalization at 37°C for 45 min, and virus internalization was terminated using a low-pH citrate buffer wash to inactivate noninternalized virus and remove the soluble proteins. The cells were then fixed and stained with monoclonal anti-pp65 followed by anti-mouse–Alexa 594 (red) secondary antibody. Nuclei were stained with DAPI (4′,6′-diamidino-2-phenylindole). (B) HS-578T cells were incubated with transferrin-conjugated Alexa 488 (green) on ice and then at 37°C for 45 min. After a low-pH buffer wash as described above, the cells were fixed and observed under the microscope. (C) HS-578T cells were incubated with both transferrin and HCMV as described for panels A and B. After the acid buffer wash, the cells were fixed and stained in the same manner as cells shown in panel A. (D and E) HS-578T cells were incubated with transferrin alone (D) or with both transferrin and HCMV (E) as described for panel C and then fixed and stained with a goat anti-THY-1 antibody followed by anti-goat–Alexa 549 (red) secondary antibody.
THY-1 colocalizes with HCMV virions on the cell surface and inside macropinosomes.
To determine if THY-1 is localized in macropinosomes and whether THY-1 contributes to the observed association of HCMV and macropinosomes, we performed cryo-immunoelectron microscopy. HCMV was allowed to bind to HS-578T cells on ice, and the temperature was raised to 37°C for 90 min to internalize the virus. The cells were then fixed in 2% paraformaldehyde and 0.25% glutaraldehyde in PBS and stained with anti-THY-1 mouse monoclonal antibody followed by gold-conjugated anti-mouse antibody. THY-1 colocalized with virions at the cell surface and in cell junctions (Fig. 5A, B, and C). In some fields, THY-1 was detected on the membrane in a morphological structure described as cup closure during macropinosome formation, together with HCMV virions (42) (Fig. 5D). Furthermore, THY-1 was present inside mature macropinosomes with virions (Fig. 6). Thus, cryo-immunoelectron microscopy provided strong evidence indicating that THY-1 is associated with HCMV virions inside macropinosomes.
FIG 5.
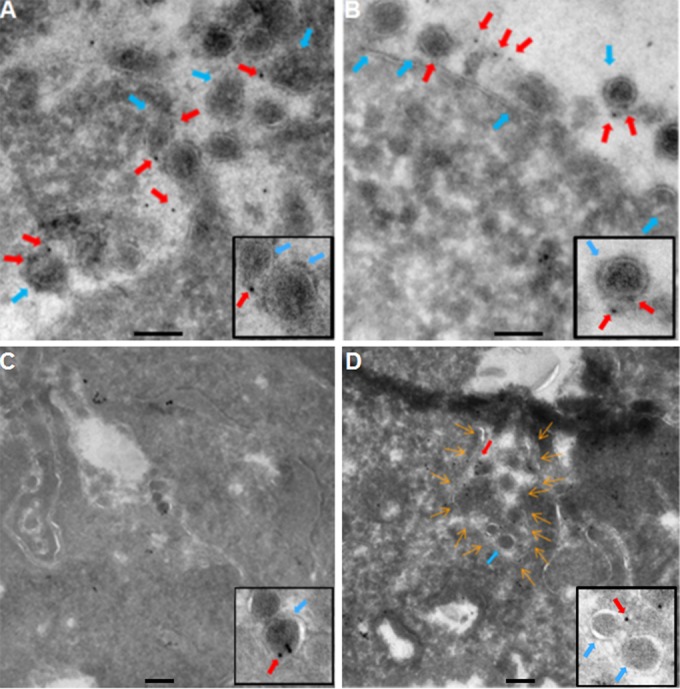
THY-1 colocalizes with HCMV virions on the cell surface and at cell junctions. HCMV was incubated with HS-578T cells and internalized as described in the legend to Fig. 2. The cells were fixed with 2% paraformaldehyde and 0.25% glutaraldehyde in PBS. Cryo-immunoelectron microscopy was performed with monoclonal anti-THY-1 antibody or isotype control antibody as described in Materials and Methods. Anti-mouse secondary antibody was conjugated with 5-nm gold particles. (A to C) HCMV virions (blue arrows) colocalized with THY-1 labeled with gold particles (red arrows) on the cell surface (A and B) and at cell junctions (C). Two representative micrographs are shown from over 20 fields in which THY-1 colocalized with virions. Insets show THY-1 associated with virus particles. (D) Both virions and THY-1 colocalized in a plasma membrane structure termed a “ruffle cup” in the process of macropinosome formation (42). The contour of the membrane invagination is outlined with orange arrows. Insets show THY-1 associated with virus particles. Bars represent 200 nm.
FIG 6.
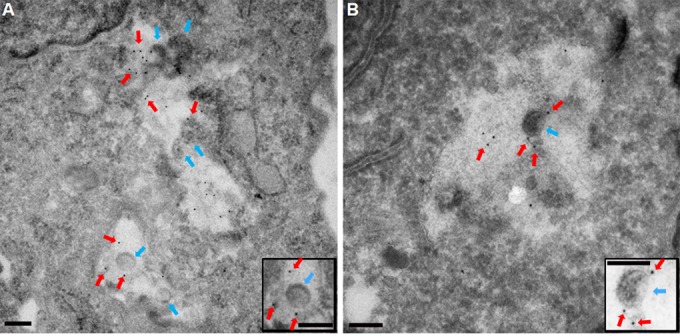
THY-1 colocalizes with HCMV virions inside macropinosomes. HCMV virions (blue arrows) colocalized with 5-nm-gold-labeled THY-1 (red arrows) inside large intracellular vesicles with diameters greater than 200 nm, a hallmark of macropinosomes. In 29 fields, we observed 70 virions and 296 labeled THY-1 particles inside macropinosomes, with 134 THY-1 particles in direct association with virus. Two representative fields are shown. Insets show THY-1 associated with virus particles. Bars represent 200 nm.
Inhibiting macropinocytosis interferes with HCMV infectivity.
A hallmark of virus entry by macropinocytosis is activation of signaling pathways following virus attachment and receptor clustering (11, 42, 51). These pathways are necessary for initiating actin remodeling that ensures membrane ruffling, invagination, and maturation of macropinosomes and primes host cells for infection (56). Inhibitors targeting different steps in the macropinosome formation process have been used to elucidate the mechanisms of virus entry. 5-(N-Ethyl-N-isopropyl)amiloride (EIPA) inhibits membrane ruffling by blocking Na+/H+ exchange, altering submembranous pH, and inhibiting signaling by Rac1 and Cdc42 (57). It is generally recognized that EIPA is the most selective inhibitor currently used for macropinocytosis (11, 58). We treated HS-578T cells with EIPA for 60 min, infected the cells with HCMV, and isolated RNA 4.5 to 5.5 h later. HCMV infectivity was then measured by RT-qPCR amplifying UL123 or by amplifying UL123 and UL55 gene products simultaneously. A previous study showed that double priming, detecting UL123 (an early gene) and UL55 simultaneously, resulted in higher sensitivity and earlier detection of HCMV RNA (59, 60). Although UL55 is a late gene, UL55 transcripts start to appear at 4 h postinfection, and expression is not strictly dependent on new viral DNA synthesis (61, 62). The copy number of HCMV UL123 RNA was then normalized against the copy number of GAPDH RNA that was simultaneously amplified in the same reaction as an internal control. EIPA abolished 90% of HCMV infectivity in HS-578T cells compared with a dimethyl sulfoxide (DMSO) control (Fig. 7A) (P < 0.0001, 3 independent experiments). The inhibitory effect of EIPA on infectivity was dose dependent (Fig. 7B). The level of GAPDH RNA was the same in cells treated with the highest dose of EIPA and DMSO (the solvent for EIPA). In addition, cell viability, determined by CytoTox-One assay (Promega, Madison, WI) which measures cell membrane integrity, was similar in EIPA-treated cells and solvent controls (Fig. 7C and D), indicating that EIPA was not cytotoxic under these conditions. Previously, we reported that soluble THY-1 (sTHY-1) blocks HCMV entry (29). Here we compared the inhibitory effects of EIPA and sTHY-1. Treatment of HS-578T cells with EIPA or sTHY-1 alone reduced HCMV infectivity by 90% and 60%, respectively (Fig. 7E). Less than 5% of the total infectivity was resistant to combined treatment with EIPA and sTHY-1. We previously showed that entry of HCMV into SNB-19 glioblastoma cells is THY-1 dependent (29). Pretreatment of SNB-19 cells with EIPA reduced HCMV infectivity by 80% in multiple independent experiments, and treatment with sTHY-1 reduced HCMV infectivity by 75% (Fig. 7F). Treatment with combined sTHY-1 and EIPA slightly reduced the HCMV infectivity compared to that with EIPA alone or sTHY-1 alone. These data suggest that macropinocytosis is an important pathway for internalization of HCMV. Since 80% of HCMV infectivity was THY-1 dependent and EIPA sensitive, the data imply that THY-1 mediates HCMV entry by macropinocytosis.
FIG 7.

Macropinocytosis inhibition of HCMV infection by EIPA is dose dependent, and EIPA and soluble THY-1 protein block HCMV infection to similar extents. (A and B) HS-578T cells were pretreated with EIPA at 215 μM (A) or at various concentrations (B), followed by HCMV infection for 4.5 to 5.5 h. RNA was extracted, and HCMV transcripts were detected using RT-qPCR and normalized against GAPDH amplified from the same reaction as an internal control. (C) To assess potential cytotoxicity, the level of GAPDH RNA was determined by RT-qPCR at the highest dose used for panel B (100 μM). (D) CytoTox-One assay was used to assess cytotoxicity based on cell membrane damage at the end of the infection. (E and F) HS-578T (E) and SNB-19 (F) cells were pretreated with 50 μM EIPA or DMSO solvent. HCMV was incubated with soluble THY-1 protein or control (filtrates that contained the same buffer composition) at room temperature for 10 min, and cells were infected for 4.5 h. RNA was extracted, and HCMV transcripts were detected using RT-qPCR and normalized against GAPDH amplified from the same reaction as an internal control.
Actin remodeling is essential for macropinosome formation, and inhibitors of actin remodeling such as jasplakinolide and cytochalasin D have been used to assess the role of macropinocytosis in virus infection (38, 40, 63–65). Treatment of HS-578T cells with jasplakinolide reduced HCMV infectivity (Fig. 8A) (P < 0.001, 6 independent experiments) at a nontoxic dose (Fig. 8B). Inhibition of actin remodeling with cytochalasin D also impaired virus infection in a dose-dependent manner (Fig. 8C). Within the dose range used, no detectable cytotoxicity was observed as assessed by monitoring the GAPDH RNA level and cell viability (Fig. 8D and E).
FIG 8.

Actin remodeling is important for HCMV-induced macropinocytosis. (A) HS-578T cells were pretreated with jasplakinolide (200 nM) for 60 min, followed by HCMV infection for 60 min. Virus internalization was then terminated by a low-pH buffer wash to inactivate any remaining extracellular virus. After an additional 5 h in culture, RNA was extracted and HCMV transcripts were detected using RT-qPCR and normalized against GAPDH amplified from the same reaction as described for Fig. 7. (B) The quantity of GAPDH RNA was determined by RT-qPCR to assess potential cytotoxicity. (C) HS-578T cells were pretreated with cytochalasin D (CytoD) at the indicated concentration, followed by HCMV infection for 4.5 h. RNA was extracted, and HCMV transcripts were detected using RT-qPCR and normalized against GAPDH amplified from the same reaction as an internal control. Infectivity was then normalized against that of the solvent control. (D) To assess potential cytotoxicity, the level of GAPDH RNA was determined by RT-qPCR at the highest CytoD concentration used for panel C (2.5 μM). (E) Cell viability was also monitored by CytoTox-One assay with 2.5 μM CytoD at the end of the infection.
The GTPase dynamin regulates endocytic vesicle formation and is generally required for clathrin- and caveola-mediated endocytosis; however, dynamin is also involved in some clathrin- and caveola-independent endocytic pathways (54). Dynasore inhibits the GTPase activity of both dynamin 1 and dynamin 2 (66). Treatment of HS-578T cells with dynasore reduced HCMV infectivity (Fig. 9A) (P = 0.006, 3 independent experiments), indicating that HCMV infection of epithelial cells is dynamin dependent. A prior study showed that dynamin is important for efficient internalization of HCMV into monocytes but not into fibroblasts (6), providing further evidence for cell type-specific entry of HCMV.
FIG 9.

Dynamin and cholesterol are important for HCMV-triggered macropinocytosis. HS-578T cells were pretreated with dynasore (71 μM) (A) or filipin (7.0 μg/ml) (B) for 60 to 90 min, followed by HCMV infection for 5.5 h. RNA was extracted, and HCMV transcripts were detected using RT-qPCR and normalized against GAPDH amplified from the same reaction. Representative data from multiple independent experiments are shown for each inhibitor.
Cholesterol is concentrated in lipid raft microdomains on the plasma membrane, is required for actin reorganization and membrane ruffling, and provides a platform for recruitment of activated signaling molecules such as Rac1 (33, 67). All of these features are required for macropinocytosis (68). THY-1 has been shown to localize in lipid rafts, and clustering of THY-1 is cholesterol dependent (69–71). Filipin is a polyene antibiotic and binds selectively to cholesterol, forming complexes in the plasma membrane that sequester cholesterol (72). In addition, treatment with filipin leads to unclustering of GPI-anchored proteins (73, 74). Depletion of cholesterol with filipin reduced HCMV infection (Fig. 9B) (P = 0.015, 2 independent experiments). These findings are consistent with a previous report that cholesterol is required for HCMV entry into fibroblasts (75).
Activation of macropinocytosis is often initiated by ligand-induced dimerization of receptor tyrosine kinases on the cell surface and the subsequent activation of signaling cascades (42, 76). PI3K and Pak1 are critical checkpoints for actin modulation, ruffle cup closure, and macropinosome trafficking (11). Treatment of the cells with a Pak1 inhibitor, IPA-3, blocked HCMV infection by ≥80% (Fig. 10A) (P = 0.0002, 2 independent experiments). Inhibition of HCMV infection by IPA-3 was dose dependent (Fig. 10B) and cytotoxicity was not observed (Fig. 10C and D). Similarly, incubation of cells with LY294002, a PI3K inhibitor, inhibited HCMV infection by 60% (Fig. 10E) (P < 0.01, 5 independent experiments). Previously, we have shown that blocking THY-1 with siRNA or antibody inhibited HCMV-induced PI3K activation within the first 45 min of infection (29). Thus, inhibiting macropinosome formation in HS-578T cells by several different mechanisms all led to impaired HCMV infection during virus entry into the cells. Similar results were also observed with MRC-5 fibroblasts, in which inhibitors targeting macropinocytosis severely impaired HCMV infectivity (Fig. 11A and B) at nontoxic doses (Fig. 11C). The inhibition was dose dependent (Fig. 11D and E). Importantly, these inhibitors were required to be present before infection, since adding the inhibitors 2 h after infection (designated “added-in” in Fig. 11B) failed to block infectivity, with the exception of IPA-3, a Pak1 inhibitor. The failure of IPA-3 to block HCMV infectivity when added after infection is likely due to the observation that p21-activated kinases (Pak) have an important role in virus replication (77). We also tested whether several other viruses, including vaccinia virus, adenovirus, respiratory syncytial virus, and human papillomavirus pseudovirions, use macropinocytosis to enter HS-578T cells. We found that the macropinocytosis inhibitors we used that blocked HCMV entry also blocked entry of the other viruses, indicating that these viruses infect HS-578T cells through macropinocytosis, consistent with prior reports that these viruses use macropinocytosis to enter other cell types (63, 64, 78, 79).
FIG 10.

Disrupting PI3K and p21-activated kinase 1 (Pak1) signaling blocks HCMV infection. HS-578T cells were pretreated with the Pak1 inhibitor IPA-3 at16.7 μM (A) or at the indicated concentration (B) or treated with the PI3K inhibitor LY294002 (12.5 μM) (E) and infected with HCMV as described for Fig. 7. Infection was monitored either by RT-qPCR for HCMV UL123 at 5.5 h postinoculation (A) or by FACS for HCMV-GFP positive cells at day 3 postinfection (E). The level of GAPDH RNA (C) and cell viability (D) were measured to assess potential cytotoxicity associated with IPA-3 as described in the legend to Fig. 7.
FIG 11.

HCMV infection of MRC-5 cells is sensitive to macropinocytosis inhibitors during entry. (A and B) MRC-5 cells were pretreated with EIPA (50 μM), IPA-3 (25 μM), dynasore (50 μM), cytochalasin D (CytoD) (2 μM), jasplakinolide (Jasp) (200 nM), or 0.1% DMSO (solvent) at 37°C for 60 min, followed by infection for 4.5 h. For the “added-in” groups (B), the cells were infected in parallel with the pretreatment groups; after infection at 37°C for 2 h, the inhibitors were added to the same concentration as in the pretreatment groups and cultured for additional 2.5 h. RNA was extracted from the cells, treated with DNase I, and repurified. HCMV UL123 expression was monitored by RT-qPCR and normalized against GAPDH amplified in the same reaction. Infectivity was then normalized against that of the DMSO group. (C) Quantitation of GAPDH RNA by RT-qPCR to monitor cytotoxicity at the end of the 4.5-h infection. (D and E) MRC-5 cells were pretreated with the indicated amount of dynasore (D) or cytochalasin D (CytoD) (E) or 0.1% DMSO (the amount used to dissolve the higher doses of the inhibitors) at 37°C for 60 min, followed by infection for 4.5 h. RNA extraction and RT-qPCR were performed to monitor infectivity as described for the previous figures.
Entry of HCMV triggers increased uptake of extracellular fluid.
Increased influx of extracellular fluid is one of the hallmarks of macropinocytosis, a result of membrane ruffling and invagination to form large fluid-filled vesicles. The process often occurs as a transient response to growth factor or virus stimulation (11, 58). However, numerous studies have shown that in antigen-presenting cells and in certain tumor cell lines, macropinocytosis is constitutively active such that efficient accumulation of fluid-phase markers is not dependent on stimulation by exogenous factors (42, 43, 80–82). Fluorophore-labeled high-molecular-mass polysaccharides, such as 10-kDa and 70-kDa dextrans, have been used to quantify extracellular fluid uptake during virus entry (63, 65, 79, 83–86). We incubated HCMV with SNB-19 cells on ice for 1 h, added FITC-conjugated 10-kDa or 70-kDa dextran to the cells, and shifted the temperature to 37°C for 1 h to allow virus internalization. Entry of HCMV was associated with increased dextran uptake compared with that in mock-infected cells (Fig. 12A [10-kDa dextran, 9 independent experiments] and B [70-kDa dextran, 4 independent experiments]). Pretreatment of the cells with EIPA, an inhibitor of macropinocytosis, blocked virus-triggered uptake of dextran (Fig. 12A and B). Interestingly, in multiple experiments with HS-578T cells, we did not observe a significantly increased fluid influx associated with HCMV (Fig. 12C) compared with the case for SNB-19 cells (Fig. 12D). Instead, HS-578T consistently demonstrated a higher level of baseline fluid influx than SNB-19 (Fig. 12C versus D), suggesting a possibility of constitutive macropinocytosis. To verify this, we stimulated the cells with EGF, which has been shown to trigger macropinocytosis through activation of receptor tyrosine kinase and EGFR (58, 81, 82, 87). While EGF stimulation resulted in increased fluid uptake in SNB-19 cells, EGF failed to increase fluid uptake further in HS-578T cells (Fig. 12E). Furthermore, the high baseline of fluid influx in HS-578T cells was macropinocytosis dependent, since treatment with EIPA totally abolished the fluid influx in HS-578T cells (Fig. 12F). Taken together, these data strongly suggest that HS-578T cells undergo constitutively active macropinocytosis.
FIG 12.

Both HCMV and EGF induce dextran uptake and activate macropinocytosis in SNB-19 cells, while HS-578T cells have high-level constitutively active macropinocytosis. (A and B) SNB-19 cells were serum starved for 5 h and then pretreated with EIPA (50 μM) or DMSO (the solvent for EIPA) at 37°C for 60 min. HCMV was added to the cells and incubated on ice for 45 to 60 min, followed by addition of 10-kDa dextran–Alexa 488 (A) or 70-kDa dextran–FITC (B) at 0.5 mg/ml. The temperature was then shifted to 37°C for 60 min. The cells were then treated with low-pH citrate buffer for 3 min and washed extensively with PBS, and uptake of dextran was quantified by FACS analysis. (C to E) HS-578T (C) or SNB-19 (D) cells were treated with 70-kDa dextran–FITC as described above. In parallel, the cells were treated with EGF at 50 ng/ml for 60 min at 37°C in the presence of 0.5 mg/ml 70-kDa dextran–FITC (E). (F) HS-578T cells were treated in the absence or presence of EIPA (50 μM) at 37°C for 60 min and then incubated with HCMV and dextran as described above.
Soluble THY-1 blocks HCMV internalization, and virus entry triggers influx of 70-kDa dextran, a marker of macropinocytosis.
Since THY-1 blocks HCMV entry in SNB-19 cells (29) (Fig. 7F), we determined whether sTHY-1 also blocks HCMV-triggered macropinocytosis. HCMV was preincubated with sTHY-1 or control protein (filtrates from the sTHY-1 preparation in which sTHY-1 was removed by a size exclusion column) at room temperature for 10 min and bound to SNB-19 cells on ice for 60 min. A marker of macropinocytosis, dextran (70 kDa)-FITC (46, 88), was then added at 0.5 mg/ml and 37°C and left for 60 min. The cells were then treated with low-pH citrate buffer for 3 min to quench any remaining extracellular dextran-FITC and inactivate virus, washed extensively with PBS, and fixed with 2% paraformaldehyde and 0.25% glutaraldehyde. HCMV was stained with a MAb against pp65 (Virusys, Taneytown, MD), followed by anti-mouse–Alexa 594-labeled secondary antibody. Confocal imaging of the cells was performed, and the mean fluorescence intensities for internalized virus particles (red) and dextran (green) were normalized against the number of cells. sTHY-1 blocked internalization of HCMV by nearly 40% (Fig. 13A). Pretreatment of HCMV with sTHY-1 diminished both internalized virus and dextran by over 50% (Fig. 13B to E). These data imply that THY-1 is important for entry of HCMV into cells by macropinocytosis.
FIG 13.

sTHY-1 blocks HCMV internalization and reduces HCMV-triggered uptake of 70-kDa dextran, a macropinocytosis marker. HCMV was bound to SNB-19 cells (nuclei were stained blue with DAPI), and 70-kDa dextran–FITC was then added at 37°C and left for 60 min (as described in the legend to Fig. 10). The cells were stained with anti-pp65 antibody followed by anti-mouse–Alexa 594. Confocal microscopy was performed; 36 fields from each sample were randomly selected, and mean fluorescence intensity for internalized virus particles (red) and dextran (green) was analyzed using Imaris imaging software. (A) The quantity of internalized HCMV particles was normalized against dextran and the cell number. (B and C) The number of internalized particles for virus (B) and dextran (C) in four fields of each sample was normalized for the number of cells. The numbers on the x axes of panels A to C indicate the number of cells examined. (D and E) Two representative fields.
Entry of HCMV in HS-578T and MRC-5 cells is pH dependent.
Most viruses that use macropinocytosis as their primary entry pathway require low pH for internalization (11). HCMV has been reported to enter dendritic cells through a macropinocytosis-like pathway, which occurs at neutral pH (40). To determine if a low pH is needed for the macropinocytic entry of HCMV into HS-578T epithelial cells, we treated the cells with several lysosomotropic agents and followed infection. Ammonium chloride (NH4Cl) is a weak base that raises the pH of acidic endosomal compartments. Pretreating HS-578T cells with NH4Cl inhibited HCMV infectivity in a dose-dependent manner (Fig. 14A and B); no loss of in cell viability was observed at the highest concentration of NH4Cl (Fig. 14C). Monensin, a carboxylic ionophore that blocks endosomal acidification, and bafilomycin A1, an inhibitor of the vacuolar H+-ATPase in the endosomal membrane which prevents acidification of endosomal vesicles, both inhibited HCMV infectivity (Fig. 14D) (4 independent experiments). Compared to NH4Cl or monensin, bafilomycin A1 had less of an inhibitory effect on HCMV infectivity. Nonetheless inhibition of infection was significant (P < 0.01 for each inhibitor versus solvent control) and reproducible in multiple independent experiments. Therefore, HCMV enters HS-578T cells in a pH-dependent manner. Similar results were observed with MRC-5 cells, and in each case the inhibitors significantly reduced the infectivity of HCMV compared with the solvent (P < 0.01) (Fig. 14E).
FIG 14.

HCMV infection is impaired when endosomal acidification is blocked. (A) HS-578T cells were pretreated with NH4Cl (50 mM) at 37°C for 60 min, followed by infection for 5.5 h. RNA was extracted, and HCMV UL123 expression was monitored by RT-qPCR and normalized against GAPDH amplified in the same reaction. (B) HS-578T cells were treated with NH4Cl at various concentrations. HCMV infection was carried out for 4.5 h, and the cells were washed with low-acid buffer for 3 min to inactivate uninternalized virus and cultured for additional 3 days without NH4Cl before FACS analysis. (C) Cell viability was monitored by CytoTox-One assay using the highest dose (100 mM) of NH4Cl. (D and E) HS-578T cells (D) and MRC-5 cells (E) were pretreated with NH4Cl (50 mM), monensin (25 nM), or bafilomycin A1 (BFLA1) (200 nM). Representative data from 3 independent experiments for each cell type are shown (P < 0.0001 for both NH4Cl and monensin versus solvent control; P < 0.01 for bafilomycin A1 versus solvent control).
DISCUSSION
Although viruses can enter host cells by direct cell surface membrane fusion, most viruses have evolved to take advantage of a number of endocytic pathways for efficient entry. Recent studies have shown that viruses from multiple families, including adenoviruses, picornaviruses, retroviruses, papillomaviruses, poxviruses, paramyxoviruses, filoviruses, and herpesviruses, use macropinocytosis (51). Macropinosomes share common features and markers with endosomal and phagocytic vesicles, and these pathways are interconnected and interdependent in complex ways in a cell type-dependent manner (11, 89). Therefore, several criteria have been proposed to define whether a virus uses macropinocytosis (51). These include morphological evidence of localization of virions in endocytic vesicles >200 nm in diameter and membrane ruffling or blebbing. Other criteria for macropinocytosis include dependence of virus infection on actin remodeling, Na+/H+ exchangers, dynamin, or signaling molecules such as p21-activated kinase 1 (Pak1) and PI3K/Akt or a transient increase in fluid uptake. We found that HCMV entry into three different cell lines of diverse tissue origins has classic features of macropinocytosis: virions were internalized into large endocytic vesicles, virus entry triggered membrane ruffling and blebbing, activation of signaling and actin remodeling were required, and inhibitors of Na+/H+ exchange (EIPA), dynamin, and cholesterol effectively blocked infectivity. Previous studies showed that HCMV enters dendritic cells, ARPE-19 epithelial cells, and primary fibroblast cells through macropinocytosis (18, 40, 41). Our results provide further evidence that macropinocytosis is a major pathway for HCMV infection in a broad spectrum of cell types.
Although macropinocytosis has often been defined as dynamin independent (90), dynamin is required for macropinocytic entry of several viruses, including HCMV entry into primary fibroblasts (41, 51, 91). In this study, we found that dynamin is needed for HCMV infection in two different cell lines. Haspot et al. reported that HCMV entry into dendritic cells is pH independent (40). However, our data, obtained using three different inhibitors to prevent endosomal acidification, clearly showed that low endosomal pH is required for HCMV infection of the cell lines used in our study. Bodaghi et al. observed that in epithelial and endothelial cells, HCMV virions localized to large endocytic vesicles, and an actin remodeling inhibitor (cytochalasin B) blocked >80% of the infectivity (92). Interestingly, infection was pH independent at high multiplicity of infection (MOI) but pH dependent at low MOI. Vanarsdall et al. reported that HCMV infects ARPE-19 epithelial cells through clathrin-independent, dynamin-dependent, and low-pH-dependent macropinocytosis (18). Recently, Hetzenecker et al. demonstrated that HCMV infects human primary fibroblasts via macropinocytosis in a pH-independent manner (41). Thus, it is clear that while macropinocytosis is a primary entry mechanism for HCMV, cell type-associated variations have been observed. Therefore, HCMV entry in these cells may be more appropriately termed as using a “macropinocytosis-like pathway.”
HCMV infection in HS-578T cells has the major hallmarks of macropinocytosis, including inhibition of infection by EIPA. Our data suggest that HS-578T cells undergo constitutively active macropinocytosis, which does not require additional exogenous stimulation. This may explain our observation that, in contrast to the case for SNB-19 cells, adding HCMV or EGF to HS-578T cells did not trigger additional fluid uptake. A previous study reported that adeno-associated virus 2 entry into HeLa and 293T cells is sensitive to EIPA and is dependent on Cdc42 signaling, cholesterol, and actin remodeling. While virus entry was associated with extensive plasma membrane blebbing and ruffling, there was no increase in fluid-phase uptake with virus infection (93).
Our previous work showed that THY-1 is important for HCMV infection during the initial stage of virus entry (29). Here, using three different cell types, we provide further evidence that HCMV utilizes THY-1 to gain entry into certain cell types through an endocytotic pathway that bears many features of macropinocytosis. THY-1 is a membrane GPI-anchored protein. In addition to its plasma membrane location, THY-1 is a major cargo content of clathrin-independent large endocytic vesicles and is excluded from clathrin-coated endosomes (39, 55). THY-1 interacts with several integrins and with growth factor receptors, such as PDGFR and EGFR, and is associated with lipid rafts, all of which have been implicated in playing important roles in HCMV entry and in activation of endocytic compartments (42, 94). Therefore, the ability of HCMV to interact with THY-1 provides a natural route for virus entry.
Interactions between virus-encoded ligands and host receptors and/or other accessory molecules serve as sorting signals for different endocytic pathways (58, 95). Cholesterol-dependent clustering of GPI-anchored proteins is critical for activation of endocytic pathways (94, 96). Binding of virus-encoded ligands cross-links GPI-anchored proteins and switches on signaling cascades that precede macropinocytosis activation. For example, group B coxsackieviruses interact with the GPI-anchored protein decay-accelerating factor, which results in clustering of the protein and leads to virus delivery to tight junctions and subsequent endocytotic entry (97, 98). Numerous studies have shown that cross-linking of THY-1 is essential for its role in signaling through Src and PI3K pathways (99, 100). Src and PI3K are critical regulators of macropinocytosis (43, 51). The putative binding of HCMV gB and/or gH to THY-1 (29) may cross-link the GPI-anchored protein. Clustering of GPI-anchored proteins has been shown to serve as a sorting signal for specific endocytic routing (76). A prior study showed that binding of αvβ3 integrin by HCMV at the cell surface results in the activation of PI3K and Src, with actin reorganization through RhoA, which correlates with translocation of HCMV capsids to the nucleus (30). Integrin plays an important role in induction of macropinocytosis by signaling through the non-receptor tyrosine kinase focal adhesion kinase and RhoA (91, 101–103). THY-1 binds to several integrins and shares common features with integrins in protein-protein interactions, including RhoA activation (48). Both THY-1 and integrins localize to lipid rafts and contribute to endocytosis. Previous work has shown that HCMV gB and/or gH interacts with THY-1 (29) and integrins (30, 104). In addition, a gH/gL/gB complex is present in HCMV particles (10). Therefore, THY-1 and integrins might cooperatively mediate HCMV internalization by bridging virus attachment with macropinocytosis.
In this study, we found that HCMV virions colocalize with THY-1 on the plasma membrane and inside macropinosomes during virus entry and that sTHY-1 simultaneously blocks virion internalization and dextran uptake (a marker of macropinocytosis). Taken together, these data strongly suggest that THY-1 may facilitate HCMV entry by the same mechanism that other GPI-anchored proteins use for selective retention in macropinosomes (105–107). This has similarities to what has been observed with adenovirus serotype 3, which utilizes CD46 and integrins as entry mediators to enter host cells by macropinocytosis. Adenovirus 3 colocalizes with CD46 and integrins inside macropinosomes (53). We found that HCMV and THY-1 did not colocalize with transferrin, indicating that HCMV and THY-1 are excluded from clathrin-coated endosomes. THY-1 is a major cargo protein of clathrin-independent endocytic vesicles (39). Thus, THY-1 may help to selectively guide its ligands to distinct endocytic pathways. Uptake of viruses by macropinocytosis may be a more efficient entry route than fusion with the plasma membrane, since macropinosomes internalize and recycle in the cell very effectively. We found that THY-1 facilities HCMV entry more efficiently during the first hour of infection than after prolonged virus incubation in certain cell types (29). These results suggest that HCMV has adapted to utilize THY-1, a cargo protein of clathrin-independent endocytotic vesicles, to facilitate efficient entry and couple virus internalization with signaling activation.
ACKNOWLEDGMENTS
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases.
We thank Sundar Ganesan for confocal microscopy.
Funding Statement
This work was funded by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health.
REFERENCES
- 1.Hahn G, Revello MG, Patrone M, Percivalle E, Campanini G, Sarasini A, Wagner M, Gallina A, Milanesi G, Koszinowski U, Baldanti F, Gerna G. 2004. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J Virol 78:10023–10033. doi: 10.1128/JVI.78.18.10023-10033.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ryckman BJ, Jarvis MA, Drummond DD, Nelson JA, Johnson DC. 2006. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J Virol 80:710–722. doi: 10.1128/JVI.80.2.710-722.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sinzger C, Digel M, Jahn G. 2008. Cytomegalovirus cell tropism. Curr Top Microbiol Immunol 325:63–83. [DOI] [PubMed] [Google Scholar]
- 4.Wang D, Shenk T. 2005. Human cytomegalovirus UL131 open reading frame is required for epithelial cell tropism. J Virol 79:10330–10338. doi: 10.1128/JVI.79.16.10330-10338.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou M, Yu Q, Wechsler A, Ryckman BJ. 2013. Comparative analysis of gO isoforms reveals that strains of human cytomegalovirus differ in the ratio of gH/gL/gO and gH/gL/UL128-131 in the virion envelope. J Virol 87:9680–9690. doi: 10.1128/JVI.01167-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nogalski MT, Chan GC, Stevenson EV, Collins-McMillen DK, Yurochko AD. 2013. The HCMV gH/gL/UL128-131 complex triggers the specific cellular activation required for efficient viral internalization into target monocytes. PLoS Pathog 9:e1003463. doi: 10.1371/journal.ppat.1003463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou M, Lanchy JM, Ryckman BJ. 2015. Human cytomegalovirus gH/gL/gO promotes the fusion step of entry into all cell types, whereas gH/gL/UL128-131 broadens virus tropism through a distinct mechanism. J Virol 89:8999–9009. doi: 10.1128/JVI.01325-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ryckman BJ, Chase MC, Johnson DC. 2008. HCMV gH/gL/UL128-131 interferes with virus entry into epithelial cells: evidence for cell type-specific receptors. Proc Natl Acad Sci U S A 105:14118–14123. doi: 10.1073/pnas.0804365105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wille PT, Wisner TW, Ryckman B, Johnson DC. 2013. Human cytomegalovirus (HCMV) glycoprotein gB promotes virus entry in trans acting as the viral fusion protein rather than as a receptor-binding protein. mBio 4:e00332-13. doi: 10.1128/mBio.00332-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vanarsdall AL, Howard PW, Wisner TW, Johnson DC. 2016. Human cytomegalovirus gH/gL forms a stable complex with the fusion protein gB in virions. PLoS Pathog 12:e1005564. doi: 10.1371/journal.ppat.1005564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mercer J, Schelhaas M, Helenius A. 2010. Virus entry by endocytosis. Annu Rev Biochem 79:803–833. doi: 10.1146/annurev-biochem-060208-104626. [DOI] [PubMed] [Google Scholar]
- 12.Nicola AV, McEvoy AM, Straus SE. 2003. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J Virol 77:5324–5332. doi: 10.1128/JVI.77.9.5324-5332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller N, Hutt-Fletcher LM. 1992. Epstein-Barr virus enters B cells and epithelial cells by different routes. J Virol 66:3409–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Compton T, Nepomuceno RR, Nowlin DM. 1992. Human cytomegalovirus penetrates host cells by pH-independent fusion at the cell surface. Virology 191:387–395. doi: 10.1016/0042-6822(92)90200-9. [DOI] [PubMed] [Google Scholar]
- 15.Helenius A. 2013. Virus entry: what has pH got to do with it? Nat Cell Biol 15:125. doi: 10.1038/ncb2678. [DOI] [PubMed] [Google Scholar]
- 16.Krummenacher C, Carfi A, Eisenberg RJ, Cohen GH. 2013. Entry of herpesviruses into cells: the enigma variations. Adv Exp Med Biol 790:178–195. doi: 10.1007/978-1-4614-7651-1_10. [DOI] [PubMed] [Google Scholar]
- 17.Akula SM, Naranatt PP, Walia NS, Wang FZ, Fegley B, Chandran B. 2003. Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) infection of human fibroblast cells occurs through endocytosis. J Virol 77:7978–7990. doi: 10.1128/JVI.77.14.7978-7990.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vanarsdall AL, Wisner TW, Lei H, Kazlauskas A, Johnson DC. 2012. PDGF receptor-alpha does not promote HCMV entry into epithelial and endothelial cells but increased quantities stimulate entry by an abnormal pathway. PLoS Pathog 8:e1002905. doi: 10.1371/journal.ppat.1002905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nanbo A, Kawanishi E, Yoshida R, Yoshiyama H. 2013. Exosomes derived from Epstein-Barr virus-infected cells are internalized via caveola-dependent endocytosis and promote phenotypic modulation in target cells. J Virol 87:10334–10347. doi: 10.1128/JVI.01310-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang HB, Zhang H, Zhang JP, Li Y, Zhao B, Feng GK, Du Y, Xiong D, Zhong Q, Liu WL, Du H, Li MZ, Huang WL, Tsao SW, Hutt-Fletcher L, Zeng YX, Kieff E, Zeng MS. 2015. Neuropilin 1 is an entry factor that promotes EBV infection of nasopharyngeal epithelial cells. Nat Commun 6:6240. doi: 10.1038/ncomms7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soroceanu L, Akhavan A, Cobbs CS. 2008. Platelet-derived growth factor-alpha receptor activation is required for human cytomegalovirus infection. Nature 455:391–395. doi: 10.1038/nature07209. [DOI] [PubMed] [Google Scholar]
- 22.Wang X, Huong SM, Chiu ML, Raab-Traub N, Huang ES. 2003. Epidermal growth factor receptor is a cellular receptor for human cytomegalovirus. Nature 424:456–461. doi: 10.1038/nature01818. [DOI] [PubMed] [Google Scholar]
- 23.Halary F, Amara A, Lortat-Jacob H, Messerle M, Delaunay T, Houles C, Fieschi F, Arenzana-Seisdedos F, Moreau JF, Dechanet-Merville J. 2002. Human cytomegalovirus binding to DC-SIGN is required for dendritic cell infection and target cell trans-infection. Immunity 17:653–664. doi: 10.1016/S1074-7613(02)00447-8. [DOI] [PubMed] [Google Scholar]
- 24.Feire AL, Koss H, Compton T. 2004. Cellular integrins function as entry receptors for human cytomegalovirus via a highly conserved disintegrin-like domain. Proc Natl Acad Sci U S A 101:15470–15475. doi: 10.1073/pnas.0406821101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feire AL, Roy RM, Manley K, Compton T. 2010. The glycoprotein B disintegrin-like domain binds beta 1 integrin to mediate cytomegalovirus entry. J Virol 84:10026–10037. doi: 10.1128/JVI.00710-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nogalski MT, Chan G, Stevenson EV, Gray S, Yurochko AD. 2011. Human cytomegalovirus-regulated paxillin in monocytes links cellular pathogenic motility to the process of viral entry. J Virol 85:1360–1369. doi: 10.1128/JVI.02090-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Isaacson MK, Feire AL, Compton T. 2007. Epidermal growth factor receptor is not required for human cytomegalovirus entry or signaling. J Virol 81:6241–6247. doi: 10.1128/JVI.00169-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan G, Nogalski MT, Yurochko AD. 2009. Activation of EGFR on monocytes is required for human cytomegalovirus entry and mediates cellular motility. Proc Natl Acad Sci U S A 106:22369–22374. doi: 10.1073/pnas.0908787106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Q, Wilkie AR, Weller M, Liu X, Cohen JI. 2015. THY-1 cell surface antigen (CD90) has an important role in the initial stage of human cytomegalovirus infection. PLoS Pathog 11:e1004999. doi: 10.1371/journal.ppat.1004999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang X, Huang DY, Huong SM, Huang ES. 2005. Integrin alphavbeta3 is a coreceptor for human cytomegalovirus. Nat Med 11:515–521. doi: 10.1038/nm1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rege TA, Hagood JS. 2006. Thy-1, a versatile modulator of signaling affecting cellular adhesion, proliferation, survival, and cytokine/growth factor responses. Biochim Biophys Acta 1763:991–999. doi: 10.1016/j.bbamcr.2006.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leyton L, Schneider P, Labra CV, Ruegg C, Hetz CA, Quest AF, Bron C. 2001. Thy-1 binds to integrin beta(3) on astrocytes and triggers formation of focal contact sites. Curr Biol 11:1028–1038. doi: 10.1016/S0960-9822(01)00262-7. [DOI] [PubMed] [Google Scholar]
- 33.Helms JB, Zurzolo C. 2004. Lipids as targeting signals: lipid rafts and intracellular trafficking. Traffic 5:247–254. doi: 10.1111/j.1600-0854.2004.0181.x. [DOI] [PubMed] [Google Scholar]
- 34.Anderson RG, Jacobson K. 2002. A role for lipid shells in targeting proteins to caveolae, rafts, and other lipid domains. Science 296:1821–1825. doi: 10.1126/science.1068886. [DOI] [PubMed] [Google Scholar]
- 35.Campadelli-Fiume G, Menotti L, Avitabile E, Gianni T. 2012. Viral and cellular contributions to herpes simplex virus entry into the cell. Curr Opin Virol 2:28–36. doi: 10.1016/j.coviro.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 36.Tang H, Mori Y. 2010. Human herpesvirus-6 entry into host cells. Future Microbiol 5:1015–1023. doi: 10.2217/fmb.10.61. [DOI] [PubMed] [Google Scholar]
- 37.Bender FC, Whitbeck JC, Ponce de Leon M, Lou H, Eisenberg RJ, Cohen GH. 2003. Specific association of glycoprotein B with lipid rafts during herpes simplex virus entry. J Virol 77:9542–9552. doi: 10.1128/JVI.77.17.9542-9552.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raghu H, Sharma-Walia N, Veettil MV, Sadagopan S, Caballero A, Sivakumar R, Varga L, Bottero V, Chandran B. 2007. Lipid rafts of primary endothelial cells are essential for Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8-induced phosphatidylinositol 3-kinase and RhoA-GTPases critical for microtubule dynamics and nuclear delivery of viral DNA but dispensable for binding and entry. J Virol 81:7941–7959. doi: 10.1128/JVI.02848-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Howes MT, Kirkham M, Riches J, Cortese K, Walser PJ, Simpson F, Hill MM, Jones A, Lundmark R, Lindsay MR, Hernandez-Deviez DJ, Hadzic G, McCluskey A, Bashir R, Liu L, Pilch P, McMahon H, Robinson PJ, Hancock JF, Mayor S, Parton RG. 2010. Clathrin-independent carriers form a high capacity endocytic sorting system at the leading edge of migrating cells. J Cell Biol 190:675–691. doi: 10.1083/jcb.201002119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haspot F, Lavault A, Sinzger C, Laib Sampaio K, Stierhof YD, Pilet P, Bressolette-Bodin C, Halary F. 2012. Human cytomegalovirus entry into dendritic cells occurs via a macropinocytosis-like pathway in a pH-independent and cholesterol-dependent manner. PLoS One 7:e34795. doi: 10.1371/journal.pone.0034795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hetzenecker S, Helenius A, Krzyzaniak MA. 2016. HCMV induces macropinocytosis for host cell entry in fibroblasts. Traffic 17:351–368. doi: 10.1111/tra.12355. [DOI] [PubMed] [Google Scholar]
- 42.Swanson JA. 2008. Shaping cups into phagosomes and macropinosomes. Nat Rev Mol Cell Biol 9:639–649. doi: 10.1038/nrm2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lim JP, Gleeson PA. 2011. Macropinocytosis: an endocytic pathway for internalising large gulps. Immunol Cell Biol 89:836–843. doi: 10.1038/icb.2011.20. [DOI] [PubMed] [Google Scholar]
- 44.West MA, Bretscher MS, Watts C. 1989. Distinct endocytotic pathways in epidermal growth factor-stimulated human carcinoma A431 cells. J Cell Biol 109:2731–2739. doi: 10.1083/jcb.109.6.2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fiorentini C, Falzano L, Fabbri A, Stringaro A, Logozzi M, Travaglione S, Contamin S, Arancia G, Malorni W, Fais S. 2001. Activation of rho GTPases by cytotoxic necrotizing factor 1 induces macropinocytosis and scavenging activity in epithelial cells. Mol Biol Cell 12:2061–2073. doi: 10.1091/mbc.12.7.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Canton J, Schlam D, Breuer C, Gutschow M, Glogauer M, Grinstein S. 2016. Calcium-sensing receptors signal constitutive macropinocytosis and facilitate the uptake of NOD2 ligands in macrophages. Nat Commun 7:11284. doi: 10.1038/ncomms11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fiore VF, Ju L, Chen Y, Zhu C, Barker TH. 2014. Dynamic catch of a Thy-1-alpha5beta1+syndecan-4 trimolecular complex. Nat Commun 5:4886. doi: 10.1038/ncomms5886. [DOI] [PubMed] [Google Scholar]
- 48.Barker TH, Hagood JS. 2009. Getting a grip on Thy-1 signaling. Biochim Biophys Acta 1793:921–923. doi: 10.1016/j.bbamcr.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herrera-Molina R, Frischknecht R, Maldonado H, Seidenbecher CI, Gundelfinger ED, Hetz C, Aylwin Mde L, Schneider P, Quest AF, Leyton L. 2012. Astrocytic alphaVbeta3 integrin inhibits neurite outgrowth and promotes retraction of neuronal processes by clustering Thy-1. PLoS One 7:e34295. doi: 10.1371/journal.pone.0034295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou Y, Hagood JS, Lu B, Merryman WD, Murphy-Ullrich JE. 2010. Thy-1-integrin alphav beta5 interactions inhibit lung fibroblast contraction-induced latent transforming growth factor-beta1 activation and myofibroblast differentiation. J Biol Chem 285:22382–22393. doi: 10.1074/jbc.M110.126227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mercer J, Helenius A. 2012. Gulping rather than sipping: macropinocytosis as a way of virus entry. Curr Opin Microbiol 15:490–499. doi: 10.1016/j.mib.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 52.Lozach PY, Huotari J, Helenius A. 2011. Late-penetrating viruses. Curr Opin Virol 1:35–43. doi: 10.1016/j.coviro.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 53.Amstutz B, Gastaldelli M, Kalin S, Imelli N, Boucke K, Wandeler E, Mercer J, Hemmi S, Greber UF. 2008. Subversion of CtBP1-controlled macropinocytosis by human adenovirus serotype 3. EMBO J 27:956–969. doi: 10.1038/emboj.2008.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Conner SD, Schmid SL. 2003. Regulated portals of entry into the cell. Nature 422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 55.Brown D, Waneck GL. 1992. Glycosyl-phosphatidylinositol-anchored membrane proteins. J Am Soc Nephrol 3:895–906. [DOI] [PubMed] [Google Scholar]
- 56.Yamauchi Y, Helenius A. 2013. Virus entry at a glance. J Cell Sci 126:1289–1295. doi: 10.1242/jcs.119685. [DOI] [PubMed] [Google Scholar]
- 57.Koivusalo M, Welch C, Hayashi H, Scott CC, Kim M, Alexander T, Touret N, Hahn KM, Grinstein S. 2010. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J Cell Biol 188:547–563. doi: 10.1083/jcb.200908086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kerr MC, Teasdale RD. 2009. Defining macropinocytosis. Traffic 10:364–371. doi: 10.1111/j.1600-0854.2009.00878.x. [DOI] [PubMed] [Google Scholar]
- 59.Bergallo M, Costa C, Terlizzi ME, Margio S, Sidoti F, Sinesi F, Cavallo R. 2008. Evaluation of two set of primers for detection of immediate early gene UL123 of human cytomegalovirus (HCMV). Mol Biotechnol 38:65–70. doi: 10.1007/s12033-007-0074-5. [DOI] [PubMed] [Google Scholar]
- 60.O'Connor CM, Shenk T. 2012. Human cytomegalovirus pUL78 G protein-coupled receptor homologue is required for timely cell entry in epithelial cells but not fibroblasts. J Virol 86:11425–11433. doi: 10.1128/JVI.05900-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smuda C, Bogner E, Radsak K. 1997. The human cytomegalovirus glycoprotein B gene (ORF UL55) is expressed early in the infectious cycle. J Gen Virol 78:1981–1992. doi: 10.1099/0022-1317-78-8-1981. [DOI] [PubMed] [Google Scholar]
- 62.Spaete RR, Thayer RM, Probert WS, Masiarz FR, Chamberlain SH, Rasmussen L, Merigan TC, Pachl C. 1988. Human cytomegalovirus strain Towne glycoprotein B is processed by proteolytic cleavage. Virology 167:207–225. doi: 10.1016/0042-6822(88)90071-2. [DOI] [PubMed] [Google Scholar]
- 63.Krzyzaniak MA, Zumstein MT, Gerez JA, Picotti P, Helenius A. 2013. Host cell entry of respiratory syncytial virus involves macropinocytosis followed by proteolytic activation of the F protein. PLoS Pathog 9:e1003309. doi: 10.1371/journal.ppat.1003309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schelhaas M, Shah B, Holzer M, Blattmann P, Kuhling L, Day PM, Schiller JT, Helenius A. 2012. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog 8:e1002657. doi: 10.1371/journal.ppat.1002657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schmidt FI, Bleck CK, Helenius A, Mercer J. 2011. Vaccinia extracellular virions enter cells by macropinocytosis and acid-activated membrane rupture. EMBO J 30:3647–3661. doi: 10.1038/emboj.2011.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. 2006. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell 10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 67.Ellis S, Mellor H. 2000. Regulation of endocytic traffic by rho family GTPases. Trends Cell Biol 10:85–88. doi: 10.1016/S0962-8924(99)01710-9. [DOI] [PubMed] [Google Scholar]
- 68.Grimmer S, van Deurs B, Sandvig K. 2002. Membrane ruffling and macropinocytosis in A431 cells require cholesterol. J Cell Sci 115:2953–2962. [DOI] [PubMed] [Google Scholar]
- 69.Madore N, Smith KL, Graham CH, Jen A, Brady K, Hall S, Morris R. 1999. Functionally different GPI proteins are organized in different domains on the neuronal surface. EMBO J 18:6917–6926. doi: 10.1093/emboj/18.24.6917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brugger B, Graham C, Leibrecht I, Mombelli E, Jen A, Wieland F, Morris R. 2004. The membrane domains occupied by glycosylphosphatidylinositol-anchored prion protein and Thy-1 differ in lipid composition. J Biol Chem 279:7530–7536. doi: 10.1074/jbc.M310207200. [DOI] [PubMed] [Google Scholar]
- 71.Chen Y, Thelin WR, Yang B, Milgram SL, Jacobson K. 2006. Transient anchorage of cross-linked glycosyl-phosphatidylinositol-anchored proteins depends on cholesterol, Src family kinases, caveolin, and phosphoinositides. J Cell Biol 175:169–178. doi: 10.1083/jcb.200512116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Awasthi-Kalia M, Schnetkamp PP, Deans JP. 2001. Differential effects of filipin and methyl-beta-cyclodextrin on B cell receptor signaling. Biochem Biophys Res Commun 287:77–82. doi: 10.1006/bbrc.2001.5536. [DOI] [PubMed] [Google Scholar]
- 73.Liu J, Oh P, Horner T, Rogers RA, Schnitzer JE. 1997. Organized endothelial cell surface signal transduction in caveolae distinct from glycosylphosphatidylinositol-anchored protein microdomains. J Biol Chem 272:7211–7222. doi: 10.1074/jbc.272.11.7211. [DOI] [PubMed] [Google Scholar]
- 74.Rothberg KG, Ying YS, Kamen BA, Anderson RG. 1990. Cholesterol controls the clustering of the glycophospholipid-anchored membrane receptor for 5-methyltetrahydrofolate. J Cell Biol 111:2931–2938. doi: 10.1083/jcb.111.6.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Juckem LK, Boehme KW, Feire AL, Compton T. 2008. Differential initiation of innate immune responses induced by human cytomegalovirus entry into fibroblast cells. J Immunol 180:4965–4977. doi: 10.4049/jimmunol.180.7.4965. [DOI] [PubMed] [Google Scholar]
- 76.Sharma P, Varma R, Sarasij RC, Ira Gousset K, Krishnamoorthy G, Rao M, Mayor S. 2004. Nanoscale organization of multiple GPI-anchored proteins in living cell membranes. Cell 116:577–589. doi: 10.1016/S0092-8674(04)00167-9. [DOI] [PubMed] [Google Scholar]
- 77.Van den Broeke C, Radu M, Chernoff J, Favoreel HW. 2010. An emerging role for p21-activated kinases (Paks) in viral infections. Trends Cell Biol 20:160–169. doi: 10.1016/j.tcb.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Meier O, Boucke K, Hammer SV, Keller S, Stidwill RP, Hemmi S, Greber UF. 2002. Adenovirus triggers macropinocytosis and endosomal leakage together with its clathrin-mediated uptake. J Cell Biol 158:1119–1131. doi: 10.1083/jcb.200112067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mercer J, Knebel S, Schmidt FI, Crouse J, Burkard C, Helenius A. 2010. Vaccinia virus strains use distinct forms of macropinocytosis for host-cell entry. Proc Natl Acad Sci U S A 107:9346–9351. doi: 10.1073/pnas.1004618107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Amyere M, Payrastre B, Krause U, Van Der Smissen P, Veithen A, Courtoy PJ. 2000. Constitutive macropinocytosis in oncogene-transformed fibroblasts depends on sequential permanent activation of phosphoinositide 3-kinase and phospholipase C. Mol Biol Cell 11:3453–3467. doi: 10.1091/mbc.11.10.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sallusto F, Cella M, Danieli C, Lanzavecchia A. 1995. Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: downregulation by cytokines and bacterial products. J Exp Med 182:389–400. doi: 10.1084/jem.182.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Swanson JA, Watts C. 1995. Macropinocytosis. Trends Cell Biol 5:424–428. doi: 10.1016/S0962-8924(00)89101-1. [DOI] [PubMed] [Google Scholar]
- 83.Krieger SE, Kim C, Zhang L, Marjomaki V, Bergelson JM. 2013. Echovirus 1 entry into polarized Caco-2 cells depends on dynamin, cholesterol, and cellular factors associated with macropinocytosis. J Virol 87:8884–8895. doi: 10.1128/JVI.03415-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Aleksandrowicz P, Marzi A, Biedenkopf N, Beimforde N, Becker S, Hoenen T, Feldmann H, Schnittler HJ. 2011. Ebola virus enters host cells by macropinocytosis and clathrin-mediated endocytosis. J Infect Dis 204(Suppl 3):S957–S967. doi: 10.1093/infdis/jir326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.de Vries E, Tscherne DM, Wienholts MJ, Cobos-Jimenez V, Scholte F, Garcia-Sastre A, Rottier PJ, de Haan CA. 2011. Dissection of the influenza A virus endocytic routes reveals macropinocytosis as an alternative entry pathway. PLoS Pathog 7:e1001329. doi: 10.1371/journal.ppat.1001329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Raghu H, Sharma-Walia N, Veettil MV, Sadagopan S, Chandran B. 2009. Kaposi's sarcoma-associated herpesvirus utilizes an actin polymerization-dependent macropinocytic pathway to enter human dermal microvascular endothelial and human umbilical vein endothelial cells. J Virol 83:4895–4911. doi: 10.1128/JVI.02498-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Norbury CC, Hewlett LJ, Prescott AR, Shastri N, Watts C. 1995. Class I MHC presentation of exogenous soluble antigen via macropinocytosis in bone marrow macrophages. Immunity 3:783–791. doi: 10.1016/1074-7613(95)90067-5. [DOI] [PubMed] [Google Scholar]
- 88.Li L, Wan T, Wan M, Liu B, Cheng R, Zhang R. 2015. The effect of the size of fluorescent dextran on its endocytic pathway. Cell Biol Int 39:531–539. doi: 10.1002/cbin.10424. [DOI] [PubMed] [Google Scholar]
- 89.Racoosin EL, Swanson JA. 1993. Macropinosome maturation and fusion with tubular lysosomes in macrophages. J Cell Biol 121:1011–1020. doi: 10.1083/jcb.121.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mercer J, Greber UF. 2013. Virus interactions with endocytic pathways in macrophages and dendritic cells. Trends Microbiol 21:380–388. doi: 10.1016/j.tim.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 91.Sandvig K, Pust S, Skotland T, van Deurs B. 2011. Clathrin-independent endocytosis: mechanisms and function. Curr Opin Cell Biol 23:413–420. doi: 10.1016/j.ceb.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 92.Bodaghi B, Slobbe-van Drunen ME, Topilko A, Perret E, Vossen RC, van Dam-Mieras MC, Zipeto D, Virelizier JL, LeHoang P, Bruggeman CA, Michelson S. 1999. Entry of human cytomegalovirus into retinal pigment epithelial and endothelial cells by endocytosis. Invest Ophthalmol Vis Sci 40:2598–2607. [PubMed] [Google Scholar]
- 93.Nonnenmacher M, Weber T. 2011. Adeno-associated virus 2 infection requires endocytosis through the CLIC/GEEC pathway. Cell Host Microbe 10:563–576. doi: 10.1016/j.chom.2011.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kirkham M, Parton RG. 2005. Clathrin-independent endocytosis: new insights into caveolae and non-caveolar lipid raft carriers. Biochim Biophys Acta 1746:349–363. doi: 10.1016/j.bbamcr.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 95.Marsh M, Helenius A. 2006. Virus entry: open sesame. Cell 124:729–740. doi: 10.1016/j.cell.2006.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mayor S, Rothberg KG, Maxfield FR. 1994. Sequestration of GPI-anchored proteins in caveolae triggered by cross-linking. Science 264:1948–1951. doi: 10.1126/science.7516582. [DOI] [PubMed] [Google Scholar]
- 97.Coyne CB, Bergelson JM. 2006. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell 124:119–131. doi: 10.1016/j.cell.2005.10.035. [DOI] [PubMed] [Google Scholar]
- 98.Coyne CB, Shen L, Turner JR, Bergelson JM. 2007. Coxsackievirus entry across epithelial tight junctions requires occludin and the small GTPases Rab34 and Rab5. Cell Host Microbe 2:181–192. doi: 10.1016/j.chom.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rege TA, Pallero MA, Gomez C, Grenett HE, Murphy-Ullrich JE, Hagood JS. 2006. Thy-1, via its GPI anchor, modulates Src family kinase and focal adhesion kinase phosphorylation and subcellular localization, and fibroblast migration, in response to thrombospondin-1/hep I. Exp Cell Res 312:3752–3767. doi: 10.1016/j.yexcr.2006.07.029. [DOI] [PubMed] [Google Scholar]
- 100.Herrera-Molina R, Valdivia A, Kong M, Alvarez A, Cardenas A, Quest AF, Leyton L. 2013. Thy-1-interacting molecules and cellular signaling in cis and trans. Int Rev Cell Mol Biol 305:163–216. doi: 10.1016/B978-0-12-407695-2.00004-4. [DOI] [PubMed] [Google Scholar]
- 101.Khandelwal P, Ruiz WG, Apodaca G. 2010. Compensatory endocytosis in bladder umbrella cells occurs through an integrin-regulated and RhoA- and dynamin-dependent pathway. EMBO J 29:1961–1975. doi: 10.1038/emboj.2010.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Veettil MV, Sharma-Walia N, Sadagopan S, Raghu H, Sivakumar R, Naranatt PP, Chandran B. 2006. RhoA-GTPase facilitates entry of Kaposi's sarcoma-associated herpesvirus into adherent target cells in a Src-dependent manner. J Virol 80:11432–11446. doi: 10.1128/JVI.01342-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.De Franceschi N, Hamidi H, Alanko J, Sahgal P, Ivaska J. 2015. Integrin traffic—the update. J Cell Sci 128:839–852. doi: 10.1242/jcs.161653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Peppenelli MA, Arend KC, Cojohari O, Moorman NJ, Chan GC. 2016. Human cytomegalovirus stimulates the synthesis of select Akt-dependent antiapoptotic proteins during viral entry to promote survival of infected monocytes. J Virol 90:3138–3147. doi: 10.1128/JVI.02879-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cortese K, Sahores M, Madsen CD, Tacchetti C, Blasi F. 2008. Clathrin and LRP-1-independent constitutive endocytosis and recycling of uPAR. PLoS One 3:e3730. doi: 10.1371/journal.pone.0003730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mayor S, Pagano RE. 2007. Pathways of clathrin-independent endocytosis. Nat Rev Mol Cell Biol 8:603–612. doi: 10.1038/nrm2216. [DOI] [PubMed] [Google Scholar]
- 107.Watarai M, Makino S, Fujii Y, Okamoto K, Shirahata T. 2002. Modulation of Brucella-induced macropinocytosis by lipid rafts mediates intracellular replication. Cell Microbiol 4:341–355. doi: 10.1046/j.1462-5822.2002.00195.x. [DOI] [PubMed] [Google Scholar]
- 108.Boeckh M, Huang M, Ferrenberg J, Stevens-Ayers T, Stensland L, Nichols WG, Corey L. 2004. Optimization of quantitative detection of cytomegalovirus DNA in plasma by real-time PCR. J Clin Microbiol 42:1142–1148. doi: 10.1128/JCM.42.3.1142-1148.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]