

Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive and lethal neurodegenerative disease characterized by loss of upper and lower motor neurons leading to muscle paralysis in affected individuals. Numerous mechanisms have been implicated in the death of these neurons. However, the pathobiology of this disease has not been completely elucidated. In the present study, we investigated to what extent tau cleavage and generation of the neurotoxic tau45-230 fragment is associated with ALS. Quantitative western blot analysis indicated that high levels of tau45-230 accumulated in lumbar and cervical spinal cord specimens obtained from ALS subjects. This neurotoxic tau fragment was also detected in ALS upper motor neurons located in the precentral gyrus. Our results also showed that tau45-230 aggregates were present in the spinal cord of ALS patients. On the other hand, this neurotoxic fragment was not generated in a mouse model of a familial form of this disease. Together, these results suggest a potential role for this neurotoxic tau fragment in the mechanisms leading to the degeneration of motor neurons in the context of sporadic ALS.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a progressive and lethal neurodegenerative disorder that affects approximately 200,000 people nationwide (reviewed in 1–3; see also references within). This disease is characterized by the selective death of motor neurons in the brain (upper motor neurons) and spinal cord (lower motor neurons), resulting in the paralysis of voluntary muscles (2–9). Gene defects are detected in ~5–10% of ALS patients (familial ALS). On the other hand, most ALS cases (~90–95%) have no apparent genetic component (sporadic ALS). Multiple potential mechanisms underlying motor neuron degeneration have been described. These mechanisms include, among others, excitotoxicity, protein misfolding and aggregation, abnormal calcium metabolism, altered axonal transport, and activation of proteases and nucleases (reviewed in 1–3, 10, 11). Several proteins have been implicated in the pathobiology of ALS, including neurofilaments, peripherin, α-internexin and the copper/zinc superoxide dismutase (SOD1). More recently, evidence has suggested that the 43 kDa transactive response (TAR) DNA-binding protein (TDP-43) and a related heterogeneous nuclear ribonucleoprotein, fused in sarcoma/translocated in liposarcoma (FUS/TLS), play an important role in the pathobiology of ALS (12–20).
Less is known about the involvement of tau in ALS. Tau is a microtubule-associated protein (MAP) highly enriched in the axons of central neurons. This MAP regulates the rate of axonal elongation by modulating the polymerization and stabilization of microtubules (21–26). Tau also plays a key role in Alzheimer’s disease (AD) and several other neurodegenerative diseases. In these diseases, hyperphosphorylated forms of tau accumulate, forming neurofibrillary tangles (27–29). Recently, tau aggregates have also been identified in ALS patients showing signs of cognitive impairment and/or frontotemporal dementia (30,31).
We have recently described tau cleavage and generation of the tau45-230 fragment as a conserved mechanism of degeneration in multiple neurodegenerative diseases (32). This calpain-mediated cleavage is an early event in the degenerative process, preceding tau phosphorylation (33). Furthermore, tau45-230 has toxic effects when expressed in neurons and other cell types (33–35). Together, these results suggest that if present, tau45-230 could lead to degeneration of motor neurons in ALS patients. In the present study, we assessed the levels of this neurotoxic fragment in upper and lower motor neurons of ALS subjects.
MATERIALS AND METHODS
Subjects
Tissue from the primary motor cortex area located in the precentral gyrus (Brodmann’s area 4) [Fr (BA4)] and the spinal cord (cervical and lumbar areas) were obtained from ALS subjects (Tables 1 and 2). Age-matched samples obtained from subjects that had no clinical history of any neurological disorders were used as controls (Tables 1 and 2). These specimens were obtained from the Cognitive Neurology and Alzheimer’s Disease Center Brain Bank at Northwestern University and the New York Brain Bank at Columbia University. The specimens were used for quantitative western blot analysis as described below.
Table 1.
Frontal cortex tissue analyzed in this study.
| Pathological Condition | Gender | Age of Death | Brain Area | Postmortem Interval (h) |
|---|---|---|---|---|
| Control | M | 64 | Fr (BA4) | 18 |
| Control | M | 88 | Fr (BA4) | 11 |
| Control | M | 71 | Fr (BA4) | 39 |
| Control | M | 77 | Fr (BA4) | 21 |
| Control | M | 52 | Fr (BA4) | 12 |
| Control | M | 41 | Fr (BA4) | 7 |
| Control | F | 52 | Fr (BA4) | 6 |
| Control | F | 67 | Fr (BA4) | 32 |
| Control | M | 89 | Fr (BA4) | 7 |
| ALS | M | 41 | Fr (BA4) | Unk |
| ALS | F | 63 | Fr (BA4) | Unk |
| ALS | F | 71 | Fr (BA4) | Unk |
| ALS | M | 67 | Fr (BA4) | 3 |
| ALS | M | 73 | Fr (BA4) | 9 |
| ALS | M | 58 | Fr (BA4) | 6 |
| ALS | M | 75 | Fr (BA4) | 7 |
| ALS | F | 52 | Fr (BA4) | 13 |
| ALS | F | 69 | Fr (BA4) | 13 |
| ALS | M | 56 | Fr (BA4) | 15 |
| ALS | M | 66 | Fr (BA4) | Unk |
| ALS | F | 80 | Fr (BA4) | 7 |
| ALS | M | 42 | Fr (BA4) | 12 |
| ALS | F | 62 | Fr (BA4) | Unk |
| ALS | M | 41 | Fr (BA4) | 7 |
| ALS | M | 42 | Fr (BA4) | 6 |
| ALS | F | 68 | Fr (BA4) | Unk |
| ALS | M | 62 | Fr (BA4) | Unk |
| ALS | F | 70 | Fr (BA4) | 16 |
| ALS | F | 53 | Fr (BA4) | 48 |
| ALS | F | 41 | Fr (BA4) | 6 |
Fr (BA4): frontal cortex Brodmann area 4; Unk: unknown.
Table 2.
Spinal cord tissue analyzed in this study.
| Pathological Condition | Gender | Age of Death | Spinal Cord Area | Postmortem Interval (h) |
|---|---|---|---|---|
| Control | M | 86 | SCL | 24 |
| Control | F | 82 | SCL | 7 |
| Control | F | 88 | SCL | 22 |
| Control | M | 41 | SCC | 7 |
| Control | F | 52 | SCC | 6 |
| Control | F | 67 | SCC | 32 |
| Control | M | 89 | SCC | 7 |
| ALS | M | 41 | SCL | Unk |
| ALS | F | 63 | SCL | Unk |
| ALS | F | 71 | SCL | Unk |
| ALS | M | 67 | SCL | 3 |
| ALS | M | 73 | SCL | 9 |
| ALS | M | 58 | SCL | 6 |
| ALS | M | 75 | SCL | 7 |
| ALS | F | 52 | SCL | 13 |
| ALS | F | 69 | SCL | 13 |
| ALS | M | 56 | SCL | 15 |
| ALS | F | 68 | SCL | Unk |
| ALS | M | 62 | SCL | Unk |
| ALS | F | 70 | SCL | 16 |
| ALS | F | 53 | SCL | 48 |
| ALS | F | 67 | SCL | 6 |
| ALS | F | 63 | SCL | 11 |
| ALS | M | 83 | SCL | 12 |
| ALS | M | 68 | SCL | 16 |
| ALS | F | 52 | SCL | 13 |
| ALS | M | 45 | SCL | 14 |
| ALS | F | 74 | SCL | 25 |
| ALS | F | 64 | SCL | 18 |
| ALS | M | 41 | SCC | Unk |
| ALS | F | 63 | SCC | Unk |
| ALS | F | 71 | SCC | Unk |
| ALS | M | 67 | SCC | 3 |
| ALS | M | 73 | SCC | 9 |
| ALS | M | 58 | SCC | 6 |
| ALS | M | 75 | SCC | 7 |
| ALS | F | 52 | SCC | 13 |
| ALS | F | 69 | SCC | 13 |
| ALS | M | 56 | SCC | 15 |
SCL: spinal cord lumbar area; SCC: spinal cord cervical area; Unk: unknown.
Preparation of Spinal Cord Samples from SOD1 Transgenic Mice
Transgenic mice expressing multiple copies of the mutated human G93A-superoxide dismutase 1 (SOD1) were obtained from the Jackson Laboratories. Male B6SJL-Tg (G93A-SOD1) 1Gur/J (G93A-SOD1) and wild-type (WT) control mice were euthanized by means of CO2 overdose 10 and 17 wks after birth. The spinal cords were removed and their anterior horns were dissected, homogenized and analyzed by means of quantitative western blot as described below.
The Northwestern University Animal Care and Use Committee approved the experimental protocol used in this study in accordance with United States Public Health Service regulations and applicable federal and local laws.
Electrophoresis and Immunoblotting
Tissue obtained from human subjects or SOD1 transgenic and WT mice was homogenized in Laemmli buffer, boiled for 10 min, and separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (32,36). Transfer of protein to Immobilon membranes (Millipore) and immunodetection were performed as previously described (37). The following primary antibodies were used: tau (clone tau5; 1:1000; BioSource), tau45-230 (1:500; 35), calpain 1 (1:500; Abcam), spectrin (1:1000; Millipore), neuron-specific Class III β-tubulin (clone TUJ1; 1:4000; R & D Systems) and α-tubulin (clone DM1A; 1:200,000; Sigma). Secondary antibodies conjugated to horseradish peroxidase (1:1000; Promega) followed by enhanced chemiluminescence reagents were used for the detection of proteins (38). Immunoreactive bands were imaged using a ChemiDoc XRS apparatus (Bio-Rad). Density of these bands was quantified using Quantity One software (Bio-Rad). Values were calculated as ratios of the target fragment to full-length tau, and of the cleaved (150 kDa) to full-length (240 kDa) spectrin in ALS specimens. These ratios were compared with those detected in controls (100%). Tubulin was used as a loading control.
For some experiments, spinal cord (anterior horn) tissue obtained from ALS subjects was homogenized in 25 mM Tris-HCl buffer plus 10% glycerol at 4°C. The homogenates were spun at 16,000 g for 30 min at 4°C and the supernatant separated under nondenaturing native discontinuous PAGE as previously described (39,40). To determine the molecular weight of tau45-230 aggregates, standard proteins were separated using gels prepared with different polyacrylamide concentrations (5%, 7.5%, 10% and 12%). The relative mobility of each marker was estimated by measuring the migration distance to the dye front and plotted against the gel concentration. The slope of the curve was determined and compared with the one obtained for the tau45-230 aggregates using the Ferguson plot analysis, as previously described (39,40).
Hippocampal Culture Preparation
Hippocampal cultures were prepared from embryonic d 16 WT mice as described previously (33–35). In brief, hippocampi were dissected and stripped of meninges. The tissue was trypsinized (0.25% for 15 min at 37°C) and neurons were dissociated by pipetting gently through a fire-polished Pasteur pipette. Cells were plated (~ 800,000 cells/60 mm dish) in minimum essential medium (MEM) containing 10% horse serum on poly-L-lysine–coated dishes. After 4 h, the medium was replaced with glia-conditioned MEM containing N2 supplements, 0.1% and 0.1 mM sodium pyruvate (N2 medium) as described (33–35).
Aβ Aggregation and Cell Treatment
Synthetic Aβ(1-40) (American Peptide) was dissolved in N2 medium to 0.5 mg/mL and incubated at 37°C for 3 d to aggregate the peptide as described (33). The aggregated peptide was added to the medium of 21 d in-culture hippocampal neurons at a final concentration of 20 μM to induce calpain-mediated tau cleavage into the tau45-230 fragment.
Statistical Analysis
The compiled data were analyzed using the Student t test. The values in the graphs represent the mean ± standard error of the mean (SEM), and statistical significance is indicated in the graphs for samples that differed from their respective controls. Correlations between the levels of tau45-230 and postmortem interval (PMI) were performed by means of the Pearson correlation test.
RESULTS
High Levels of Tau45-230 Were Present in the Spinal Cords of ALS Subjects
Data recently obtained in our laboratory indicated that the neurotoxic tau45-230 fragment accumulated in affected brain areas in AD and other tauopathies (32). To assess whether this 17 kDa molecular weight tau fragment was also generated in the context of ALS, we performed quantitative Western blot analysis of spinal cord samples obtained from ALS subjects and age-matched controls. For the first set of experiments, homogenates were prepared from the anterior horn of the spinal cord at both the lumbar and cervical levels. Immunoblotting was performed using a phosphorylation-independent tau antibody directed to an epitope present in both full-length tau and tau45-230 (clone tau5). Strong immunoreactive bands corresponding to full-length tau were detected in control samples (Figures 1 A and B). Full-length tau was also easily detectable in ALS specimens. However, a greater variation in the levels of this MAP was detected in samples obtained from ALS subjects as compared with age-matched controls (Figures 1A and B). While no tau45-230 immunoreactive band was observed in control samples (Figures 1A and B), immunoreactive bands corresponding to the tau45-230 fragment were detected in ALS lumbar and cervical spinal cord extracts (Figures 1A and B). Quantification of these immunoreactive bands showed a significant increase (two-to-four-fold) in the ratio of tau45-230/full-length tau in ALS samples when compared with age-matched controls (Figures 1C and D).
Figure 1.

High levels of tau45-230 were detected in ALS spinal cords. (A and B) Quantitative Western blot analysis of tau content in representative homogenates prepared from postmortem samples of the anterior horn of lumbar (A) and cervical (B) spinal cords from control (C) and ALS (ALS) subjects. Immunoreactive bands at 17 kDa apparent molecular weight corresponding to tau45-230 were readily detectable in ALS samples. (C and D) Graphs show the ratios of tau45-230/full-length tau in control and ALS subjects. Numbers represent the mean ± SEM of control (n = 7) and ALS (lumbar n = 22, cervical n = 10) samples. Values are expressed as percentage of controls, considering the values obtained in control subjects as 100%. Class III β tubulin (TUJ1) was used as loading control. *Differs from control subjects; P < 0.01.
We next determined whether this fragment was also present in upper motor neurons located in the precentral gyrus of ALS subjects. Western blot analysis of lysates prepared from the Fr (BA4) area revealed the presence of a tau45-230 immunoreactive band in approximately half of the samples analyzed (Figure 2A). In these samples, densitometric analysis of tau immunoreactive bands demonstrated an increase (~ five-fold) in the tau45-230/full-length tau ratio when compared with age-matched controls (Figure 2B).
Figure 2.

Tau45-230 was also detected in the precentral gyrus of ALS subjects. (A) Quantitative Western blot analysis of tau content in representative homogenates prepared from postmortem samples of precentral gyrus from control (C) and ALS (ALS) subjects. Immunoreactive bands at 17 kDa apparent molecular weight corresponding to tau45-230 were readily detectable in some ALS samples. (B) Graphs show the ratios of tau45-230/full-length tau in control and ALS subjects. Numbers represent the mean ± SEM of control (n = 9) and ALS tau45-230 positive (n = 10) samples. Values are expressed as percentage of controls, considering the values obtained in control subjects as 100%. ALS Class III β tubulin (TUJ1) was used as loading control. *Differs from control subjects; P < 0.01.
To rule out a potential effect of the PMI in the generation of this fragment, we first determined the mean ± SEM PMI for the ALS samples and compared them to age-matched controls (Tables 1 and 2). No significant differences were detected in the PMI of lumbar spinal cord samples from ALS subjects when compared with age-matched controls (14.1 ± 2 h versus 17.6 ± 6 h, respectively). Similar results were obtained when PMIs of ALS samples obtained from cervical spinal cord (9.4 ± 1 h) and precentral gyrus cortex (12 ± 11 h) were compared with their respective age-matched controls (13 ± 7 h and 17 ± 4 h, respectively).
We also calculated a correlation coefficient between PMI and tau45-230 levels for all samples used in this study. We found no significant correlation between PMI and tau45-230 levels in either the spinal cord or cortex samples analyzed (R2 = 0.0057 and R2 = 0.0052). These results suggest that the increased tau45-230 levels observed in the ALS samples could not be explained by variation in the PMI.
Tau45-230 Aggregates Were Detected in the Spinal Cords of ALS Subjects
One of the pathological characteristics of ALS is the formation of protein aggregates (reviewed in 1–3; see also references within). We determined next whether tau45-230 aggregates were present in ALS, since we have previously shown that this tau fragment aggregated in vitro (32). For these experiments, ALS spinal cord samples were analyzed by means of native/nondenaturing polyacrylamide electrophoresis followed by Western blot analysis as previously described (39,40). Samples were separated by native discontinuous electrophoresis under conditions that precluded full-length tau from entering the separating gel (pH 8.2). Western blot analysis using a tau antibody detected two tau immunoreactive bands in these samples. To confirm that these aggregates were formed by tau45-230, these membranes were reprobed using a specific tau45-230 antibody recently generated and characterized in our laboratory (35). As shown in Figure 3, both bands were highly immunoreactive with this tau45-230 antibody. The separation of standard proteins at four different concentrations of acrylamide was used to construct a molecular weight standard curve without the use of SDS (Ferguson plot analysis). Using the slope of this curve, we estimated that the molecular weight of the bands recognized by the tau45-230 antibody were ~148 and ~178 kDa, respectively (Figure 3).
Figure 3.
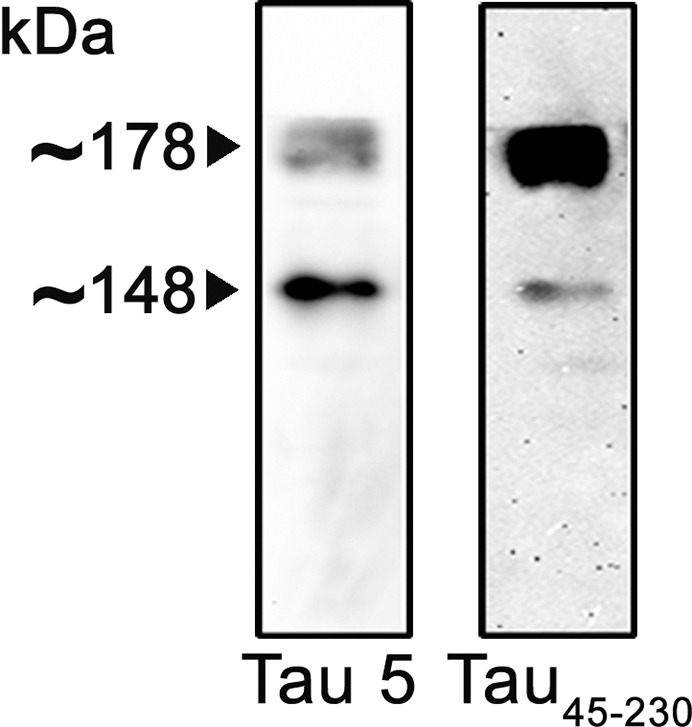
Detection of tau45-230 aggregates in ALS spinal cord samples. Homogenates of lumbar spinal cord samples obtained from ALS subjects were analyzed by means of native/nondenaturing PAGE followed by Western blot. Membranes were reacted with a nonphosphorylation-dependent tau antibody (clone tau5) and stripped and reprobed with a tau45-230 specific antibody. Two tau45-230 immunoreactive bands were detected in ALS samples.
Generation of Tau45-230 in the Context of ALS was Accompanied by Changes in Calpain
We have previously shown that generation of the tau45-230 fragment was mainly a result of the dysregulation of calpain activity and not due to changes in the expression levels of this protease in AD and related tauopathies (32). To assess to what extent the increase in tau45-230 levels detected in ALS patients was also associated with calpain changes, we first performed a quantitative Western blot analysis of homogenates prepared from ALS subjects using a calpain antibody. Immunoblotting revealed strong calpain-reactive bands at ~80 kDa in spinal cord (lumbar and cervical regions) and precentral gyrus in both control and ALS subjects (Figures 4A and C). Quantification of these bands did not detect significant differences in calpain levels when ALS lysates prepared from lumbar spinal cord or Fr (BA4) cortex were compared with age-matched controls (Figures 4A and C). On the other hand, a small increase in calpain levels was detected in cervical spinal cord samples obtained from ALS subjects when compared with controls (Figure 4B).
Figure 4.

Determination of calpain content and activity in spinal cord and precentral gyrus in ALS subjects. (A–C) Western blot analysis of calpain content and (D–F) calpain activity, by means of spectrin degradation, in homogenates prepared from postmortem samples of lumbar (A and D) and cervical (B and E) spinal cords, and prefrontal cortex (C and F) samples obtained from ALS and age-matched control subjects. Graphs show calpain levels and calpain activity, as the ratio 150/240 kDa spectrin bands, in control and ALS subjects. Numbers represent the mean ± SEM of control and ALS samples as described in Figures 1 and 2 legends. Values are expressed as percentage of controls, considering the values obtained in control subjects as 100%. Class III β tubulin (TUJ1) was used as loading control. *Differs from control subjects; P < 0.05.
We next examined calpain activity by determining spectrin degradation. For these experiments, Western blot analysis using a spectrin antibody was performed using ALS and control specimens, and the ratio of cleaved (150 kDa) to full-length (240 kDa) spectrin immunoreactive bands was calculated. This ratio has been routinely used to assess calpain activity in the central nervous system (33,41). Full-length and cleaved spectrin immunoreactive bands were detected in all the samples analyzed (Figures 4D–F). Quantitative analysis showed that the ratios cleaved to full-length spectrin were consistently higher in ALS samples when compared with age-matched controls (Figures 4D–F). Thus, calpain-mediated spectrin cleavage was higher in both the lumbar and cervical spinal cord regions (increases ranging from 3% to 230% and from 10% to 69%, respectively) of ALS subjects when compared with controls (Figures 4D and F). An increase of 5% to 45% in calpain activity was also detected in precentral gyrus samples obtained from ALS subjects compared with controls (Figure 4F).
Tau45-230 Was Not Generated in SOD1 Transgenic Mice
While the majority of ALS cases are sporadic, 5% of the familial cases of this disease are associated with a mutated SOD1 (G93A). Thus, transgenic mice expressing this SOD1 mutation have been extensively studied as a model system of familial ALS. To determine whether the neurotoxic tau45-230 fragment also accumulated in the lower motor neurons in the context of the most common familial ALS, we prepared lysates from the spinal cords of SOD1 transgenic mice at postnatal weeks 10 and 17. These mice were in the presymptomatic and symptomatic phases of the disease process, respectively (42). Samples obtained from SOD1 transgenic mice and WT controls were analyzed by SDS-PAGE followed by quantitative Western blot analysis. Strong immunoreactive bands corresponding to full-length tau were detected in both transgenic and WT samples at both ages studied (Figures 5A and B). On the other hand, no immunoreactive bands were detected at the 17 kDa molecular weight corresponding to the tau45-230 fragment in either SOD1 transgenic or WT controls at any of the ages analyzed (Figures 5A and B). To further rule out the presence of tau45-230 in these SOD1 transgenic mouse samples, we performed western blot analysis using the specific tau45-230 antibody. As a positive control, we included whole-cell homogenates obtained from 21 d in-culture hippocampal neurons incubated in the presence of aggregated Aβ, the experimental condition in which this fragment was originally identified (33). As previously described, a tau45-230 immunoreactive band (~17 kDa) was detected in Aβ-treated hippocampal neurons (Figure 5C). In contrast, no tau45-230 immunoreactive bands were detected in WT or SOD1 cervical and lumbar spinal cord samples (Figure 5C).
Figure 5.
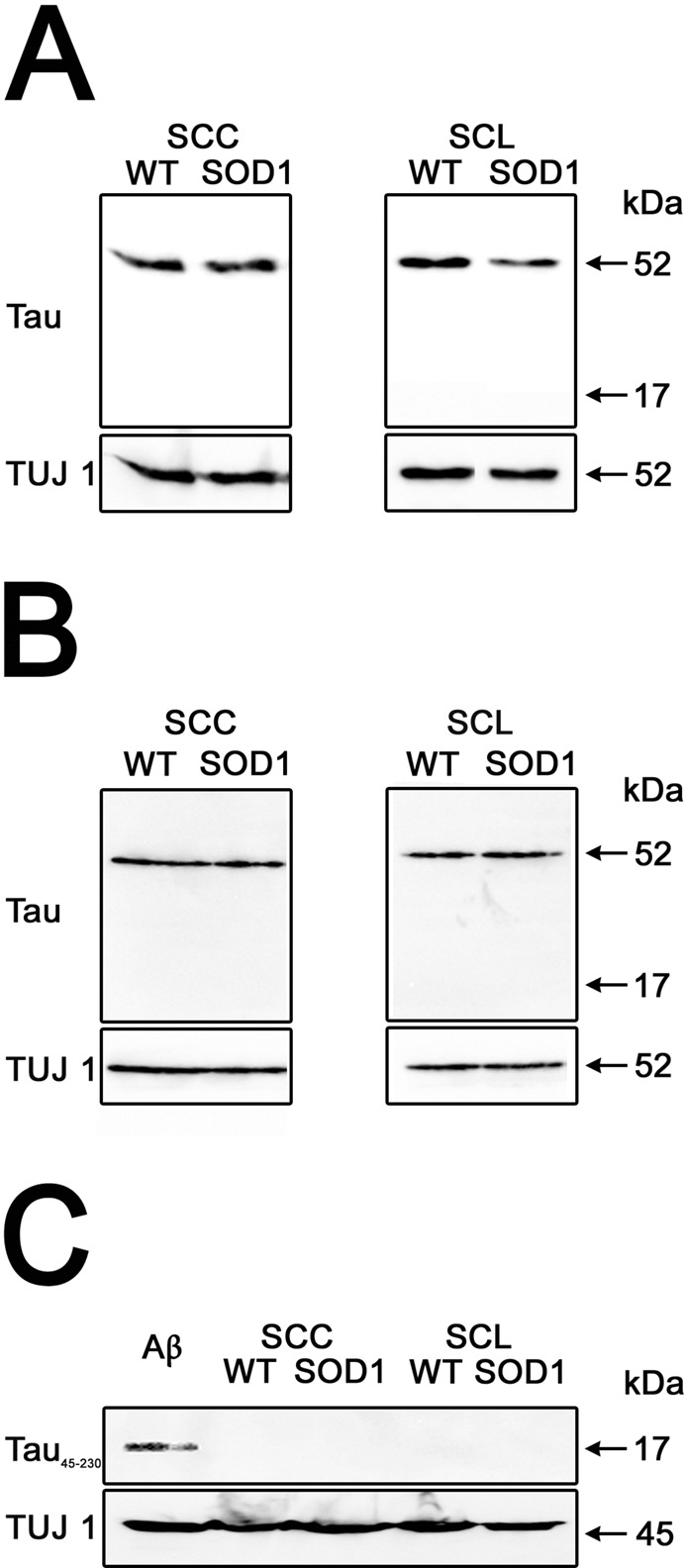
Tau45-230 was not detected in the spinal cord of SOD1 transgenic mice. (AC) Western blot analysis of cervical (SCC) and lumbar (SCL) spinal cord samples from wild-type (WT) and SOD1 transgenic (SOD1) mice obtained 10 and 17 wks after birth reacted with tau (clone tau5) (A and B) and tau45-230 (C) antibodies. No immunoreactive bands of 17 kDa apparent molecular weight were detected in any of the samples analyzed. A tau45-230-immunoreactive band was only detected in whole-cell extracts prepared from hippocampal cultures treated with aggregated β amyloid (Aβ) used as a positive control (C). Class III β tubulin (TUJ1) was used as loading control.
DISCUSSION
Tau plays an important role in the mechanisms underlying neuronal death in a number of neurodegenerative diseases known as tauopathies. These diseases are characterized by the presence of hyperphosphorylated forms of tau in affected areas (reviewed in 43,44). More recently, we have identified tau cleavage leading to generation of the tau45-230 fragment as a conserved mechanism of degeneration in these diseases (32). Although ALS has not been considered a tauopathy, tau hyperphosphorylation has been reported in the spinal cords of SOD1 transgenic mice (45). On the other hand, little is known regarding tau cleavage in the motor neurons of ALS patients. Our results show that tau45-230 accumulated in the anterior horn of spinal cords obtained from ALS subjects. The presence of this fragment was accompanied by variations in the levels of full-length tau, especially in the cervical region of ALS spinal cords. Variations in full-length tau could reflect changes in its expression level or turnover of this MAP in motor neurons undergoing degeneration in these patients. Alternatively, they could parallel the extent of cleavage into tau45-230 and/or other fragments. Tau45-230 was also present in some (~ 50%) of the ALS precentral gyrus samples analyzed. These results suggest that accumulation of this neurotoxic fragment in upper motor neurons takes place only in a subset of ALS patients. Similarly, several reports have identified aggregates of phosphorylated tau in approximately half of the ALS subjects analyzed. These aggregates were mainly located in the amygdala, entorhinal cortex, anterior cingulate gyrus, superior frontal cortex and substantia nigra (31,46). The presence of this tau pathology in ALS has been associated with a syndrome of frontotemporal dysfunction characterized as frontotemporal lobe degeneration (31,46). It is tempting to speculate that the accumulation of tau45-230 might not be limited to upper motor neurons in this group of ALS patients. This fragment could also be generated in other brain areas responsible for cognitive functions and may contribute to the cognitive symptoms in this subgroup of patients. Further studies on a larger cohort of ALS patients will be needed to address the relationship between tau cleavage into this tau fragment in upper motor neurons and its presence in brain areas affected by tau pathology in ALS patients diagnosed with frontotemporal dementia.
We detected not only soluble tau45-230 but also two sets of aggregates containing this fragment in ALS samples. The apparent molecular weight of these tau45-230 aggregates indicated that they could contain eight or ten molecules of the fragment, respectively. Aggregates of recombinant tau45-230 generated in vitro were slightly smaller (up to four to six molecules of tau45-230) than those isolated from the spinal cords of ALS subjects (Rubino and Ferreira, unpublished observations). These observations could reflect differential aggregation properties of endogenously generated tau45-230 in the context of neurons that develop in situ versus those detected when the recombinant tau fragment is incubated in the presence of an inducer of aggregation. Alternatively, they could be due to the presence of other proteins and/or posttranslationally modified tau45-230 in these aggregates. These potential posttranslational modifications may not include phosphorylation, since we have previously shown that this fragment was not phosphorylated even under experimental conditions that induced hyperphosphorylation of full-length tau (33). Regardless of the composition of tau45-230 aggregates, these results are in agreement with experimental evidence demonstrating that one of the pathological characteristics of ALS is the formation of protein aggregates (reviewed in 1–3; see also references within). Aggregates composed of neurofilaments, peripherin, mutated SOD1 or TDP-43 have already been described in this neurodegenerative disease (49–53). The role of these protein aggregates during the pathological process has yet to be elucidated. However, it is worth noting that the toxic effects of small aggregates of full-length tau have already been described in AD and other tauopathies (54–57). Furthermore, these oligomeric forms of tau have been implicated in the propagation of the disease process (56,59). Thus, the presence of tau45-230 aggregates could also have some deleterious effects in motor neurons in the context of ALS.
A growing body of evidence indicates that tau45-230 does induce neuronal death and is not merely a marker of neuronal degeneration. The neurotoxic toxic effects of this fragment were first studied in cultured cells (33,34). Thus, expression of this fragment in otherwise healthy cultured central neurons and several non-neuronal cell types induced neuronal death (33,47). Conversely, preventing generation of this fragment resulted in enhanced neuronal survival of β-amyloid–treated hippocampal neurons (48). More recently, we have characterized the phenotype of tau45-230 transgenic mice (35). Selective expression of this fragment in the hippocampus of transgenic mice resulted in increased neuronal death, synapse loss and behavioral deficits (35). Taken together, these data suggest that the presence of high levels of tau45-230 in motor neurons of ALS patients might contribute to their degeneration.
The mechanisms underlying generation of tau45-230 in tauopathies have been the focus of extensive research. Data obtained in our laboratory indicate that tau cleavage into tau45-230 is the result of enhanced calpain activity associated with little or no change in calpain levels (32). This seems to be the case also in ALS, at least in the lumbar spinal cord and precentral gyrus areas, as described above. On the other hand, a significant increase in calpain levels was detected in ALS cervical spinal cord samples. This regional difference in calpain levels could be due to differential protein expression in the cervical versus lumbar spinal cord in ALS as previously described for other proteins, including transcription factors, growth factors and cytokines (60). Alternatively, it could reflect a temporally regulated response associated with progression of the neurodegenerative disease from motor neurons innervating muscles of the leg toward those innervating the upper limb. Our results also showed an increase in calpain activity using spectrin cleavage as a surrogate marker for this protease activity in ALS. Increases in calpain activity ranged from 3% to 230% in ALS samples. This variability among samples diminished the statistical power of our analysis. Nevertheless, these results strongly suggest that the activity of this protease is enhanced in ALS subjects and seems to parallel accumulation of this tau fragment being more pronounced in lower than upper motor neurons.
The calpain dysregulation observed in AD culture and animal models was induced by enhanced extracellular calcium influx mediated by glutamate N-methyl-D-aspartate (NMDA) receptors (61,62). Glutamate excitotoxicity has also been described in ALS. This toxicity could decrease glutamate uptake by astrocytes and/or alter mitochondria function in this disease (10,63). The accumulation of tau45-230 in ALS motor neurons provided evidence of an alternative molecular mechanism by which excitotoxicity could lead to neurodegeneration of motor neurons. In contrast to what has been described in AD and other tauopathies, calpain dysregulation in the context of ALS does not lead to aberrant cleavage of dynamin 1 (Vintilescu and Ferreira, unpublished observations, see also 61,62). This protein is involved in the recycling of synaptic vesicles and could be responsible for synaptic dysfunction underlying the cognitive deficits characteristic of these diseases. However, calpain cleavage is not unique to tau in ALS. Thus, calpain dysregulation is responsible for cleavage of the TDP-43 in this disease (12–20). This pathological calpain-mediated TDP-43 cleavage generated aggregation-prone small C-terminal fragments (25-35 kDa) (12,64). These fragments also served as seeds for the formation of TDP-43 inclusions and its cytosolic sequestration. Both the pathological localization and posttranslational modifications of TDP-43 are consistently present in sporadic and familial forms of the disease, except in those associated with mutations of SOD1 (65–67). As in the case of TDP-43, tau cleavage into tau45-230 was detected in sporadic forms of ALS and not in SOD1 animal models. These results provide further support for the prevailing view that sporadic and familial forms of ALS share many but not all of the molecular mechanisms leading to degeneration of the motor system.
CONCLUSION
Our results suggest that tau cleavage into the neurotoxic tau45-230 fragment could have some bearing on the pathobiology of ALS. The mechanisms underlying the toxic effects of this tau fragment and its aggregates in the degeneration of motor neurons and/or propagation of the ALS disease process await further investigation.
ACKNOWLEDGEMENTS
The authors thank the Cognitive Neurology and Alzheimer’s Disease Center Brain Bank at Northwestern University and the New York Brain Bank at Columbia University for providing the human specimens used in this study.
Footnotes
Online address: http://www.molmed.org
Disclosure
This work was supported in part by grants from the Amyotrophic Lateral Sclerosis Foundation, ALSA 1522 and NIH R01NS090993, to AF.
Cite this article as: Vintilescu CR, Afreen S, Rubino AE, Ferreira A. (2016) The neurotoxic tau45-230 fragment accumulates in upper and lower motor neurons in amyotrophic lateral sclerosis subjects. Mol. Med. 22:477–86.
REFERENCES
- Strong MJ, Kesavapany S, Pant HC. The pathobiology of amyotrophic lateral sclerosis: a proteinopathy. J Neuropathol Exp Neurol. 2005;64:649–64. doi: 10.1097/01.jnen.0000173889.71434.ea. [DOI] [PubMed] [Google Scholar]
- Boillee S, Velde CV, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Mitchell JD, Borasio GD. Amyotrophic lateral sclerosis. Lancet. 2007;369:2031–41. doi: 10.1016/S0140-6736(07)60944-1. [DOI] [PubMed] [Google Scholar]
- Bento-Abreu A, Van Damme P, Den Bosch L, Robberecht W. The neurobiology of amyotrophic lateral sclerosis. Eur J Neurosci. 2010;31:2247–65. doi: 10.1111/j.1460-9568.2010.07260.x. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, et al. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–4. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–49. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- De Carvhalo M, Swash M. Amyotrophic lateral sclerosis: an update. Curr Opin Neurol. 2011;24:497–503. doi: 10.1097/WCO.0b013e32834916a9. [DOI] [PubMed] [Google Scholar]
- Van Damme P, Robberecht W. Recent advances in motor neuron disease. Curr Opin Neurol. 2009;22:486–92. doi: 10.1097/WCO.0b013e32832ffbe3. [DOI] [PubMed] [Google Scholar]
- Simpson CL, Al-Chalabi A. Amyotrophic lateral sclerosis as a complex genetic disease. Biochem Biophys Acta. 2006;1762:973–85. doi: 10.1016/j.bbadis.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Van Den Bosch L, Van Damme P, Bogaert E, Robberecht W. The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochem Bioyphys Acta. 2006;1762:1068–82. doi: 10.1016/j.bbadis.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Chevalier-Larsen E, Holzbaur ELF. Axonal transport and neurodegenerative disease. Biochem Biophys Acta. 2006;1762:1094–108. doi: 10.1016/j.bbadis.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Gendron TF, Petrucelli L. Rodent models of TDP-43 proteinopathy: Investigating the mechanisms of TDP-43-mediated neurodegeneration. J Mol Neurosci. 2011;45:486–99. doi: 10.1007/s12031-011-9610-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Cleveland DW. Rethinking ALS: The FUS about TDP-43. Cell. 2009;136:1001–4. doi: 10.1016/j.cell.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Cleveland DW. An expansion in ALS genetics. Nature. 2010;466:1052–3. doi: 10.1038/4661052a. [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Human Mol Gen 19:Review Issue 1. 2010:R46–R64. doi: 10.1093/hmg/ddq137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling SC, et al. ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc. Natl. Acad. Sci. U.S.A. 2010;107:13318–23. doi: 10.1073/pnas.1008227107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IRA, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis. Lancet Neurol. 2010;9:995–1007. doi: 10.1016/S1474-4422(10)70195-2. [DOI] [PubMed] [Google Scholar]
- Shiina Y, Arima K, Tabunoki H, Satoh JI. TDP-43 dimerizes in human cells in culture. Cell Mol. Neurobiol. 2010;30:641–52. doi: 10.1007/s10571-009-9489-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai KJ, et al. Elevated expression of TDP-43 in the forebrain of mice is sufficient to cause neurological and pathological phenotypes mimicking FTLD-U. J Exp Med. 2007;207:1661–73. doi: 10.1084/jem.20092164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youmans KL, Wolozin B. TDP-43: a new player on the AD field? Experimental Neurol. 2012;237:90–5. doi: 10.1016/j.expneurol.2012.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas PW, Pienkowski TP, Kosik KS. Processes induced by tau expression in Sf9 cells have an axon-like microtubule organization. J Cell Biol. 1991;115:1333–44. doi: 10.1083/jcb.115.5.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas PW, et al. Tau confers drug-stability but not cold stability to microtubules in living cells. J Cell Sci. 1994;107:135–43. doi: 10.1242/jcs.107.1.135. [DOI] [PubMed] [Google Scholar]
- Caceres A, Kosik KS. Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature. 1990;343:461–3. doi: 10.1038/343461a0. [DOI] [PubMed] [Google Scholar]
- Drubin D, Feinstein SC, Shooter EM, Kirschner MW. Nerve growth factor induced neurite outgrowth in PC12 cells involves the coordinate induction of microtubule assembly-promoting factors. J Cell Biol. 1985;101:1790–807. doi: 10.1083/jcb.101.5.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knops J, et al. Overexpression of tau in a non-neuronal cell induces long cellular processes. J Cell Biol. 1991;114:725–33. doi: 10.1083/jcb.114.4.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles R, Leclerc N, Kosik KS. Organization of actin and microtubules during process formation in tau-expressing Sf9 cells. Cell Motil Cytoskel. 1994;28:256–64. doi: 10.1002/cm.970280308. [DOI] [PubMed] [Google Scholar]
- Kosik KS, Joachim CL, Selkoe DJ. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci U.S.A. 1986;83:4044–8. doi: 10.1073/pnas.83.11.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JG, Mirra SS, Pollock NJ, Binder LI. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau) Proc Natl Acad Sci U.S.A. 1986;83:4040–3. doi: 10.1073/pnas.83.11.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo J, et al. The carboxyl third of tau is tightly bound to paired helical filaments. Neuron. 1988;1:827–34. doi: 10.1016/0896-6273(88)90130-4. [DOI] [PubMed] [Google Scholar]
- Strong MJ, et al. Tau protein hyperphosphorylation in sporadic ALS with cognitive impairment. Neurology. 2006;66:1770–1. doi: 10.1212/01.wnl.0000218161.15834.db. [DOI] [PubMed] [Google Scholar]
- Yang W, Sopper MM, Leystra-Lantz C, Strong MJ. Microtubule associated tau protein positive neuronal and glial inclusions in amyotrophic lateral sclerosis. Neurology. 2003;61:1766–73. doi: 10.1212/01.wnl.0000099372.75786.f8. [DOI] [PubMed] [Google Scholar]
- Ferreira A, Bigio EH. Calpain-mediated tau cleavage: a mechanism leading to neurodegeneration shared by multiple tauopathies. Mol Med. 2011;17:676–85. doi: 10.2119/molmed.2010.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SY, Ferreira A. The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration. J Neurosci. 2005;25:5365–75. doi: 10.1523/JNEUROSCI.1125-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S-Y, Tournell CE, Sinjoanu RC, Ferreira A. Caspase 3- and calpain-mediated tau cleavage are differentially prevented by estrogen and testosterone in beta-amyloid-treated hippocampal neurons. Neurosci. 2007;144:119–27. doi: 10.1016/j.neuroscience.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang AE, Riherd Methner DN, Ferreira A. Neuronal degeneration, synaptic defects and behavioral deficits in tau45-230 transgenic mice. Neurosci. 2014;275:322–39. doi: 10.1016/j.neuroscience.2014.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–85. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U.S.A. 1979;76:4350–54. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakunin AF, Hallenbeck PC. A luminol/iodophenol chemiluminescent detection system for western immunoblots. Anal Biochem. 1998;258:146–9. doi: 10.1006/abio.1998.2571. [DOI] [PubMed] [Google Scholar]
- Gallagher SR. One-dimensional electrophoresis using nondenaturing conditions. Curr Protoc Mol Biol Chapter 10:Unit 10 2B. 2001 doi: 10.1002/0471142727.mb1002bs47. [DOI] [PubMed] [Google Scholar]
- Guttmann RP, Erickson AC, Johnson GV. Tau self-association: stabilization with a chemical cross-linker and modulation by phosphorylation and oxidation state. J Neurochem. 1995;64:1209–15. doi: 10.1046/j.1471-4159.1995.64031209.x. [DOI] [PubMed] [Google Scholar]
- Czogalla A, Sikorski AF. Spectrin and calpain: a ‘target’ and a ‘sniper’ in the pathology of neuronal cells. Cell Mol Life Sci. 2005;62:1913–24. doi: 10.1007/s00018-005-5097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niessen HG, et al. Metabolic progression markers of neurodegeneration in the transgenic G93A-SOD1 mouse model of amyotrophic lateral sclerosis. Eur J Neurosci. 2007;25:1669–77. doi: 10.1111/j.1460-9568.2007.05415.x. [DOI] [PubMed] [Google Scholar]
- Iqbal K, et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005;1739:198–210. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Frost B, Gotz J, Feany MB. Connecting the dots between tau dysfunction and neuronal degeneration. Trends in Cell Biol. 2015;25:46–53. doi: 10.1016/j.tcb.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farah CA, Nguyen MD, Julien JP, Leclerc N. Altered levels and distribution of microtubule-associated proteins before disease onset in a mouse model of amyotrophic lateral sclerosis. J Neurochem. 2003;84:77–86. doi: 10.1046/j.1471-4159.2003.01505.x. [DOI] [PubMed] [Google Scholar]
- Yang W, Strong MJ. Widespread neuronal and glial hyperphosphorylated tau deposition in ALS with cognitive impairment. Amyotroph Lateral Sc. 2012;13:178–93. doi: 10.3109/17482968.2011.622405. [DOI] [PubMed] [Google Scholar]
- Reinecke JB, et al. Implicating calpain in tau-mediated toxicity in vivo. PloS One d.o.i: 10.1371/journal.pone.0023865. 2011 doi: 10.1371/journal.pone.0023865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinjoanu RC, et al. The novel calpain inhibitor A-705253 potently inhibits oligomeric beta-amyloid-induced dynamin 1 and tau cleavage in hippocampal neurons. Neurochem Intl. 2008;53:79–88. doi: 10.1016/j.neuint.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migheli A, Pezzulo T, Attanasio A, Schiffer D. Peripherin immunoreactive structures in amyotrophic lateral sclerosis. Lab Invest. 1993;68:185–91. [PubMed] [Google Scholar]
- Wong N, He BP, Strong MJ. Characterization of neuronal intermediate filament protein expression in cervical spinal cord motor neurons in sporadic amyotrophic lateral sclerosis (ALS) J Neuropathol Exp Neurol. 2000;59:972–82. doi: 10.1093/jnen/59.11.972. [DOI] [PubMed] [Google Scholar]
- Strong MJ. Neurofilament metabolism in sporadic amyotrophic lateral sclerosis. J Neurol Sci. 1999;169:170–7. doi: 10.1016/s0022-510x(99)00241-5. [DOI] [PubMed] [Google Scholar]
- Arai T, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–11. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Dalton MJ, Gurney ME, Kopito RR. Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U.S.A. 2000;97:12571–6. doi: 10.1073/pnas.220417997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, et al. Aggregation of detergent-insoluble tau is involved in neuronal loss but not in synapse loss. J Biol Chem. 2010;49:38692–9. doi: 10.1074/jbc.M110.136630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, et al. Preparation and characterization of neurotoxic tau oligomers. Biochem. 2010;49:10039–41. doi: 10.1021/bi1016233. [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, et al. Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. Faseb J. 2012;26:1946–59. doi: 10.1096/fj.11-199851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson KR, et al. Characterization of prefibrillar tau oligomers in vitro and in Alzheimer Disease. J Biol Chem. 2011;286:23063–76. doi: 10.1074/jbc.M111.237974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usenovic M, et al. Internalized tau oligomers cause neurodegeneration by inducing accumulation of pathogenic tau in human neurons derived from induced pluripotent stem cells. J Neurosci. 2015;35:14234–50. doi: 10.1523/JNEUROSCI.1523-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirbaha H, Holmes BB, Sanders DW, Bieschke J, Diamond MI. Tau trimmers are the minimal propagation unit spontaneously internalized to seed intracellular aggregation. J Biol Chem. 2015;290:14893–903. doi: 10.1074/jbc.M115.652693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers DR, et al. Neuroinflamation modulates distinct regional and temporal clinical response in ALS mice. Brain Beh Immun. 2011;25:1025–35. doi: 10.1016/j.bbi.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly B, Ferreira A. Beta-amyloid-induced dynamin 1 degradation is mediated by NMDA receptors in hippocampal neurons. J Biol Chem. 2006;281:28079–89. doi: 10.1074/jbc.M605081200. [DOI] [PubMed] [Google Scholar]
- Kelly BL, Ferreira A. Beta-amyloid disrupted synaptic vesicle endocytosis in cultured hippocampal neurons. Neurosci. 2007;147:60–70. doi: 10.1016/j.neuroscience.2007.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalloni A, Nutini M, Longone L. Role of N-methyl-D-aspartate receptors complex in amyotrophic lateral sclerosis. Biochim Biophys Acta. 2013;1832:312–22. doi: 10.1016/j.bbadis.2012.11.013. [DOI] [PubMed] [Google Scholar]
- Yamashita T, et al. A role for calpain-dependent cleavage of TDP-43 in amyotrophic lateral sclerosis pathology. Nat Commun. 2012;3:1307–21. doi: 10.1038/ncomms2303. [DOI] [PubMed] [Google Scholar]
- Joyce PI, Fratta P, Fisher EMC, Acevedo-Arozena A. SOD1 and TDP-43 animal models of amyotrophic lateral sclerosis: recent advances in understanding disease toward the development of clinical treatments. Mamm Genome. 2011;22:420–48. doi: 10.1007/s00335-011-9339-1. [DOI] [PubMed] [Google Scholar]
- Kabashi E, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–4. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- Sreedharan J, et al. TDP-43 mutation in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–72. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]