

Abstract
Long bones and the cranial base are both formed through endochondral ossification. Elongation of long bones is primarily through the growth plate, which is a cartilaginous structure at the end of long bones made up of chondrocytes. Growth plate chondrocytes are organized in columns along the longitudinal axis of bone growth. The cranial base is the growth center of the neurocranium. Synchondroses, consisting of mirror-image growth plates, are critical for cranial base elongation and development. Over the last decade, considerable progress has been made in determining the roles of the parathyroid hormone–related protein, Indian hedgehog, fibroblast growth factor, bone morphogenetic protein, and Wnt signaling pathways in various aspects of skeletal development. Furthermore, recent evidence indicates the important role of the primary cilia signaling pathway in bone elongation. Here, we review the development of the growth plate and cranial synchondrosis and the regulation by the above-mentioned signaling pathways, highlighting the similarities and differences between these 2 structures.
Keywords: chondrocyte, PTHrP, Ihh, FGF, Wnt, primary cilia
Introduction
The skeleton is mainly formed through endochondral ossification, with the exception of a few areas—most notably the flat bones of the skull, which are formed through intramembranous ossification (Kronenberg 2003). Endochondral bone formation starts with the formation of a cartilage template from condensed mesenchymal cells. Some chondrocytes in the center of the cartilage template become hypertrophic and subsequently undergo apoptotic cell death. The primary ossification center is formed after osteoblast invasion. A portion of the chondrocytes form a column-like structure with an orientation that directs the lengthening of the bone along a particular axis. Long bones and the cranial base are both similarly formed through endochondral ossification (Nie 2005). However, developmental regulation of the cranial base has some unique features compared with long bones. This review presents current views on the signaling pathways involved in the regulation of bone elongation, highlighting the similarities and differences between long bones and the cranial synchondrosis. An overview of development of the growth plate and cranial synchondrosis precedes the discussion of signaling pathways. Of note, transcription factors such as SRY-box 9 (SOX9) and runt-related transcription factor 2 (RUNX2) play central roles in growth plate regulation (Kronenberg 2003). However, they are not included in this review because there is no report to date about their roles in synchondrosis development.
Development of the Growth Plate and Cranial Synchondrosis
Long bones function to support body weight and facilitate movement. In the long bone of a limb, a secondary ossification center forms near the end of the cartilage template with a modality similar to the primary ossification center. Cartilage between the primary and secondary ossification centers is the so-called growth plate, which is responsible for longitudinal bone growth until the end of adolescence. The growth plate has very unique cellular architecture with 3 histologically distinct cell layers: resting, proliferative and hypertrophic zones (Kronenberg 2003). Cells in the proliferative zone line up in columns parallel to the long axis of the bone. The characteristic column-like chondrocyte orientation in the growth plate is critical for bone elongation. The growth plate chondrocytes contribute to bone elongation through proliferation, extracellular matrix secretion, and hypertrophy. Bone elongation stops when growth plate cartilage disappears as a consequence of exhausted proliferative potential of the chondrocytes.
The cranial base functions as a supporting platform for the development of the brain, provides unique space to organs such as the pituitary, and contains multiple growth centers to drive both cranial and upper facial growth. The prechondral, hypophyseal, and parachondral cartilaginous plates form the central region of the cranial base. These plates lie below the developing brain and fuse to form an uninterrupted cartilaginous structure spanning the region from the foramen magnum to the interorbital junction (Thorogood 1988). As development progresses, primary ossification centers emerge and form various bones such as the ethmoidal bone, presphenoid bone, basisphenoid bone, and occipital bone. The cartilaginous segments persisting between the ossification centers represent the synchondroses, including the sphenoethmoidal synchondrosis, intersphenoid synchondrosis (ISS), spheno-occipital synchondrosis (SOS), and intraoccipital synchondrosis, according to their anatomical locations. In the middle line of the cranial base, the SOS joins the basisphenoid and occipital bones, and the ISS joins the presphenoid and basisphenoid bones (Fig. 1).
Figure 1.

The cranial base and synchondroses. (A) Micro–computed tomography image of the skeletal structures of the mouse cranial base at 2 mo of age. From the rostral side (right) to the caudal side (left), the cranial base of the mouse is composed of presphenoid, basisphenoid, and basioccipital bone. Between the adjacent bones are cartilaginous synchondroses, namely the intersphenoid synchondrosis (ISS) and the spheno-occipital synchondrosis (SOS). (B) Histological structures of the cranial base in a newborn mouse. Three ossification centers (occipital, basisphenoid, and presphenoid) are separated by 2 synchondroses: the ISS and the SOS. The synchondrosis is composed of mirror-image growth plates with a central resting zone (R), proliferative zones (P), and hypertrophic zones (H) on both sides.
The synchondrosis is composed of mirror-image growth plates with a central resting zone and proliferative and hypertrophic zones on both sides (Fig. 1B). Similar to the growth plate in long bones, proliferation and hypertrophy of chondrocytes contribute to the elongation of the cranial base. However, the cranial synchondrosis is different from the long bone growth plate in the following aspects. First, given the orientation of chondrocytes, the synchondrosis produces growth in opposing directions. Second, the synchondrosis is not overlaid by an articular synovial layer, which is typical of the growth plate in developing long bones. Third, the secondary ossification center is absent in the cranial synchondrosis.
Because cranial synchondroses are important centers of longitudinal growth in the human skull and they play a critical role in upper face and cranial vault development, premature fusion of synchondroses can lead to midface hypoplasia, as seen in patients with syndromic craniosynostosis (Goldstein et al. 2014). Among several synchondroses of the cranial base, the SOS has received almost exclusive attention in clinical studies because it is the last synchondrosis to fuse in humans (Madeline and Elster 1995). Complete fusion occurs between 2 and 3 y for the ISS, between 9 and 15 y for the sphenoethmoidal synchondrosis, and between 16 and 18 y for the SOS (Madeline and Elster 1995). It is assumed that the role of the SOS in cranial growth is proportionally greater than the roles of other synchondroses. However, 60% of cranial base growth occurs during the embryonic stage and 40% of postnatal cranial base growth occurs during the first 2 y of life (Scott 1958). Thus, the contribution from other synchondroses such as the ISS should not be neglected.
Signaling Pathways That Control the Development of the Growth Plate and Cranial Synchondrosis
Bone growth in length is regulated by many signaling pathways. Because of space limitations, this review focuses on the most well-studied pathways, including the parathyroid hormone–related protein (PTHrP), Indian hedgehog (Ihh), fibroblast growth factor (FGF), bone morphogenetic protein (BMP), and Wnt/β-catenin pathways. The noncanonical Wnt signaling pathway plays important roles in growth plate development (Usami et al. 2016) but is not discussed here because there are currently no reports about its function in the synchondrosis. In addition, the primary cilia signaling pathway is included because recent studies have demonstrated its important roles in the development of both the growth plate and cranial synchondrosis (Koyama et al. 2007; Ruiz-Perez et al. 2007; Song et al. 2007; Ochiai et al. 2009; Pacheco et al. 2012; Yuan and Yang 2015).
PTHrP Signaling
During fetal growth plate development in long bones, PTHrP is secreted from cells in the resting zone near the end of the bone and signals through PTH1R to keep chondrocytes proliferating and inhibit their differentiation toward hypertrophic chondrocytes (Kronenberg 2003). When the proliferating chondrocytes are sufficiently distant from PTHrP-producing cells, the local PTHrP concentration reaches a critically low level to allow the proliferating chondrocyte to stop proliferating and start hypertrophic differentiation. PTHrP regulates chondrocytes through multiple molecular mechanisms (Fig. 2). RUNX2, RUNX3, and myocyte enhancer factor 2C and 2D (MEF2C/D) are important regulators of chondrocyte maturation. PTHrP can down-regulate RUNX2 expression through the protein kinase A signaling pathway (Li et al. 2004) or it can induce degradation of RUNX2 and RUNX3 in a cyclin-D1–dependent manner (Zhang et al. 2009). PTHrP can promote dephosphorylation of histone deacetylase 4 (HDAC4) to facilitate its nuclear localization and subsequently inhibit MEF2C transcription (Kozhemyakina et al. 2009). PTHrP can increase the expression of zinc finger transcriptional coregulator 521 (Zfp521). Zfp521 associates with and antagonizes RUNX2 in chondrocytes via an HDAC4-dependent mechanism (Correa et al. 2010).
Figure 2.
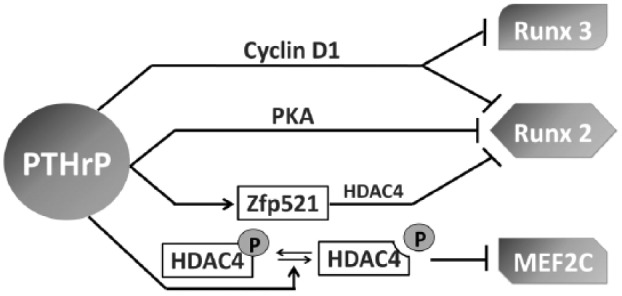
Mechanisms of parathyroid hormone–related protein (PTHrP) signaling in chondrocyte maturation. PTHrP can cyclin-D1–dependently induce degradation of runt-related transcription factor 2 and 3 (Runx2/3); down-regulate Runx2 expression through the protein kinase A (PKA) pathway; increase expression of zinc finger transcriptional coregulator 521 (Zfp521), which antagonizes Runx2 through histone deacetylase 4 (HDAC4); and promote dephosphorylation of HDAC4 and thus inhibit myocyte enhancer factor 2C (MEF2C) transcription. P represents phosphorylation.
Similar to the growth plate of long bones, PTHrP signaling in the synchondrosis can maintain chondrocyte proliferation and prevent chondrocyte hypertrophic differentiation (Fig. 3A). However, chondrocyte differentiation is markedly more accelerated in the ISS and SOS of PTHrP−/− mice, compared with the growth plate (Ishii-Suzuki et al. 1999). This accelerated chondrocyte differentiation is associated with more accelerated endochondral bone formation. In contrast with the more localized expression of PTHrP in the resting zone of the growth plate with a gradient effect, PTHrP is expressed in both the resting and proliferative zones of the synchondrosis (Young et al. 2006; Nagayama et al. 2008). This may explain the phenotypic difference between the growth plate and synchondroses in PTHrP−/− mice.
Figure 3.

Regulation of parathyroid hormone–related protein (PTHrP)/Indian hedgehog (Ihh) signaling and fibroblast growth factor receptor 3 (FGFR3) signaling on cranial synchondrosis development. (A) PTHrP can maintain chondrocyte proliferation and inhibit their hypertrophic differentiation. Ihh promotes chondrocyte proliferation and maturation and also stimulates PTHrP expression. It is unclear whether Ihh stimulates PTHrP expression as in the growth plate of long bones. (B) Active mutation of FGFR3 suppresses chondrocyte proliferation and down-regulates expression of the PTHrP receptor and Ihh. Premature fusion of synchondroses in FGFR3 mutant mice may be mediated by the mitogen-activated protein kinase (MAPK) pathway.
Ihh Signaling
Ihh is predominantly produced by prehypertrophic chondrocytes and promotes chondrocyte proliferation. Through a not-yet-clear mechanism, Ihh stimulates PTHrP expression in periarticular chondrocytes. Subsequently, PTHrP maintains chondrocyte proliferation and inhibits their differentiation toward hypertrophic chondrocytes (Kronenberg 2003) (Fig. 3A). Ihh can also regulate growth plate chondrocyte differentiation in a PTHrP-independent manner (Kobayashi et al. 2005b; Mak et al. 2008; Maeda et al. 2010). Ihh can directly promote the transition from periarticular chondrocytes to proliferating chondrocytes, although Ihh-PTHrP signaling is required to prevent premature hypertrophy (Kobayashi et al. 2005b).
Ihh expression can be regulated by several mechanisms (Fig. 4). Runx2 may interact with CCAAT/enhancer binding protein beta (C/EBPβ) at the putative C/EBPβ responsive element in the Ihh promoter and may thus cooperatively stimulate Ihh expression (Ushijima et al. 2014). Embryos that are double heterozygous for Wnt9a and β-catenin have reduced Ihh expression; there is direct interaction between the β-catenin/lymphoid enhancer binding factor 1 (Lef1) complex and the Ihh promoter, suggesting that Ihh expression is regulated by the canonical Wnt signaling pathway (Spater et al. 2006). δ-EF1, a two-handed zinc finger/homeodomain transcriptional repressor, can bind to the putative Ihh regulatory elements and down-regulate Ihh expression (Bellon et al. 2009). On the contrary, activating transcription factor 4 (ATF4) can activate the transcription of Ihh by binding to its promoter (Wang et al. 2009). Sirtuin 6 may regulate Ihh expression by increasing the affinity of ATF4 for the Ihh promoter (Piao et al. 2013).
Figure 4.
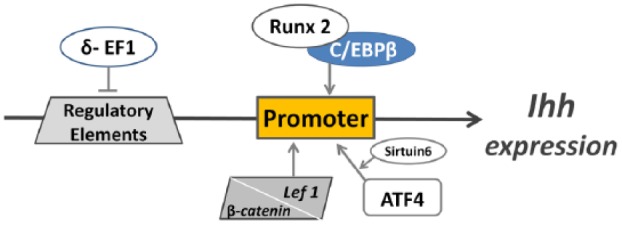
Regulatory mechanisms of Indian hedgehog (Ihh) expression. Both the CCAAT/enhancer binding protein beta (C/EBPβ)/runt-related transcription factor 2 (Runx2) complex and the β-catenin/lymphoid enhancer binding factor 1 (Lef1) complex can bind the Ihh promoter to stimulate Ihh expression. Activating transcription factor 4 (ATF4) can activate Ihh transcription by binding to its promoter. Sirtuin 6 may increase the affinity of ATF4 to the Ihh promoter. δ–EF1, a two-handed zinc finger/homeodomain transcriptional repressor, can inhibit Ihh transcription by binding to Ihh regulatory elements.
Ihh is expressed in the prehypertrophic zone in the synchondrosis (Young et al. 2006; Nagayama et al. 2008). Ihh signaling promotes chondrocyte proliferation and maturation in the synchondrosis (Fig. 4). In Ihh−/− mice, synchondroses are disorganized, chondrocyte proliferation is significantly decreased, and chondrocyte maturation is initially delayed before becoming widespread (Young et al. 2006). Hypertrophic chondrocytes in the synchondroses of Ihh−/− mice have a distinct abnormal distribution. In the growth plate, hypertrophic chondrocytes in Ihh−/− mice have been observed close to the ends of the skeletal element (St-Jacques et al. 1999). In the synchondroses, the mutant hypertrophic chondrocytes are widespread and occupy a central location throughout the synchondroses flanked by less mature collagen II–expressing chondrocytes. In addition, the hypertrophic chondrocytes in the SOS are much less affected than those in the ISS. The different topographic disturbance of hypertrophic chondrocyte distribution suggests the differential roles of Ihh in the growth plate and synchondroses. It has been proposed that the Ihh/PTHrP axis has a role in the synchondrosis similar to that in the long bone because PTHrP expression is greatly reduced, chondrocyte proliferation is decreased, and hypertrophy is enhanced in the synchondrosis of Ihh−/− mice (Young et al. 2006; Nagayama et al. 2008). However, the synchondrosis does not have an overlaid periarticular/articular layer, and the Ihh/PTHrP relay must function between Ihh-producing prehypertrophic chondrocytes and PTHrP-producing reserve/proliferating chondrocytes. As mentioned previously, PTHrP is expressed in both resting and proliferating zones of the synchondroses, which might also contribute to this phenotypic difference. In addition, notochord remnants express Sonic hedgehog (Shh) near the incipient SOS but not the ISS. Because Shh has a redundant role with Ihh, this likely explains the more severe phenotype in the ISS than in the SOS of Ihh−/− mice (Young et al. 2006). Chondrocyte-specific deletion of Ihh with Col2aCreER at the neonatal stage leads to shortening of the cranial base and disorganized synchondroses; moreover, the synchondroses prematurely fuse by postnatal day 15 (Koyama et al. 2007) (Fig. 5).
Figure 5.

Mutations leading to the premature fusion of cranial synchondroses. Chondrocyte-specific deletion of Indian hedgehog (Ihh), fibroblast growth factor receptor (FGFR) mutations (FGFR3369/369, FGFR3G374R/+, FGFR3P244R+/+, FGFR3365/+, FGFR2IIIcP253R, FGFR2IIIc−/−, FGFR2P253R/+), and primary cilia signaling dysregulation (loss of Ellis-van Creveld syndrome protein homolog [EVC] and chondrocyte-specific deletion of Kif3a [a component of the Kinesin-II motor complex] or intraflagellar transport 88 [IFT88]) can all lead to the premature fusion of synchondroses in mice.
FGF Signaling
Since the discovery of the causative role of fibroblast growth factor receptor 3 (FGFR3) mutations in achondroplasia (the most common form of human dwarfism), FGF signaling through FGFR3 has been extensively investigated. Decreased chondrocyte proliferation and decreased hypertrophy contribute to the decreased bone growth caused by activating mutations in FGFR3, but these 2 phenotypes are regulated by different branches of FGFR3 signaling. The proliferative branch is likely mediated through signal transducer and activator of transcription 1 (STAT1) (Murakami et al. 2004). Activation of mitogen-activated protein kinase (MAPK) signaling (Murakami et al. 2004), suppression of SOX9 down-regulation (Zhou et al. 2015), and activation of the protein phosphatase 2A containing B55αsubunit by FGFR3 (Kolupaeva et al. 2013) may all contribute to decreased chondrocyte hypertrophy. In addition, FGFR3 signaling can regulate growth plate development by suppressing bone morphogenetic protein receptor BMPR1A (Qi et al. 2014) in postnatal growth plate chondrocytes. FGF18−/− mice exhibit expanded proliferating and hypertrophic zones similar to FGFR3−/− mice, suggesting that FGF18 can signal through FGFR3 (Liu et al. 2002). FGF9−/− mice and FGFR1 conditional knockout mice with Dermo1-Cre showed a similar decrease in hypertrophic zone size at midembryonic stages, suggesting that FGF9 likely signals through FGFR1 to promote chondrocyte maturation at the early stage of chondrogenesis (Hung et al. 2007).
Activation mutations in FGFR3 result in several human skeletal dysplasias associated with craniofacial abnormalities. Recent research with transgenic mouse models revealed the important roles of FGFR3 signaling in the development of synchondroses of the cranial base. FGFR3369/369 (Chen et al. 1999), FGFR3P244R+/+ (Laurita et al. 2011), FGFR3365/+ (Chen et al. 2001), and FGFR3G374R/+ (Matsushita et al. 2009) mice all present with a prematurely fused cranial synchondrosis (Fig. 5). An activating mutation in FGFR3 leads to decreased proliferation and enhanced osteoblast differentiation in both the growth plate and synchondrosis, but with an obvious phenotypic difference. In FGFR3369/369 mice, the proliferation and maturation zones of the growth plate are disorganized and are associated with dramatic expansion of the resting zone and a significant decrease in other zones (Chen et al. 1999). In contrast with the growth plate, FGFR3369/369 synchondroses have decreased length in all zones prior to the premature fusion. Similarly, FGFR3G374R/+ synchondroses also present with premature loss of both resting and proliferating zones. The phenotypic difference between the growth plate and synchondrosis is likely attributable to the site-specific downstream signaling mediators. Increased Stat protein activation and up-regulation of cell-cycle inhibitors is observed in the growth plate of FGFR3369/369 mice. In cranial synchondroses, it was suggested that MAPK pathway alteration may contribute to the abnormality because chondrocyte-specific expression of constitutively active mitogen-activated protein kinases kinase (MAPKK or MEK1) also leads to premature fusion of cranial synchondroses (Matsushita et al. 2009) (Fig. 4).
FGFR2 mutations are also associated with premature fusion of cranial synchondroses (Fig. 5). FGFR2IIIcP253R mice carry the mutation identified in Apert syndrome, which leads to constitutive activation of FGFR2. The mutant mice have precocious ossification in the SOS at birth and subsequently in the ISS (Nagata et al. 2011). This premature fusion of the cranial synchondroses is associated with accelerated maturation and hypertrophy of chondrocytes. In FGFR2P253R/+ mice, the cranial base is shortened and the ISS is prematurely fused at postnatal day 9 (Yin et al. 2008). FGFR2IIIC−/− mice have reduced growth of the cranial base and premature fusion of the SOS (Eswarakumar et al. 2002).
BMP Signaling
BMPs have multiple roles during growth plate development. BMP2, BMP3, BMP4, BMP5, and BMP7 are expressed in the perichondrium, BMP7 is primarily expressed in the proliferative zone, and BMP2 and BMP6 are highly expressed in hypertrophic chondrocytes (Nilsson et al. 2007). Type 1 receptor BMPR1A is expressed broadly in the embryonic mesenchyme and type 1 receptor BMPR1B is expressed in cartilage condensation (Kronenberg 2003).
Compared with FGFR3 signaling, BMP signaling has an opposite effect on chondrocyte proliferation. BMP signaling increases Ihh expression in prehypertrophic chondrocytes and thus increases chondrocyte proliferation (Kronenberg 2003). Overexpression of a constitutively active form of BMPR1A results in the shortening of the long bone and growth plate associated with a decrease in proliferating chondrocyte columnar stacks and up-regulation of maturation markers, suggesting that BMP signaling can promote hypertrophic differentiation (Kobayashi et al. 2005a). BMP2+ /−; BMP6−/− mice have normal growth plate chondrocyte differentiation and expression of hypertrophic chondrocyte markers, suggesting the functional redundancy of BMPs (Kugimiya et al. 2005).
In an organ culture system, exogenous BMP4 protein stimulates growth of the cranial synchondrosis in a dose-dependent manner, with the enhancement of both proliferation and differentiation of chondrocytes (Shum et al. 2003). The increase in hypertrophy is associated with increased Ihh and PTHrP receptor expression but not PTHrP expression. Reversely, noggin, a BMP inhibitor, inhibits growth of the cranial base. Further in vivo study of BMP signaling in cranial synchondrosis is needed to reconcile the phenotypic difference between the growth plate observed in vivo and the synchondrosis observed ex vivo when BMP signaling is enhanced.
Wnt/β-catenin Signaling
Wnt proteins comprise 19 members and signal through β-catenin–dependent or β-catenin–independent pathways, which are also called canonical and noncanonical pathways, respectively. During embryogenesis, Wnt/β-catenin signaling can inhibit the differentiation of mesenchymal progenitor cells toward chondrocytes. During later stages, however, Wnt/β-catenin signaling is needed for the proliferation and differentiation of chondrocytes (Liu et al. 2008).
Conditional inhibition of Wnt/β-catenin signaling by overexpressing catenin beta interacting protein 1 in chondrocytes leads to reduced chondrocyte proliferation and differentiation as well as increased chondrocyte apoptosis (Chen et al. 2008). The mutant mice have a decreased length of proliferating and hypertrophic zones in the growth plate, delayed formation of the secondary ossification center, and overall reduced skeletal growth. Similarly, conditional inhibition of Wnt/β-catenin signaling in Col2a1-expressing chondrocytes by tamoxifen-induced β-catenin deletion at E13.5 with Col2CreERT2 results in a delayed onset of chondrocyte hypertrophy and maturation (Dao et al. 2012) or a disorganized growth plate and decreased hypertrophic zone (Yuasa et al. 2009). In contrast with the Col2CreERT2 model, conditional deletion of β-catenin by Col2a-Cre (which is expressed in Col2a1-expressing chondrocytes as early as E12.5) leads to perinatal death with ectopic cartilage formation in short, thick, and bowed long bones (Day et al. 2005). These abnormalities are the result of diminished osteoblast differentiation and ectopic chondrocyte differentiation associated with an expanded hypertrophic zone. Finally, deletion of Wntless, a trafficking protein that is essential for Wnt protein secretion, by Col2-Cre blocks all Wnt signaling (including canonical and noncanonical) in chondrocytes, leading to delayed chondrocyte hypertrophy as well as disturbed morphology and division orientation of proliferating chondrocytes (Lu et al. 2013).
The gain of function of Wnt/β-catenin signaling has both overlapping and different consequences compared with loss-of-function models. Conditional activation of Wnt/β-catenin signaling in chondrocytes achieved by expressing a constitutively active β-catenin using a Col2CreERT2 system leads to accelerated hypertrophy and disorganization of the columnar structure in the growth plate (Dao et al. 2012). Postnatal activation of Wnt/β-catenin signaling in cartilage by overexpressing a constitutively active form of β-catenin leads to decreased hypertrophic chondrocyte numbers and a shorter/disorganized and prematurely fused growth plate (Yuasa et al. 2009).
Cartilage-specific deletion of β-catenin with Col2a-Cre in mice leads to similar but different abnormalities in cranial synchondroses compared with those in the growth plate (Day et al. 2005; Nagayama et al. 2008). In both locations, decreased bone formation is documented. Ectopic chondrocyte differentiation is reported in the growth plate but not in the synchondrosis. Mutant cranial synchondroses have ill-defined chondrocyte zones with many immature, small, and collagen II–expressing chondrocytes that fail to form an organized growth plate. The molecular mechanisms that account for the phenotypic differences between the growth plate and the synchondrosis in mice with cartilage-specific β-catenin deletion are largely unknown. The difference in the downstream effectors of Wnt signaling in the 2 structures may contribute to this disparity. In normal development, β-catenin, Patched1, and sFRP1 are all expressed in the perichondrium of the synchondrosis flanking the prehypertrophic and hypertrophic zones, where perichondrial ossification initiates. However, expression of these molecules as well as Ihh and PTHrP is reduced in β-catenin–deficient synchondroses. It is currently unknown how the expression levels of these signaling molecules are altered in the β-catenin–deficient growth plate. In a reciprocal mouse model in which CA-LEF1 is expressed in chondrocytes, constitutive Wnt/β-catenin signaling is elicited (Nagayama et al. 2008). Mutant synchondroses have accelerated chondrocyte maturation. Precocious maturation and topographical disorganization result in intermingling of chondrocytes at different stages of differentiation. Excessive bone collar formation along the synchondroses also occurs in the mutant mice. The synchondroses abnormalities in both loss-of-function and gain-of-function models indicate that the Wnt/β-catenin signaling pathway plays an important role in cranial synchondrosis development, and tight regulation in this pathway is crucial for the normal progression of the chondrocyte maturation process.
Primary Cilia
Primary cilia are microtubule-based organelles that can transduce extracellular signals into cells through a variety of molecular signaling pathways (Veland et al. 2009). Recent evidence, as summarized below, demonstrates that primary cilia play important roles in regulating development of the growth plate and synchondrosis.
Intraflagellar transport (IFT) is the process by which primary cilia are generated and maintained. Kif3a, a component of the Kinesin-II motor complex, plays essential roles in IFT. Disruption of Kif3a can disable IFT and lead to failure in the formation and maintenance of primary cilia (Hirokawa 2000). Chondrocyte-specific deletion of Kif3a with Col2a-Cre leads to significant depletion of chondrocyte cilia and postnatal dwarfism owing to premature loss of the growth plate (Song et al. 2007). The premature loss of the growth plate is likely explained by reduced proliferation and accelerated hypertrophic differentiation of chondrocytes. In addition, Kif3a-deficient chondrocytes have an altered cell shape and fail to rotate properly, which results in an altered orientation and disrupted columnar structure. This is correlated with disruptions in the organization of the actin cytoskeleton and alterations in the localization of activated focal adhesion kinase.
IFT88/Polaris/Tg737 is an IFT protein that is essential for ciliogenesis. Chondrocyte-specific deletion of IFT88 with Col2a-Cre causes a similar phenotype compared with Kif3a deletion (Song et al. 2007). Mechanistically, it has been proposed that IFT88 and primary cilia regulate Sfrp5 and Wnt signaling pathways in the growth plate through the regulation of Ihh signaling (Chang and Serra 2013). Tg737orpk (orpk) mice carry a hypomorphic mutation in IFT88. In orpk mice, primary cilia are significantly shorter than wild-type cilia and are often difficult to detect (McGlashan et al. 2007). The growth plate of orpk mice is significantly smaller. Organization of the proliferating chondrocyte columns is less ordered and cells are less flattened, although the abnormality is less severe than that in Kif3a-deficient mice.
IFT80 is a core protein in IFT complex B. Hypomorphic IFT80 knockout results in embryonic lethality (Rix et al. 2011). The rare survivors have growth retardation and shortening of the long bones, and growth plate chondrocytes are disorganized. Chondrocyte-specific deletion of IFT80 with Col2a-Cre causes cilia loss and chondrodysplasia (Yuan and Yang 2015). In these mutant mice, growth plate length is shortened, with the proliferating zone being most affected. The columnar structure in mutant growth plates is mildly disrupted, and the proliferating zone is less organized.
Ellis-van Creveld syndrome protein homolog (EVC) and EVC2/LIMBIN are localized at the base of cilia (Takeda et al. 2002; Ruiz-Perez et al. 2007). Loss of EVC does not affect cilia formation but Ihh signal transduction is compromised and is associated with advanced chondrocyte maturation and delayed bone collar formation at the growth plate in mice with a C57BL/6;129 mixed background (Ruiz-Perez et al. 2007). The same group also demonstrated compromised bone collar formation in Evc−/− mice in the C57BL/6 homogenous background but showed that both proliferation and differentiation of chondrocytes are compromised in mutant mice (Pacheco et al. 2012). EVC and EVC2 are mutually required for their ciliary localization and loss of Evc2/Limbin results in similar skeletal phenotypes found in Evc−/− mice (Caparrós-Martín et al. 2013; Zhang et al. 2015).
Primary cilia and associated signaling pathways also play important roles in synchondrosis development. Chondrocyte-specific deletion of Kif3a with Col2a-Cre results in a shorter and wider cranial base associated with abnormal cranial synchondroses (Koyama et al. 2007). Kif3a-deficient synchondroses are highly disorganized and most of the synchondroses have prematurely fused by postnatal day 15 (Fig. 5). In these mutant synchondroses, hedgehog signaling has an aberrant topography. In particular, expression of Patched1 and Gli1 is markedly decreased and scattered, whereas Ihh expression is more diffused in mutant synchondroses. Chondrocyte-specific deletion of IFT88/Polaris with Col2a-Cre results in a deformed cranial base and disorganized synchondroses (Ochiai et al. 2009). In IFT88-deficient mice, the cranial synchondroses are prematurely fused (Fig. 5) with reduced expression of Ihh, Patched1, collagen X, and MMP-13. However, there is excessive intramembranous ossification along the perichondrium, and Patched-1 expression is abnormally high at this site. In both Kif3a-deficient and IFT88-deficient synchondroses, Ihh signaling is disrupted. It has been proposed that abnormally high Ihh signaling in the perichondrium triggers excessive intramembranous bone deposition and subsequent premature fusion of synchondrosis (Ochiai et al. 2009). However, although EVC−/− mice also have premature fusion of the ISS (Fig. 5) associated with dysregulated Ihh signaling in both the ISS and SOS at the neonatal stage, Patched1 expression is decreased in the perichondrium (Pacheco et al. 2012), indicating that EVC and Kif3a/IFT88 regulate the synchondrosis with different mechanisms. Finally, EVC2−/− mice also have impaired ISS development (Caparrós-Martín et al. 2013).
Summary
The cranial base and long bones are both formed through endochondral ossification. Their development has similar yet unique features. Growth plates and synchondroses play critical roles in the elongation of the endochondrally formed long bones and cranial base, respectively. PTHrP, Ihh, FGF, BMP, Wnt, and primary cilia signaling pathways play important roles in the developmental regulation of the growth plate and cranial synchondrosis. The disruption of these signaling pathways leads to both similar and distinctly different abnormalities in the development of the growth plate and synchondrosis. Alterations in Ihh, FGF, and primary cilia signaling pathways can all cause the premature fusion of cranial synchondroses. Differences in the expression pattern of signaling molecules, location-specific downstream signaling targets, and unique anatomic structures are contributors to the disparity of abnormalities between the growth plate and synchondrosis. However, the detailed molecular mechanisms accountable for the differences remain to be fully deciphered. Given the critical roles of the growth plate and cranial base in bone development, it is important to fully elucidate the developmental regulation by the aforementioned signaling pathways and factors.
Author Contributions
X. Wei, F. Liu, contributed to conception, design, data acquisition, analysis, and interpretation, drafted the manuscript; M. Hu, Y. Mishina, contributed to conception, critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Acknowledgments
We thank Dr. Ernestina Schipani for critical review of the manuscript, and Neil Thomas for help in editing the manuscript. We apologize to researchers whose work could not be cited because of space limitations.
Footnotes
This work was supported by the National Institutes of Health (grant R01AR062030 to F.L. and grants R01DE020843 and R01DE023538 to Y.M.).
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Bellon E, Luyten FP, Tylzanowski P. 2009. Delta-EF1 is a negative regulator of Ihh in the developing growth plate. J Cell Biol. 187(5):685–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caparrós-Martín JA, Valencia M, Reytor E, Pacheco M, Fernandez M, Perez-Aytes A, Gean E, Lapunzina P, Peters H, Goodship JA, et al. 2013. The ciliary Evc/Evc2 complex interacts with Smo and controls Hedgehog pathway activity in chondrocytes by regulating Sufu/Gli3 dissociation and Gli3 trafficking in primary cilia. Hum Mol Genet. 22(1):124–139. [DOI] [PubMed] [Google Scholar]
- Chang CF, Serra R. 2013. Ift88 regulates Hedgehog signaling, Sfrp5 expression, and β-catenin activity in post-natal growth plate. J Orthop Res. 31(3):350–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Adar R, Yang X, Monsonego EO, Li C, Hauschka PV, Yayon A, Deng CX. 1999. Gly369Cys mutation in mouse FGFR3 causes achondroplasia by affecting both chondrogenesis and osteogenesis. J Clin Invest. 104(11):1517–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Li C, Qiao W, Xu X, Deng C. 2001. A Ser(365)–>Cys mutation of fibroblast growth factor receptor 3 in mouse downregulates Ihh/PTHrP signals and causes severe achondroplasia. Hum Mol Genet. 10(5):457–465. [DOI] [PubMed] [Google Scholar]
- Chen M, Zhu M, Awad H, Li TF, Sheu TJ, Boyce BF, Chen D, O’Keefe RJ. 2008. Inhibition of beta-catenin signaling causes defects in postnatal cartilage development. J Cell Sci. 121(Pt 9): 1455–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa D, Hesse E, Seriwatanachai D, Kiviranta R, Saito H, Yamana K, Neff L, Atfi A, Coillard L, Sitara D, et al. 2010. Zfp521 is a target gene and key effector of parathyroid hormone-related peptide signaling in growth plate chondrocytes. Dev Cell. 19(4):533–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao DY, Jonason JH, Zhang Y, Hsu W, Chen D, Hilton MJ, O’Keefe RJ. 2012. Cartilage-specific β-catenin signaling regulates chondrocyte maturation, generation of ossification centers, and perichondrial bone formation during skeletal development. J Bone Miner Res. 27(8):1680–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day TF, Guo X, Garrett-Beal L, Yang Y. 2005. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 8(5):739–750. [DOI] [PubMed] [Google Scholar]
- Eswarakumar VP, Monsonego-Ornan E, Pines M, Antonopoulou I, Morriss-Kay GM, Lonai P. 2002. The IIIc alternative of Fgfr2 is a positive regulator of bone formation. Development. 129(16):3783–3793. [DOI] [PubMed] [Google Scholar]
- Goldstein JA, Paliga JT, Wink JD, Bartlett SP, Nah HD, Taylor JA. 2014. Earlier evidence of spheno-occipital synchondrosis fusion correlates with severity of midface hypoplasia in patients with syndromic craniosynostosis. Plast Reconstr Surg. 134(3):504–510. [DOI] [PubMed] [Google Scholar]
- Hirokawa N. 2000. Stirring up development with the heterotrimeric kinesin KIF3. Traffic. 1(1):29–34. [DOI] [PubMed] [Google Scholar]
- Hung IH, Yu K, Lavine KJ, Ornitz DM. 2007. FGF9 regulates early hypertrophic chondrocyte differentiation and skeletal vascularization in the developing stylopod. Dev Biol. 307(2):300–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii-Suzuki M, Suda N, Yamazaki K, Kuroda T, Senior PV, Beck F, Hammond VE. 1999. Differential responses to parathyroid hormone-related protein (PTHrP) deficiency in the various craniofacial cartilages. Anat Rec. 255(4):452–457. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Lyons KM, McMahon AP, Kronenberg HM. 2005a. BMP signaling stimulates cellular differentiation at multiple steps during cartilage development. Proc Natl Acad Sci U S A. 102(50):18023–18027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Soegiarto DW, Yang Y, Lanske B, Schipani E, McMahon AP, Kronenberg HM. 2005b. Indian hedgehog stimulates periarticular chondrocyte differentiation to regulate growth plate length independently of PTHrP. J Clin Invest. 115(7):1734–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolupaeva V, Daempfling L, Basilico C. 2013. The B55α regulatory subunit of protein phosphatase 2A mediates fibroblast growth factor-induced p107 dephosphorylation and growth arrest in chondrocytes. Mol Cell Biol. 33(15):2865–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama E, Young B, Nagayama M, Shibukawa Y, Enomoto-Iwamoto M, Iwamoto M, Maeda Y, Lanske B, Song B, Serra R, et al. 2007. Conditional Kif3a ablation causes abnormal hedgehog signaling topography, growth plate dysfunction, and excessive bone and cartilage formation during mouse skeletogenesis. Development. 134(11):2159–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozhemyakina E, Cohen T, Yao TP, Lassar AB. 2009. Parathyroid hormone-related peptide represses chondrocyte hypertrophy through a protein phosphatase 2A/histone deacetylase 4/MEF2 pathway. Mol Cell Biol. 29(21):5751–5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenberg HM. 2003. Developmental regulation of the growth plate. Nature. 423(6937):332–336. [DOI] [PubMed] [Google Scholar]
- Kugimiya F, Kawaguchi H, Kamekura S, Chikuda H, Ohba S, Yano F, Ogata N, Katagiri T, Harada Y, Azuma Y, et al. 2005. Involvement of endogenous bone morphogenetic protein (BMP) 2 and BMP6 in bone formation. J Biol Chem. 280(42):35704–35712. [DOI] [PubMed] [Google Scholar]
- Laurita J, Koyama E, Chin B, Taylor JA, Lakin GE, Hankenson KD, Bartlett SP, Nah HD. 2011. The Muenke syndrome mutation (FgfR3P244R) causes cranial base shortening associated with growth plate dysfunction and premature perichondrial ossification in murine basicranial synchondroses. Dev Dyn. 240(11):2584–2596. [DOI] [PubMed] [Google Scholar]
- Li TF, Dong Y, Ionescu AM, Rosier RN, Zuscik MJ, Schwarz EM, O’Keefe RJ, Drissi H. 2004. Parathyroid hormone-related peptide (PTHrP) inhibits Runx2 expression through the PKA signaling pathway. Exp Cell Res. 299(1):128–136. [DOI] [PubMed] [Google Scholar]
- Liu F, Kohlmeier S, Wang CY. 2008. Wnt signaling and skeletal development. Cell Signal. 20(6):999–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Xu J, Colvin JS, Ornitz DM. 2002. Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes Dev. 16(7):859–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Wan Y, Cao J, Zhu X, Yu J, Zhou R, Yao Y, Zhang L, Zhao H, Li H, et al. 2013. Wnt-mediated reciprocal regulation between cartilage and bone development during endochondral ossification. Bone. 53(2):566–574. [DOI] [PubMed] [Google Scholar]
- Madeline LA, Elster AD. 1995. Suture closure in the human chondrocranium: CT assessment. Radiology. 196(3):747–756. [DOI] [PubMed] [Google Scholar]
- Maeda Y, Schipani E, Densmore MJ, Lanske B. 2010. Partial rescue of postnatal growth plate abnormalities in ihh mutants by expression of a constitutively active PTH/PTHrP receptor. Bone. 46(2):472–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak KK, Kronenberg HM, Chuang PT, Mackem S, Yang Y. 2008. Indian hedgehog signals independently of PTHrP to promote chondrocyte hypertrophy. Development. 135(11):1947–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita T, Wilcox WR, Chan YY, Kawanami A, Bukulmez H, Balmes G, Krejci P, Mekikian PB, Otani K, Yamaura I, et al. 2009. FGFR3 promotes synchondrosis closure and fusion of ossification centers through the MAPK pathway. Hum Mol Genet. 18(2):227–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlashan SR, Haycraft CJ, Jensen CG, Yoder BK, Poole CA. 2007. Articular cartilage and growth plate defects are associated with chondrocyte cytoskeletal abnormalities in Tg737orpk mice lacking the primary cilia protein polaris. Matrix Biol. 26(4):234–246. [DOI] [PubMed] [Google Scholar]
- Murakami S, Balmes G, McKinney S, Zhang Z, Givol D, de Crombrugghe B. 2004. Constitutive activation of MEK1 in chondrocytes causes Stat1-independent achondroplasia-like dwarfism and rescues the Fgfr3-deficient mouse phenotype. Genes Dev. 18(3):290–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata M, Nuckolls GH, Wang X, Shum L, Seki Y, Kawase T, Takahashi K, Nonaka K, Takahashi I, Noman AA, et al. 2011. The primary site of the acrocephalic feature in Apert Syndrome is a dwarf cranial base with accelerated chondrocytic differentiation due to aberrant activation of the FGFR2 signaling. Bone. 48(4):847–856. [DOI] [PubMed] [Google Scholar]
- Nagayama M, Iwamoto M, Hargett A, Kamiya N, Tamamura Y, Young B, Morrison T, Takeuchi H, Pacifici M, Enomoto-Iwamoto M, et al. 2008. Wnt/beta-catenin signaling regulates cranial base development and growth. J Dent Res. 87(3):244–249. [DOI] [PubMed] [Google Scholar]
- Nie X. 2005. Cranial base in craniofacial development: developmental features, influence on facial growth, anomaly, and molecular basis. Acta Odontol Scand. 63(3):127–135. [DOI] [PubMed] [Google Scholar]
- Nilsson O, Parker EA, Hegde A, Chau M, Barnes KM, Baron J. 2007. Gradients in bone morphogenetic protein-related gene expression across the growth plate. J Endocrinol. 193(1):75–84. [DOI] [PubMed] [Google Scholar]
- Ochiai T, Nagayama M, Nakamura T, Morrison T, Pilchak D, Kondo N, Hasegawa H, Song B, Serra R, Pacifici M, et al. 2009. Roles of the primary cilium component polaris in synchondrosis development. J Dent Res. 88(6):545–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco M, Valencia M, Caparros-Martin JA, Mulero F, Goodship JA, Ruiz-Perez VL. 2012. Evc works in chondrocytes and osteoblasts to regulate multiple aspects of growth plate development in the appendicular skeleton and cranial base. Bone. 50(1):28–41. [DOI] [PubMed] [Google Scholar]
- Piao J, Tsuji K, Ochi H, Iwata M, Koga D, Okawa A, Morita S, Takeda S, Asou Y. 2013. Sirt6 regulates postnatal growth plate differentiation and proliferation via Ihh signaling. Sci Rep. 3:3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi H, Jin M, Duan Y, Du X, Zhang Y, Ren F, Wang Y, Tian Q, Wang X, Wang Q, et al. 2014. FGFR3 induces degradation of BMP type I receptor to regulate skeletal development. Biochim Biophys Acta. 1843(7):1237–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rix S, Calmont A, Scambler PJ, Beales PL. 2011. An Ift80 mouse model of short rib polydactyly syndromes shows defects in hedgehog signalling without loss or malformation of cilia. Hum Mol Genet. 20(7):1306–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Perez VL, Blair HJ, Rodriguez-Andres ME, Blanco MJ, Wilson A, Liu YN, Miles C, Peters H, Goodship JA. 2007. Evc is a positive mediator of Ihh-regulated bone growth that localises at the base of chondrocyte cilia. Development. 134(16):2903–2912. [DOI] [PubMed] [Google Scholar]
- Scott JH. 1958. The cranial base. Am J Phys Anthropol. 16(3):319–348. [DOI] [PubMed] [Google Scholar]
- Shum L, Wang X, Kane AA, Nuckolls GH. 2003. BMP4 promotes chondrocyte proliferation and hypertrophy in the endochondral cranial base. Int J Dev Biol. 47(6):423–431. [PubMed] [Google Scholar]
- Song B, Haycraft CJ, Seo HS, Yoder BK, Serra R. 2007. Development of the post-natal growth plate requires intraflagellar transport proteins. Dev Biol. 305(1):202–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spater D, Hill TP, O’Sullivan R J, Gruber M, Conner DA, Hartmann C. 2006. Wnt9a signaling is required for joint integrity and regulation of Ihh during chondrogenesis. Development. 133(15):3039–3049. [DOI] [PubMed] [Google Scholar]
- St-Jacques B, Hammerschmidt M, McMahon AP. 1999. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 13(16):2072–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda H, Takami M, Oguni T, Tsuji T, Yoneda K, Sato H, Ihara N, Itoh T, Kata SR, Mishina Y, et al. 2002. Positional cloning of the gene LIMBIN responsible for bovine chondrodysplastic dwarfism. Proc Natl Acad Sci U S A. 99(16):10549–10554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorogood P. 1988. The developmental specification of the vertebrate skull. Development. 103(Suppl):141–153. [DOI] [PubMed] [Google Scholar]
- Usami Y, Gunawardena AT, Iwamoto M, Enomoto-Iwamoto M. 2016. Wnt signaling in cartilage development and diseases: lessons from animal studies. Lab Invest. 96(2):186–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushijima T, Okazaki K, Tsushima H, Ishihara K, Doi T, Iwamoto Y. 2014. CCAAT/enhancer binding protein β regulates expression of Indian hedgehog during chondrocytes differentiation. PLoS One. 9(8):e104547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veland IR, Awan A, Pedersen LB, Yoder BK, Christensen ST. 2009. Primary cilia and signaling pathways in mammalian development, health and disease. Nephron Physiol. 111(3):39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Lian N, Li L, Moss HE, Perrien DS, Elefteriou F, Yang X. 2009. Atf4 regulates chondrocyte proliferation and differentiation during endochondral ossification by activating Ihh transcription. Development. 136(24):4143–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L, Du X, Li C, Xu X, Chen Z, Su N, Zhao L, Qi H, Li F, Xue J, et al. 2008. A Pro253Arg mutation in fibroblast growth factor receptor 2 (Fgfr2) causes skeleton malformation mimicking human Apert syndrome by affecting both chondrogenesis and osteogenesis. Bone. 42(4):631–643. [DOI] [PubMed] [Google Scholar]
- Young B, Minugh-Purvis N, Shimo T, St-Jacques B, Iwamoto M, Enomoto-Iwamoto M, Koyama E, Pacifici M. 2006. Indian and sonic hedgehogs regulate synchondrosis growth plate and cranial base development and function. Dev Biol. 299(1):272–282. [DOI] [PubMed] [Google Scholar]
- Yuan X, Yang S. 2015. Deletion of IFT80 impairs epiphyseal and articular cartilage formation due to disruption of chondrocyte differentiation. PLoS One. 10(6):e0130618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuasa T, Kondo N, Yasuhara R, Shimono K, Mackem S, Pacifici M, Iwamoto M, Enomoto-Iwamoto M. 2009. Transient activation of Wnt/{beta}-catenin signaling induces abnormal growth plate closure and articular cartilage thickening in postnatal mice. Am J Pathol. 175(5):1993–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Takeda H, Tsuji T, Kamiya N, Rajderkar S, Louie K, Collier C, Scott G, Ray M, Mochida Y, et al. 2015. Generation of Evc2/Limbin global and conditional KO mice and its roles during mineralized tissue formation. Genesis. 53(9):612–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Xie R, Hou W, Wang B, Shen R, Wang X, Wang Q, Zhu T, Jonason JH, Chen D. 2009. PTHrP prevents chondrocyte premature hypertrophy by inducing cyclin-D1-dependent Runx2 and Runx3 phosphorylation, ubiquitylation and proteasomal degradation. J Cell Sci. 122(Pt 9): 1382–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou ZQ, Ota S, Deng C, Akiyama H, Hurlin PJ. 2015. Mutant activated FGFR3 impairs endochondral bone growth by preventing SOX9 downregulation in differentiating chondrocytes. Hum Mol Genet. 24(6):1764–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]