

ABSTRACT
Although melanoma progression and staging is clinically well characterized, a large variation is observed in pathogenesis, progression, and therapeutic responses. Clearly, intrinsic characteristics of melanoma cells contribute to this variety. An important factor, in both progression of the disease and response to therapy, is the tumor-associated vasculature. We postulate that melanoma cells communicate with endothelial cells (ECs) in order to establish a functional and supportive blood supply. We investigated the angiogenic potential of human melanoma cell lines by monitoring the survival of ECs upon exposure to melanoma conditioned medium (CM), under restrictive conditions. We observed long-term (up to 72 h) EC survival under hypoxic conditions upon treatment with all melanoma CMs. No such survival effect was observed with the CM of melanocytes. The CM of pancreatic and breast tumor cell lines did not show a long-term survival effect, suggesting that the survival factor is specific to melanoma cells. Furthermore, all size fractions (up to < 1 kDa) of the melanoma CM induced long-term survival of ECs. The survival effect observed by the < 1 kDa fraction excludes known pro-angiogenic factors. Heat inactivation and enzymatic digestion of the CM did not inactivate the survival factor. Global gene expression and pathway analysis suggest that this effect is mediated in part via the AKT and p38 MAPK/ ERK-1/2 signaling axis. Taken together, these data indicate the production of (a) survival factor/s (< 1 kDa) by melanoma cell lines, which enables long-term survival of ECs and promotes melanoma-induced angiogenesis.
KEYWORDS: Angiogenesis, endothelial cells, hypoxia, melanoma
Introduction
Malignant melanoma is a highly metastatic disease, with an increasing rate of incidence, poor prognosis, and high resistance to therapeutic intervention.1 Although early diagnosis and surgical resection of the primary lesion could significantly improve survival, the high propensity of melanomas to disseminate through intradermal, haematogenous, and lymphatic routes to regional and visceral sites leads to poor prognosis and high mortality rates.2,3 Metastatic melanoma is by large refractory to conventional therapies and until recently patients presenting with advanced disease had low life expectancies, with a median overall survival (OS) of 6 to 9 mo and 5 y survival rates as low as 5–10%.1,4,5 Promising recent therapeutic advances in the treatment of malignant melanoma, such as the use of targeted therapies6-8 and immunological approaches,9-12 have demonstrated substantial clinical benefit and significantly improved survival in patients with advanced disease. These results, while encouraging, highlight the necessity to better characterize disease biology as a considerable fraction of melanoma patients still remain, or become, incurable.
Melanomas evolve through distinct sequential neoplastic transformations and tumor progression is intimately associated with a high degree of angiogenic activity, facilitated by the constitutive expression of multiple growth factors.13,14 Angiogenesis is an imperative feature of melanoma development as evidenced by the correlation of degree of angiogenesis with aggressiveness, risk of recurrence, and clinical outcome.15 Furthermore, the increased expression of proangiogenic ligands and their receptors have been observed in melanomas compared to benign nevi16-18 and elevated levels detected in the blood of melanoma patients.19-21 The clinical relevance and therapeutic potential of targeting angiogenesis have led to the development of several targeting modalities. Despite promising preclinical results, clinical trials evaluating single antiangiogenic agent use as well as combinations with chemotherapy have thus far shown marginal response rates (reviewed in refs.22-24). Translating the targeting of this pivotal pathophysiological feature of melanomas into a curative therapeutic modality has remained challenging, warranting a better insight into the angiogenic cascade of melanomas.
A critical component in eliciting and sustaining the angiogenic phenotype of tumors is hypoxia.25 Heterogeneity in oxygenation is a constant feature of the tumor microenvironment as neoplastic vasculature development cannot keep up with the metabolic demands of proliferating tumor cells, resulting in an architecturally and functionally compromised vasculature. The hypoxic microenvironment of melanomas is associated with tumor progression and metastatic dissemination26,27 and serves as a stimulus for the induction of several pro-angiogenic modulators. Endothelial cells (ECs) are known to undergo cell death in response to adverse stimuli such as hypoxia.28 As EC survival is a crucial step in the angiogenic cascade, we sought to evaluate the angiogenic potential of melanoma cell lines, differing in their degree of aggressiveness and clinical staging,29 by investigating their ability to mediate EC survival under restrictive culture conditions.
Results
Melanoma conditioned medium (CM) prevents endothelial cell (EC) death under restrictive culture conditions
Conditioned medium (CM) derived from six melanoma cell lines, differing in angiogenic profiles,29,30 were tested for their ability to promote EC survival under normoxic and hypoxic conditions. All melanoma CMs were capable of promoting long-term (up to 72 h) human umbilical vein endothelial cell (HUVEC) survival under hypoxia. Basal medium treatment did not prevent cell death as no viable population was observed at 48 and 72 h (Fig. 1a). All melanoma CMs and basal medium were capable of promoting HUVEC survival under normoxic conditions (Fig. S1a). To determine if the observed survival effect was restricted to an EC subtype, we tested the ability of melanoma CMs to promote human dermal microvascular endothelial cell (HMVEC) survival under hypoxia. As with HUVECs, all melanoma CMs were capable of mediating HMVEC survival under normoxic and hypoxic conditions, whereas basal medium only mediated survival under normoxic conditions (Fig. 1b and Fig. S1b). These data suggest that the pro-survival effect induced by melanoma CMs is not restricted to a specific EC subtype. Next, we sought to determine if CM generated from melanoma cells cultured under hypoxic conditions were capable of inducing a similar pro-survival effect in ECs. Conditioned media derived from all melanoma cell lines under hypoxic conditions were capable of sustaining HUVEC (Fig. 1c) and HMVEC (Fig. 1d) survival under hypoxia. Survival of ECs under normoxic conditions are shown in Fig. S1c and d.
Figure 1.

Melanoma-conditioned medium (CM) prevents endothelial cell (EC) death under severe hypoxia. (a) HUVEC and (b) HMVEC survival under hypoxia upon treatment with melanoma CMs collected under normoxic conditions. (c) HUVEC and (d) HMVEC survival under hypoxia upon treatment with melanoma CMs collected under hypoxic conditions. Subconfluent ECs were treated with various melanoma CMs (BLM:  ; M14:
; M14:  ; Mel57:
; Mel57:  ; 530:
; 530:  ; 1F6:
; 1F6:  ; Mel57-VEGF165:
; Mel57-VEGF165:  ) and basal medium (
) and basal medium ( ). Cells were fixed at 24, 48, and 72 h and the percentage of total surviving cells was determined. Normalized cell viability (%) data in (a–d) are expressed relative to treatment with basal medium (DMEM + 10% FBS) at 24 h normoxia and represent mean ± SEM of three independent experiments, conducted in triplicate. *p < 0.05. (e) Morphology of ECs under various treatments at 48 h hypoxia. Image in the dotted black box shows EC morphology with EC culture medium under normoxic conditions. Scale bar, 100 µm.
). Cells were fixed at 24, 48, and 72 h and the percentage of total surviving cells was determined. Normalized cell viability (%) data in (a–d) are expressed relative to treatment with basal medium (DMEM + 10% FBS) at 24 h normoxia and represent mean ± SEM of three independent experiments, conducted in triplicate. *p < 0.05. (e) Morphology of ECs under various treatments at 48 h hypoxia. Image in the dotted black box shows EC morphology with EC culture medium under normoxic conditions. Scale bar, 100 µm.
Although the tested melanoma cell lines have been documented to vary in the expression of angiogenic factors and tumor vascular density in corresponding xenografts,29,31 we did not observe a clear correlation with the extent to which the melanoma CMs promoted EC survival under hypoxia. Near confluent cultures of ECs treated with undiluted melanoma CMs showed the classic cobblestone morphology under restrictive culture conditions (Fig. 1e).
We used the sulphorhodamine B (SRB) assay to measure EC survival in our experiments. As this assay measures cell density based on cellular protein content, we also performed the colorimetric XTT assay that measures metabolically active cells, under the same experimental conditions (Fig. S2). Both SRB and XTT assays yielded similar results and indicated that CM from melanoma cells protects ECs from cell death under hypoxia.
Melanoma conditioned medium (CM) prevents hypoxia induced apoptotic cell death
As non-physiological variances such as hypoxia are known to provoke cell death,28,32 we sought to determine if the observed survival effect was a result of inhibition of apoptotic or necrotic cell death. ECs treated with undiluted and diluted melanoma CMs for 48 h under hypoxia were subjected to a bivariate FACS analysis using FITC-Annexin V and PI. This aided in the discrimination of viable cells (FITC−PI−), early apoptotic (FITC+PI−), and late apoptotic or necrotic cells (FITC+PI+). Representative dot-plots are shown in Fig. 2a. Percentages of apoptotic and necrotic populations at 48 h hypoxia under various treatments are depicted in Fig. 2b. Although over 95% of the cells underwent apoptosis or became necrotic when cultured in basal medium under hypoxia, treatment with melanoma CMs rendered the majority of the cells viable (85% for BLM CM and 83% for 1F6 CM) (Fig. 2a and b). A shift toward apoptosis and necrosis was observed upon 2-fold and 5-fold dilutions of the melanoma CMs. Furthermore, the presence of apoptotic nuclei in treated ECs was visualized by staining with the nuclear dye, YOPRO-1 at 48 h of hypoxia treatment. Melanoma CM-treated ECs under hypoxia did not show the presence of apoptotic nuclei, indicating viable ECs, whereas treatment with the cytotoxic agent Etoposide showed a marked increase in the presence of apoptotic nuclei. Basal medium treated ECs were not viable and showed fewer apoptotic nuclei indicating that cell detachment preceded cell death (Fig. 2c).
Figure 2.

Melanoma conditioned medium (CM) prevents hypoxia induced apoptosis of endothelial cells (EC). (a,b) Bivariate FACS analysis of melanoma CM treated ECs (a) Representative dot plots of ECs treated with undiluted, twice diluted (2*dil) and five times diluted (5*dil) melanoma CMs, and basal medium (DMEM + 10% FBS) at 48 h of hypoxia. Treated cells were stained with Annexin V (X-axis) and Propidium Iodide (PI, Y-axis) and subjected to flow cytometry analysis to determine percentages of apoptotic, necrotic and viable cells. (b) Relative percentages of apoptotic and necrotic ECs with different treatments at 48 h hypoxia. Treatment with diluted melanoma CMs showed a gradual shift toward apoptotic cells. Data are derived from three independent experiments. *p < 0.05. (c) Visualization of apoptotic nuclei in ECs subjected to melanoma CM or basal medium (DMEM+10% FBS) treatments at 48 h of hypoxia. As controls, ECs were treated with EC culture medium with or without 25 µg/mL Etoposide (positive and negative control for apoptosis, respectively) under normoxic conditions. Scale bar, 100 µm.
Melanoma conditioned medium prevents endothelial cell death independent of the VEGF signaling pathway
The role of vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) as potent regulators of angiogenesis has been sturdily established in melanomas and other human tumors.33-35 We therefore evaluated the ability of VEGF and bFGF in sustaining survival of ECs under restrictive culture conditions. Although both mitogens were capable of promoting EC growth under normoxia (Fig. 3a), these factors were unable to rescue ECs from cell death when cultured under hypoxia (Fig. 3b). Next, we evaluated the VEGF profile of melanoma cells by measuring the levels of VEGF in serum-free melanoma conditioned medium (SF-CM). BLM produced the highest levels of VEGF (3.6 ng/106 cells), whereas Mel57 and 1F6 hardly produced any VEGF (Fig. 3c). Additionally, we blocked VEGF signaling by the addition of either VEGF-neutralizing antibody (Fig. 3d) or VEGFR kinase inhibitor (Fig. 3e) to undiluted and 2-fold diluted melanoma CM. Although EC proliferation was reduced upon VEGFR inhibition (Fig. 3e), the ability of melanoma CMs to promote EC survival under hypoxia was not hindered by blocking members of the VEGF signaling pathway, suggesting the involvement of alternate signaling pathways in mediating the observed survival effect.
Figure 3.

Melanomas mediate endothelial cell (EC) survival under hypoxia, independent of the VEGF signaling pathway. (a,b) ECs were treated with 10 ng/mL VEGF ( ) or 200 ng/mL bFGF (
) or 200 ng/mL bFGF ( ) alone, or in combination (
) alone, or in combination ( ). Cell survival was monitored under normoxia (a) and hypoxia (b) for up to 72 h. (c) Levels of VEGF (pg/ 1 × 106 cells) in serum-free melanoma conditioned medium (SF-CM) as detected by ELISA. Data were normalized to the number of cells used to generate the SF-CM and are expressed as mean ± SD of quadruplicate samples. (d, e) Effect of VEGF pathway inhibition on EC survival under hypoxia. ECs were treated with undiluted and twice diluted melanoma conditioned medium in the presence of (d) VEGF neutralizing antibody (0.3 µg/mL) or (e) VEGFR kinase inhibitor (0.3 µg/mL) (undiluted melanoma CM:
). Cell survival was monitored under normoxia (a) and hypoxia (b) for up to 72 h. (c) Levels of VEGF (pg/ 1 × 106 cells) in serum-free melanoma conditioned medium (SF-CM) as detected by ELISA. Data were normalized to the number of cells used to generate the SF-CM and are expressed as mean ± SD of quadruplicate samples. (d, e) Effect of VEGF pathway inhibition on EC survival under hypoxia. ECs were treated with undiluted and twice diluted melanoma conditioned medium in the presence of (d) VEGF neutralizing antibody (0.3 µg/mL) or (e) VEGFR kinase inhibitor (0.3 µg/mL) (undiluted melanoma CM:  ; undiluted melanoma CM with neutralizing antibody/inhibitor:
; undiluted melanoma CM with neutralizing antibody/inhibitor:  ; twice diluted melanoma CM:
; twice diluted melanoma CM:  ; twice diluted melanoma CM with neutralizing antibody/inhibitor:
; twice diluted melanoma CM with neutralizing antibody/inhibitor:  ; and basal medium:
; and basal medium:  ). Normalized cell viability (%) data in (a, b, d, and e) is expressed relative to treatment with basal medium (DMEM + 10% FBS) at 24 h normoxia and represent mean ± SEM of three independent experiments, conducted in triplicate. *p < 0.05.
). Normalized cell viability (%) data in (a, b, d, and e) is expressed relative to treatment with basal medium (DMEM + 10% FBS) at 24 h normoxia and represent mean ± SEM of three independent experiments, conducted in triplicate. *p < 0.05.
Delineation and specificity of the melanoma induced survival effect
As the melanoma CM has the presence of serum, we queried the contribution of serum to the observed survival effect. Additionally, as the growth rates of the tested melanoma cell lines used to generate the CM varied, and therefore the consumption of serum components, we reasoned that the final concentration of serum might differ among the melanoma CMs. Therefore, we prepared serum-free melanoma conditioned medium (SF-CM) and monitored long-term survival of ECs under hypoxic conditions. SF-CMs were capable of inducing a similar survival response in ECs as the CMs, and likewise, this effect was lost upon dilution of the SF-CM (Fig. 4a). In order to further characterize the melanoma-induced survival effect, we size fractionated the SF-CMs with several molecular weight cutoffs. Interestingly, no long-term survival effect was induced by the > 50 kDa fraction, whereas all small molecule fractions tested (< 50 kDa, < 5 kDa, < 3 kDa, < 1 kDa) resulted in a survival effect comparable to that of the unfractionated SF-CM (Fig. 4b). In order to determine if the observed pro-survival effect is an acquired trait during melanoma development, we assayed for the ability of primary and immortalized human melanocytes to promote EC survival under normoxic and hypoxic conditions. Both unfractionated and fractionated serum-free melanocyte conditioned media were not able to induce EC survival under hypoxia (Fig. 4c). Under normoxic conditions, EC survival was observed upon treatment with melanocyte CM (Fig. S3a).
Figure 4.

Delineation of melanoma-induced survival effect. (a) Effect of serum on endothelial cell (EC) survival. ECs were grown to near confluence and treated with undiluted ( ), twice diluted (
), twice diluted ( ), and five times diluted (
), and five times diluted ( ) serum-free melanoma-conditioned medium (SF-CM) and basal medium (
) serum-free melanoma-conditioned medium (SF-CM) and basal medium ( ) under hypoxia for up to 72 h. (b) Effect of melanoma SF-CM size fractionation on EC survival. SF-CMs were size fractionated and the various fractions (> 50 kDa:
) under hypoxia for up to 72 h. (b) Effect of melanoma SF-CM size fractionation on EC survival. SF-CMs were size fractionated and the various fractions (> 50 kDa:  ;< 50 kDa:
;< 50 kDa:  ; < 5 kDa:
; < 5 kDa:  ; < 3 kDa:
; < 3 kDa:  ; < 1 kDa:
; < 1 kDa:  ; and basal medium:
; and basal medium:  ) were added to near confluent EC cultures. (c) Effect of melanocyte conditioned medium on EC survival. Unfractionated and fractionated serum-free melanocyte conditioned medium was added to near confluent EC cultures. Melanocyte conditioned medium was not capable of promoting EC survival under hypoxic conditions (unfractionated hMEL SF-CM:
) were added to near confluent EC cultures. (c) Effect of melanocyte conditioned medium on EC survival. Unfractionated and fractionated serum-free melanocyte conditioned medium was added to near confluent EC cultures. Melanocyte conditioned medium was not capable of promoting EC survival under hypoxic conditions (unfractionated hMEL SF-CM:  ; < 1 kDa hMEL SF-CM:
; < 1 kDa hMEL SF-CM:  ; unfractionated NHEM SF-CM:
; unfractionated NHEM SF-CM:  ; < 1 kDa NHEM SF-CM:
; < 1 kDa NHEM SF-CM:  ; and basal medium:
; and basal medium:  ). (d) Effect of breast and pancreatic cell conditioned medium on EC survival. Size fractionated < 1 kDa fractions of serum-free conditioned medium derived from breast cancer cell lines (MCF-7:
). (d) Effect of breast and pancreatic cell conditioned medium on EC survival. Size fractionated < 1 kDa fractions of serum-free conditioned medium derived from breast cancer cell lines (MCF-7:  , SKBR3:
, SKBR3:  ), pancreatic cancer cell lines (BxPc-3:
), pancreatic cancer cell lines (BxPc-3:  , Panc-1:
, Panc-1:  , MiaPaCa:
, MiaPaCa:  ) and basal medium (
) and basal medium ( ) were added to near confluent EC cultures. Breast and pancreatic SF-CM were not capable of inducing long-term EC survival under hypoxia. Normalized cell viability (%) in (a–d) is expressed relative to treatment with basal medium (serum-free DMEM) at 24 h normoxia. Data represent mean ± SEM of four independent experiments, conducted in triplicate. *p < 0.05.
) were added to near confluent EC cultures. Breast and pancreatic SF-CM were not capable of inducing long-term EC survival under hypoxia. Normalized cell viability (%) in (a–d) is expressed relative to treatment with basal medium (serum-free DMEM) at 24 h normoxia. Data represent mean ± SEM of four independent experiments, conducted in triplicate. *p < 0.05.
Furthermore, we proceeded to determine if the survival effect mediated by the < 1 kDa fraction was specific to melanomas or a common feature among other tumor types. To this end, we prepared serum-free conditioned medium from breast and pancreatic tumor-cell lines, varying in their growth profiles and genomic abnormalities.36,37 Unfractionated (data not shown) and fractionated (< 1 kDa) tumor conditioned media were tested for their ability to promote EC survival under hypoxia. Interestingly, although some cell lines were capable of inducing a short-term survival response in ECs under hypoxia, no long-term survival was seen (Fig. 4d). Thus, the survival effect mediated by the < 1 kDa fraction seems to be specific for melanoma cells. Under normoxic conditions, EC survival was observed with all breast and pancreatic tumor CM tested (Fig. S3b).
Characterization of the melanoma specific survival effect
In order to further characterize the melanoma-specific induction of survival responses in ECs under hypoxia, we subjected the SF-CM < 1 kDa fraction to heat inactivation (Fig. 5a), trypsin (Fig. 5b), and chymotrypsin digestion (data not shown). No difference was observed between pro-survival responses generated by treated and untreated < 1 kDa SF-CM fractions. Additionally, the protein and non-protein fractions of the < 1 kDa SF-CM were separated using acetone precipitation. We observed that although the reconstituted protein pellet was not able to mediate EC survival, the non-protein fraction sustained long-term EC survival under hypoxia. As a control, basal medium was subjected to the same treatment. No EC survival was observed (Fig. 5c). We also performed a BSA back exchange assay, with increasing concentrations of fatty acid-free BSA, to separate the lipid and non-lipid components present in the < 1 kDa SF-CM fraction. Post-treatment, samples were size fractionated to obtain BSA-rich (retentate) and BSA-poor (flowthrough) fractions. We observed EC survival under hypoxia when treated with the BSA-poor fractions (flowthrough) of the 1% and 3% BSA-treated samples. Interestingly, treatment of the < 1 kDa SF-CM fraction with 10% BSA resulted in the removal of the observed survival effect from the flowthrough, suggesting that BSA selectively adsorbs the survival factor/s at a higher concentration (Fig. 5d, e). This selective separation of the melanoma-specific survival factor/s suggests that it might be a lipid/lipophilic molecule.
Figure 5.

Characterization of the melanoma-specific survival effect. (a) Effect of heat treatment on endothelial cell (EC) survival. Size fractionated< 1 kDa fraction of serum-free melanoma conditioned medium (SF-CM) was heat inactivated at 56°C, 80°C and 100°C, respectively, for 30 min. Untreated ( ), heat-inactivated (56°C:
), heat-inactivated (56°C:  ; 80°C:
; 80°C:  ; 100 °C:
; 100 °C:  ) media and basal medium (
) media and basal medium ( ) were added to near confluent EC cultures and long-term survival under hypoxia was monitored. (b) Effect of enzymatic digestion on EC survival. Size fractionated < 1 kDa fraction of melanoma SF-CM was treated with 100 and 200 µg/mL of trypsin for 30 min at 37 °C. Untreated (
) were added to near confluent EC cultures and long-term survival under hypoxia was monitored. (b) Effect of enzymatic digestion on EC survival. Size fractionated < 1 kDa fraction of melanoma SF-CM was treated with 100 and 200 µg/mL of trypsin for 30 min at 37 °C. Untreated ( ), trypsin treated samples (100 µg/mL:
), trypsin treated samples (100 µg/mL:  ; 200 µg/mL:
; 200 µg/mL:  ) and basal medium (
) and basal medium ( ) were added to near confluent EC cultures and monitored for long-term survival under hypoxia. (c) Effect of protein precipitation on EC survival. Size fractionated < 1 kDa fractions of melanoma SF-CM and basal medium were subjected to protein precipitation using acetone. Reconstituted protein and non-protein fractions were added to subconfluent EC cultures and survival under hypoxia was monitored (< 1 kDa SF-CM untreated:
) were added to near confluent EC cultures and monitored for long-term survival under hypoxia. (c) Effect of protein precipitation on EC survival. Size fractionated < 1 kDa fractions of melanoma SF-CM and basal medium were subjected to protein precipitation using acetone. Reconstituted protein and non-protein fractions were added to subconfluent EC cultures and survival under hypoxia was monitored (< 1 kDa SF-CM untreated:  ; < 1 kDa SF-CM protein fraction:
; < 1 kDa SF-CM protein fraction:  ; < 1 kDa SF-CM non-protein fraction:
; < 1 kDa SF-CM non-protein fraction:  ; basal medium:
; basal medium:  ; basal medium protein fraction:
; basal medium protein fraction:  ; basal medium non-protein fraction:
; basal medium non-protein fraction:  ). (d,e) Effect of lipid extraction on EC survival. Size fractionated < 1 kDa SF-CM fractions of melanoma SF-CM were treated with 1%, 3%, or 10% fatty-acid free BSA. BSA-poor (flowthrough) fractions were obtained as described under ‘Materials and Methods’. (d) Untreated (
). (d,e) Effect of lipid extraction on EC survival. Size fractionated < 1 kDa SF-CM fractions of melanoma SF-CM were treated with 1%, 3%, or 10% fatty-acid free BSA. BSA-poor (flowthrough) fractions were obtained as described under ‘Materials and Methods’. (d) Untreated ( ), BSA treated flowthroughs (1%:
), BSA treated flowthroughs (1%:  ; 3%:
; 3%:  ; 10%:
; 10%:  ; ) and basal medium (
; ) and basal medium ( ) were added to subconfluent ECs to monitor cell survival under hypoxia. (e) Morphology of ECs under various treatments at 48 h hypoxia. Scale bar, 100 µm. Normalized cell viability (%) in (a–d) is expressed relative to survival of basal medium (serum-free DMEM) treated ECs at 24 h normoxia and represent mean ± SEM of four independent experiments, conducted in triplicate. *p < 0.05.
) were added to subconfluent ECs to monitor cell survival under hypoxia. (e) Morphology of ECs under various treatments at 48 h hypoxia. Scale bar, 100 µm. Normalized cell viability (%) in (a–d) is expressed relative to survival of basal medium (serum-free DMEM) treated ECs at 24 h normoxia and represent mean ± SEM of four independent experiments, conducted in triplicate. *p < 0.05.
Global changes in gene expression upon treatment with melanoma conditioned medium
To characterize the molecular mechanisms underlying the survival effect, we performed a whole genome gene expression study and analyzed the changes in relative mRNA abundance induced by treating ECs with melanoma CM or basal medium for 12 h under hypoxic and normoxic conditions. The 12 h time point was chosen, as basal medium-treated ECs under hypoxia maintain substantial morphological integrity and cell viability; prolonging the treatment results in extensive cell death. Based on the confidence level of their expression measurements (Affymetrix presence calls), 27,410 probesets passed the set filtering threshold and were used for further statistical analysis. Unsupervised multidimensional scaling (MDS) analysis identifies hypoxic/normoxic growth conditions and melanoma CM treatment as the main sources of variability in our dataset. Samples treated with melanoma CM clustered apart from basal medium-treated control samples, indicating that CM treatment induces a consistent modification in the pattern of expressed genes. This modification appears to be minimally modified by the hypoxic/normoxic culturing condition, consistent with the observation that variations in cell morphology and viability is minimal at the 12 h time point (Fig. 6a and Fig. S4). To identify the genes regulating the observed survival effect, we compared the gene expression profile of ECs treated with melanoma CM to that of ECs grown in basal medium, under hypoxia. Using a random variance t-test, 694 probesets were significantly modulated as a consequence of the melanoma CM treatment. The 694 probesets represent 524 individual transcripts, of which 296 were induced and 228 were repressed. A table reporting all the genes passing the test along with p values, fold change, and per gene FDR estimates is posted as Table S1. Probesets passing the test were clustered and displayed as a Heatmap using the clustering tool in BRB ArrayTools. Additionally, we visualized the expression values of the same probesets in ECs treated for 12 h under normoxia (Fig. S4) and observed a similar pattern of gene expression modulation.
Figure 6.

Global changes in gene expression upon treatment with melanoma conditioned medium (a–c). Changes in relative mRNA abundance induced by treating endothelial cells (EC) with melanoma CM or basal medium for 12 h under hypoxic and normoxic conditions. (a) Unsupervised Multidimensional scaling (MDS) analysis of the 27,410 probesets passing Affymetrix presence calls (green sphere: basal medium, hypoxia; green cube: basal medium, normoxia; blue sphere: melanoma CM hypoxia; blue cube: melanoma CM, normoxia). Probesets were filtered out if called “absent” in > 80% of the samples. (b) Heatmaps of pathways significantly altered as a consequence of melanoma CM treatment at 12 h of hypoxia; KEGG “proteasome” (hsa03050), GO “regulation of glycolysis” (GO:0006110). p < 0.005. (c) Heatmaps of genes constituting pro-apoptotic and pro-survival signaling pathways obtained from pathway analysis (p < 0.005). A consistent downregulation of genes involved in pro-apoptotic signaling and an upregulation of genes involved in pro-survival signaling was observed with melanoma CM-treated ECs, as compared to basal medium treatment, both under hypoxic and normoxic conditions. (d,e) Differential expression of select genes from the microarray experiment was verified using real-time quantitative PCR. ECs were treated with (d) unfractionated or (e) fractionated (< 1 kDa) serum-free melanoma conditioned medium under hypoxia for 12 h. Changes in transcript abundance were normalized to B2M expression and are expressed as log2 ratio relative to basal medium (serum-free DMEM) treatment. *p < 0.05.
The gene expression changes identified appear consistent with the observed survival activity of melanoma CM. Among the genes more differentially expressed, we observed increased expression of transcripts encoding cytokines and other gene products involved in cytokine signaling (CXCL2, CCL2, IL32, A2M, JAK3, STAT6, CXCR7, CASP1), cell metabolism, and survival (INSR, IGF1R, AKT3, MAP2K5, JUNB). A number of transcripts encoding proteins involved in apoptosis and inhibition of transcription (ID2, EID3, FAS) were among the most repressed ones. To gain further insight into the biological functions altered as a consequence of the gene expression changes induced by CM treatment, we performed pathway analysis and interrogated different databases using the pathway analysis tool in BRB ArrayTools. The threshold of determining significant gene sets was set at p < 0.005. Different pathways were significant under the test conditions used including KEGG “proteasome” (hsa03050) showing coordinated downregulation of multiple proteasome subunits, and GO “regulation of glycolysis” (GO:0006110) showing augmented expression of transcripts involved in glucose metabolism and energy production. Heatmaps displaying expression of genes in relevant pathways are shown in Fig. 6b. Furthermore, we clustered and imaged the expression values of genes constituting the “Apoptosis,” “MAPK kinase signaling,” “Insulin signaling,” and “Cytokine-cytokine receptor signaling” pathways of the KEGG database under hypoxic and normoxic conditions. We could clearly identify two main clusters of genes showing consistent modulation upon melanoma CM treatment. Consistent with the observed survival effect induced by melanoma CM, genes involved in the proapoptotic signaling cluster were downregulated in CM-treated group, whereas genes involved in the pro-survival signaling cluster were upregulated (Fig. 6c).
The gene expression changes observed in the microarray experiments were validated by real-time PCR. For all the transcripts tested, the results of the real-time PCR validation experiments were in good agreement with the microarray analysis. Of note, this validation experiment was performed with both unfractionated (Fig. 6d) and fractionated < 1 kDa (Fig. 6e) SF-CM to rule out the effect of growth factors and other large mass bioactive molecules present in unfractionated CM. Gene expression changes under normoxic conditions are provided in Fig. S5.
Melanoma conditioned medium induces a pro-survival signal transduction cascade in endothelial cells
As a robust survival response was generated in hypoxic ECs upon treatment with melanoma conditioned media, we proceeded to investigate the signal transduction events mediating this effect. Caspases are a group of endoproteases that play a central role in the regulation of programmed cell death in response to environmental stresses, such as hypoxia.38 We therefore evaluated the levels of active caspases 3/7 in ECs treated with the < 1 kDa fraction of SF-CMs from a panel of cell lines. At 16 h of hypoxia treatment, levels of active caspases were 2.5-fold higher in melanocyte < 1 kDa SF-CM-treated samples, and about 3-fold higher in breast and pancreatic tumor < 1 kDa SF-CM-treated samples, as compared to the melanoma < 1 kDa SF-CM treatment (Fig. 7a). Furthermore, we evaluated the phosphorylation status of AKT, ERK-1/2, and p38 MAPK that are known to be central mediators of the survival signaling cascade in ECs. We observed pronounced AKT and ERK-1/2 activation upon treatment with both fractionated and unfractionated melanoma SF-CM under hypoxia, as compared to other SF-CMs and basal medium. Consistent with the microarray observations, phosphorylated p38 MAPK levels were downregulated in both fractionated and unfractionated melanoma SF-CM treatments, indicating the induction of a pro-survival signaling cascade (Fig. 7b).
Figure 7.
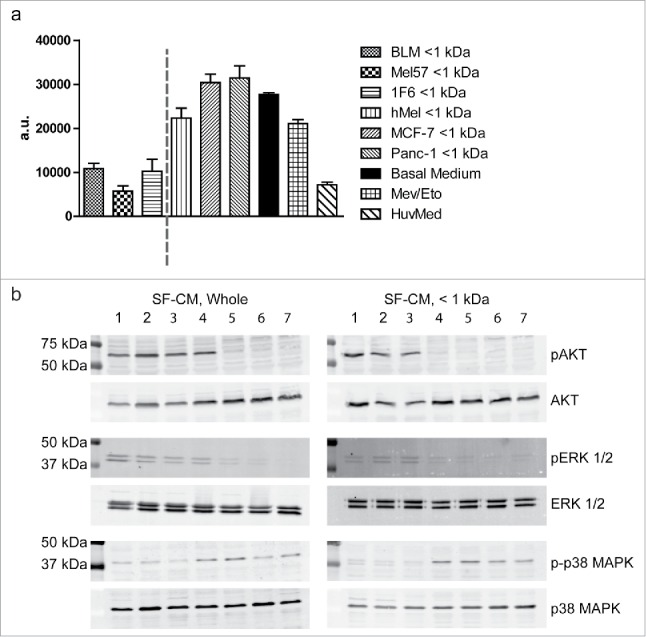
Melanoma conditioned medium induces a pro-survival signal transduction cascade (a) Caspase induction in endothelial cells (ECs) under hypoxia. ECs were treated with < 1 kDa fractions of various serum-free melanoma conditioned media (SF-CM), under hypoxic conditions. ECs cultured with HUVEC medium under normoxic conditions were used as a negative control for apoptosis and the addition of Etoposide (25 µg/mL) served as the positive control for apoptosis induction. Levels of active caspase 3/7 were measured at 16 h of treatment. Data are represented as mean ± SD of three independent experiments, conducted in triplicate. (b) Serum-starved ECs were treated with unfractionated or fractionated (< 1 kDa) SF-CM under hypoxic conditions. Cells were lysed and western blotting was performed to detect total and activated forms of AKT (MW, 60 kDa), ERK-1/2 (MW, 42.44 kDa), and p38 MAPK (MW, 43 kDa). SF-CM treatment lanes: 1. BLM, 2. Mel57, 3. 1F6, 4. PANC-1, 5. MCF-7, 6. hMEL, 7. Basal medium.
Melanoma specific endothelial cell survival effect is mediated via the AKT and p38 MAPK/ ERK-1/2 signaling pathway
To corroborate our observation of activation of AKT and ERK-1/2 signaling when treated with the < 1 kDa fraction of the melanoma SF-CM, we blocked the respective signaling pathways using specific inhibitors. We observed that blocking AKT signaling, using the AKT inhibitor LY294002, reduced long-term EC survival under hypoxic conditions. Alternatively, blocking ERK-1/2 signaling using the inhibitor PD98059 also reduced EC survival under hypoxia. In contrast, inhibiting p38 MAPK signaling with the inhibitor SB203580 resulted in viable ECs similar to untreated < 1 kDa SF-CM (Fig. 8a and b). FACS analysis, performed at 12 h of hypoxia treatment, corroborated these results (Fig. 8c and d). These results suggest that the survival specific factor produced by the melanoma cells induces a pro-survival effect in ECs under restrictive hypoxic conditions, and is mediated by the AKT and p38 MAPK/ ERK-1/2 signaling pathway.
Figure 8.

Inhibition of the melanoma-specific survival effect (a,b). Effect of pathway inhibitors on endothelial cell (EC) survival. ECs were treated with < 1 kDa fractions of (a) Mel57 or (b) 1F6 SF-CM, in the presence of PI3 kinase inhibitor (LY294002), MEK inhibitor (PD98059) or p38 MAPK inhibitor (SB203580) and long-term survival under hypoxia was monitored (< 1 kDa SF-CM:  ; + LY294002, 50 µM :
; + LY294002, 50 µM :  ; + PD98059, 20 µM:
; + PD98059, 20 µM:  ; + SB203580, 10 µM:
; + SB203580, 10 µM:  ). Normalized cell viability (%) in (a,b) is expressed relative to survival of basal medium (serum-free DMEM) treated ECs at 24 h normoxia and represent mean ± SEM of three independent experiments, conducted in triplicate. All inhibitor treated samples were compared to treatment with melanoma SF-CM < 1 kDa treatment. *p < 0.05. (c,d) Bivariate FACS analysis of ECs treated with pathway inhibitors. (c) Representative dot plots of ECs treated with < 1 kDa melanoma SF-CM fraction with or without pathway inhibitors, and basal medium (serum-free DMEM) at 12 h of hypoxia. Treated cells were stained with Annexin V (X-axis) and Propidium Iodide (PI, Y-axis) and subjected to flow cytometry analysis to determine percentages of apoptotic, necrotic, and viable cells. (d) Relative percentages of apoptotic and necrotic ECs with different treatments at 12 h hypoxia. Data are derived from three independent experiments. *p < 0.05.
). Normalized cell viability (%) in (a,b) is expressed relative to survival of basal medium (serum-free DMEM) treated ECs at 24 h normoxia and represent mean ± SEM of three independent experiments, conducted in triplicate. All inhibitor treated samples were compared to treatment with melanoma SF-CM < 1 kDa treatment. *p < 0.05. (c,d) Bivariate FACS analysis of ECs treated with pathway inhibitors. (c) Representative dot plots of ECs treated with < 1 kDa melanoma SF-CM fraction with or without pathway inhibitors, and basal medium (serum-free DMEM) at 12 h of hypoxia. Treated cells were stained with Annexin V (X-axis) and Propidium Iodide (PI, Y-axis) and subjected to flow cytometry analysis to determine percentages of apoptotic, necrotic, and viable cells. (d) Relative percentages of apoptotic and necrotic ECs with different treatments at 12 h hypoxia. Data are derived from three independent experiments. *p < 0.05.
Discussion
In this study, we analyzed the angiogenic potential of a panel of human melanoma cell lines with respect to their ability to promote EC survival under tumor-associated hypoxic conditions. Although the melanoma cell lines differed in their clinical staging, in vitro and in vivo growth properties,29 all tested melanoma conditioned supernatants were capable of eliciting a survival response in ECs under hypoxic conditions. Moreover, the survival response was not EC subtype specific as melanoma CM-treated HMVECs were also capable of long-term survival under hypoxia. The survival response was determined to be regulated by prevention of apoptotic cell death. Interestingly, however, all diluted melanoma CMs induced a shift in the EC populations to an apoptotic phenotype, suggesting dose dependency. We also observed that all size fractions (up to and including < 1 kDa) of the melanoma conditioned media were capable of generating pro-survival responses in ECs. Furthermore, this effect seemed to be an attribute specific to melanoma pathophysiology as neither melanocytes nor aggressive breast and pancreatic cell lines were capable of preventing EC apoptosis under hypoxia.
The clinical relevance and therapeutic potential of targeting angiogenesis have led to the development of several targeting modalities including neutralization of pro-angiogenic ligands using monoclonal antibodies, soluble decoy traps to inhibit receptor signaling, broad blocking with tyrosine kinase inhibitors, and vascular disrupting agents that target the established tumor vasculature. Much of these efforts in melanoma have focused on VEGF and bFGF.22,23 The role of both mitogens in supporting melanoma growth in vivo has been established.29,31,39 Graeven et al. demonstrated that bFGF is critical for melanoma growth in vivo, whereas VEGF is dispensible.40 Additionally, several in vivo studies report the role of these mitogens in promoting survival of tumor endothelium.41,42 However, VEGF and bFGF were not capable of inducing EC survival under hypoxic conditions. Moreover, the melanoma cell lines tested vary in VEGF expression profile.31 Thus, although BLM produced the highest levels of VEGF, Mel57 and 1F6 cell lines recorded low VEGF production. Furthermore, inhibition of the VEGF signaling axis, either by ligand neutralization or receptor blocking, in melanoma CM-treated ECs did not induce apoptosis under hypoxia. These data suggest that angiogenic events during melanoma development are not restricted to VEGF and bFGF signaling.
As the < 1 kDa melanoma-specific fraction excludes known angiogenic factors, we attempted to further characterize this factor/s. Heat inactivation, trypsin, and chymotrypsin digestion did not alter the survival promoting attributes of the melanoma-specific fraction. Global changes in endothelial gene expression were visualized by comparing melanoma CM treatment and basal medium treatment. Of the transcripts most differentially regulated, proangiogenic modulators such as chemokine (C-C motif) ligand 2 (CCL2), angiopoetin-like-4 (ANGPTL4), v-akt murine thymoma viral oncogene homolog 3 (AKT3), and insulin receptor (INSR) were upregulated in the melanoma CM-treated samples. Furthermore, genes involved in pro-apoptotic cascade such as (FAS) and (ID2) were downregulated upon melanoma CM treatment. The gene expression data were further verified with the small molecule fraction treatment, and similar changes in transcript abundance were observed.
In response to soluble factors secreted by tumor cells, several signaling cascades are activated in angiogenic ECs. Central components of the activated signaling pathways in ECs include the mitogen-activated protein kinases (MAPK) and the serine/threonine-specific protein kinase AKT that regulate diverse cellular functions including cell growth, proliferation, survival, and migration.43 A previous study has demonstrated the activation of the MAPK-ERK and PI3-K/AKT survival pathways in immortalized and primary rat brain ECs upon treatment with CM derived from human melanoma cells.44 These signaling events contributed to an increase in proliferative and migratory responses of ECs and could be attenuated with specific pathway inhibitors. The signal transduction pathways triggered by the melanoma-specific fraction in our study, also showed the involvement of the AKT and p38 MAPK/ ERK-1/2 signaling axis. Under hypoxic conditions, activation of AKT and ERK-1/2 was visible in all melanoma CM-treated samples. The levels of active caspases, the downstream effectors of apoptosis, were also lower in melanoma CM-treated samples as compared to other conditioned media treatments.
Taken together, our data demonstrate the presence of small molecule melanoma-specific factor/s capable of promoting long-term EC survival under hypoxic conditions. This observation draws a parallel with a previous study where a small molecule fraction (< 3 kDa) of CM from malignant colon cancer cells was capable of preventing EC apoptosis.45 Elucidation and further characterization of the survival-specific molecule/s could assist in the development of new anti-angiogenic therapies to target the aggressive attributes of malignant melanoma.
Materials and methods
Reagents
Tissue culture reagents, unless otherwise specified, were obtained from Biowhittaker (Walkersville, MD). Human recombinant VEGF, bFGF and epidermal growth factor (EGF) were purchased from PrepoTech (Rocky Hill, NJ). Sugen 5416 was obtained from Cayman Chemical Company (Ann Arbor, MI) and human VEGF neutralizing antibody was purchased from R&D Systems Europe (Abingdon, UK). Inhibitors LY294002, PD98059, and SB203580 were purchased from Cell Signaling Technology, Inc. (Bioke, Leiden, NL). Antibodies against Akt, Phospho-Akt (S473), p44/42 MAPK (ERK1/2), Phospho-p44/42 MAPK (ERK1/2), p38 MAPK, and Phospho- p38 MAPK (Thr180/Tyr182) were from Cell Signaling and mouse anti-human β actin monoclonal antibody was from Abcam (Cambridge, UK). Gelatin was obtained from Sigma-Aldrich and Fibronectin from Roche Diagnostics. SDS-PAGE reagents were obtained from BioRad (Hercules, CA). TaqMan Gene Expression assays were purchased from Applied Biosystems (Carlsbad, CA), TRIzol reagent and First Strand cDNA synthesis kits were purchased from Invitrogen (Carlsbad, CA). All other reagents were from Sigma-Aldrich Chemie B.V. (Zwijndrecht, NL), unless stated otherwise.
Cell lines and culture conditions
The five human melanoma cell lines BLM, M14, Mel57, 530, and 1F6 were kindly donated by Dr. van Muijen (Department of Pathology, University of Nijmegen, The Netherlands) and were maintained in Dulbecco's modified Eagle's medium (DMEM) with glutamine supplemented with 10% FBS. The modified human melanoma cell line, Mel57-VEGF165 was maintained in DMEM supplemented with 10% FBS and 1 mg/mL G418. Normal human epidermal neonatal melanocytes (NHEM-neo) were maintained in MBM-4 medium supplemented with the MGM-4 Bulletkit (Clonetics Melanocyte Cell Systems, Lonza Benelux BV, Breda, NL). Immortalized human melanocytes (Hermes 1), a gift from Dr. Sviderskaya (Department of Basic Medical Sciences, St George's Hospital Medical School, London, UK), were grown in RPMI medium with glutamine supplemented with 10% FBS, 12-O-tetradecanoylphorbol-13-acetate (TPA, 200 nM), cholera toxin (200 pM), human stem cell factor (SCF, 10 ng/mL), and endothelin 1 (10 nM) and maintained under conditions of 10% CO2, as previously described.46 Human breast carcinoma cells (MCF-7, Cama-1 and SKBr3) were gifts from Dr. M. Schutte (Department of Medical Oncology, Josephine Nefkens Institute, Erasmus MC, The Netherlands) and were cultured in RPMI medium with glutamine supplemented with 10% FBS. Pancreatic cancer cells (PANC-1, MIAPaCa-2, and BxPC-3) were donated by Dr. W. Dinjens (Department of Pathology, Josephine Nefkens Institute, Erasmus MC, The Netherlands) and were grown in RPMI 1640 medium with glutamine supplemented with 10% FBS.
Primary cultures of HUVEC were established by isolating ECs from umbilical cords with collagenase digestion, as described.47 Primary EC cultures from eight separate donors were used for this study. Adult Human Dermal Microvascular Endothelial cells (HMVECs) were obtained from Clonetics (Lonza Benelux BV, Breda, NL). EC's were routinely cultured in Human Endothelial-SFM (Invitrogen) supplemented with 20% heat inactivated new born calf serum, 10% heat inactivated human serum, 20 ng/mL bFGF, and 100 ng/mL EGF, on 0.1% gelatin coated flasks. All experiments and assays were performed on fibronectin (10 µg/mL)-coated plates, with ECs between passages 3 and 6. NHEM-neo, ECs, and tumor cells were routinely cultured in a well-humidified incubator (20% O2, 5% CO2 37°C; referred throughout as normoxia) and passaged when confluent. To inflict profound cell death within 24–48 h, severe hypoxic treatments were conducted by placing cultures in a well-humidified hypoxic chamber (Pro-ox-110, Biospherix, Redfield, NY) maintained at 1% O2, 1% CO2, 37°C (referred throughout as hypoxia).
Preparation and fractionation of melanoma conditioned medium (CM)
Tumor cells and melanocytes were maintained in standard culture medium. Upon 70–80% confluency, the cells were washed twice and CM was prepared by incubation in either DMEM supplemented with 10% FBS or serum-free DMEM to generate CM and SF-CM, respectively, under normoxic or hypoxic conditions. For all experiments where CM was used, DMEM+ 10% FBS was used as the control and for experiments with SF-CM, serum-free DMEM was used as the control. Controls are referred throughout as basal medium. The conditioned media were collected after 96 h, centrifuged at 1,500 rpm for 5 min to remove cellular components and stored at −20°C. Where applicable, conditioned media were size fractionated using ultrafiltration devices with specific molecular weight cutoffs of 50,000 Da, 5,000 Da, 3,000 Da (Amicon Inc., Beverly, MA), and 1,000 Da (Microsep, Pall Corporation, Ann Harbor, MI) and stored at −20°C until use.
Cell survival assay
ECs were seeded at a density of 6 × 103 cells per well in 96-well cluster plates and grown to 60% confluence. After 24 h in endothelial culture medium, tumor CM was added to cultured cells, and incubations continued for 24–72 h under normoxic or hypoxic conditions. After 24–72 h, cells were fixed with 10% trichloroacetic acid, washed under tap water, and stained with SRB. After washing with 1% acetic acid, plates were dried at 50°C and the dye was solubilized in 10 mM Tris buffer. The absorbance was measured using a microplate reader (Victor 1420, Wallac, Turku, Finland) at 510 nm. All reported EC survival data have been normalized to EC survival with basal medium treatment at 24 h under normoxic conditions and are denoted as normalized cell viability (%).
Cell morphology
ECs were seeded at a density of 3 × 104 cells per well in 24-well cluster plates and grown to 60% confluence. After 24 h in endothelial culture medium, CM was added to the ECs, and incubations continued for 24–72 h under normoxic or hypoxic conditions. After 24–72 h, cells were analyzed microscopically using an Axiovert 100 M inverted microscope with a 10X/0.30 Plan-Neofluar objective (Carl Zeiss) and images were captured with an Axiocam MRC digital camera using AxioVision 4.5 software (Carl Zeiss B.V., Sliedrecht, NL).
Flow cytometry
ECs cultured under hypoxic conditions in the presence of melanoma CM or basal medium were stained with FITC Annexin V/Dead Cell Apoptosis Kit (Molecular Probes, Carlsbad, CA), as per the manufacturer's instructions, and analyzed by flow cytometry (FACScan, Becton Dickinson, Palo Alto, CA). Data analysis was performed using the FlowJo software (TreeStar Inc., Ashland, OR).
Detection of adherent apoptotic cells
Yo-Pro-1 (Molecular Probes) staining was used for microscopic detection of adherent apoptotic cells. Endothelial culture medium supplemented with 25 µg/mL Etoposide was used as a positive control for apoptosis. Briefly, after 24 h of incubation with melanoma CM or basal medium under hypoxia, detached cells were removed and ECs were incubated with 0.5 µM Yo-Pro-1 for 15 min at 37°C. Fluorescence was visualized using an Axiovert 100 M inverted microscope with a 10X/0.30 Plan-Neofluar objective (Carl Zeiss) and an ORCA II ER camera (Hamamatsu Photonics Systems).
Growth factors and inhibitors
Recombinant human VEGF (10 ng/mL) or bFGF (200 ng/mL) was added to ECs and survival under normoxia and hypoxia was monitored. Alternatively, VEGFR kinase inhibitor Sugen 5416 (5 µM) or VEGF-neutralizing antibody (0.3 µg/mL) was added to the melanoma CM. For pathway inhibition studies, ECs were treated with LY294002 (50 µM), PD98059 (20 µM), and SB203580 (10 µM) for 12 h in the presence of melanoma-CM and cell viability was analyzed by flow cytometry. Alternatively, EC survival was assayed up to 72 h.
ELISA
To determine the presence of VEGF in the melanoma supernatants, a commercial ELISA kit for human VEGF165 (Quantikine, R&D Systems) was used, and assays were performed according to the manufacturer's specifications.
Heat inactivation of the melanoma conditioned media
Conditioned media were heat inactivated for 30 min at 56, 80, and 100°C.
Trypsin and chymotrypsin digestion of the melanoma conditioned media
Conditioned media were subjected to trypsin (100 and 200 μg/mL) and chymotrypsin (100 and 200 μg/mL) treatments (Sigma-Aldrich). Digestions were carried out at 37°C for 30 min and the reaction was terminated by the addition of 1 mg/mL soybean trypsin inhibitor (Sigma-Aldrich) or L-1-Tosylamide-2-phenylethyl chloromethyl ketone (TPCK)-treated inhibitor. Alternatively, for smaller size fractions (< 1 kDa) of CM treated with trypsin and chymotrypsin, the reaction was terminated by the removal of trypsin and chymotrypsin with a 3 kDa cutoff fractionation column (MW trypsin = 24 kDa, MW chymotrypsin = 25 kDa).
Acetone precipitation
In order to determine if the survival factor/s were present in the protein or non-protein phase, conditioned media were treated with ice-cold acetone in a 1:2 (v/v) ratio. The reaction was vortexed, incubated for 1 h at −20°C and pelleted by spinning at 14,000 rpm for 10 min at 4°C. The upper aqueous non-protein phase was collected and evaporated to a film in a rotary evaporator and subsequently dissolved in water. The air-dried protein pellet was reconstituted in serum-free basal medium. Both fractions were filter sterilized and assayed for their ability to promote EC survival under normoxic and hypoxic conditions.
BSA back exchange assay
Bovine serum albumin (BSA) back exchange assay was performed to separate the lipid and non-lipid components of the < 1 kDa SF-CM fraction. Briefly, the < 1 kDa SF-CM fractions were treated with 1, 3, or 10% (w/v) fatty acid free BSA (Sigma-Aldrich) for 30 min at 37°C. Treated samples were fractionated using a 30 kDa cutoff fractionation column (MW BSA = 66 kDa) to obtain the BSA rich fraction (retentate) and BSA poor fraction (flowthrough). Both fractions were filter sterilized and assayed for their ability to promote EC survival under normoxic and hypoxic conditions. Cell viability was measured using the XTT (2,3-Bis-(2-Methoxy-4-Nitro-5-Sulfophenyl)-2H-Tetrazolium-5-Carboxanilide) assay, as described elsewhere.
RNA isolation
Total RNA was extracted using TRIzol (Invitrogen) following manufacturer's instructions. Purified RNA was quantified by spectophotometric analysis using a NanoDrop 2000 (Thermo Fisher Scientific Inc., CA) and analyzed on an Agilent 2100 Bioanalyzer (Agilent Technologies, CA). Only RNA with RIN >8.0 were used for the microarray experiment.
Affymetrix GeneChips
In this study, we used the Affymetrix HG-U133-plus 2.0 GeneChip (Affymetrix Inc., Santa Clara, CA). Target synthesis was performed using 5 µg total RNA as template, as described in the Affymetrix Gene Expression Manual. GeneChips were washed and stained using the Affymetrix fluidic station 430 and analyzed using Affymetrix 3000 7 G GeneChip scanner. Gene expression values were summarized from probesets using RMA as implemented in Affymetrix Gene Expression Console. The same software was used to quality control (QC) the GeneChips. Chips not meeting QC criteria were excluded from further analysis. RMA expression summaries were merged with the Affymetrix MAS 5.0 Presence calls into a single matrix, filtered in Excel and imported into BRB ArrayTools for further analysis.48 Probesets were filtered out if called “absent” in > 80% of the samples. Correlation among samples was assessed using the MDS and the hierarchical clustering tools in BRB ArrayTools. We identified genes that are differentially expressed among the two classes using a random variance t-test as implemented in the class comparison tool in BRB ArrayTools (developed by Dr. Richard Simon and BRB-ArrayTools Development Team). An estimate of the associated false discovery rate (FDR) was computed per gene using the method of Benjamini and Hochberg. Genes were considered significant if p < 0.001. A table reporting all the genes passing the test along with p values, fold change, and per gene FDR estimates is posted under supplementary information. Gene clustering and visualization were performed using BRB ArrayTools. Pathway analysis was performed using the GeneSet Class Comparison tool in BRB ArrayTools. For GeneSet enrichment, Gene Ontology, BioCarta, and KEGG databases were independently queried. The microarray data from this publication have been submitted to the GEO database (http://www.ncbi.nlm.nih.gov/geo/) and assigned the identifier GSE33115.
Real-time RT-PCR
Microarray gene expression data were validated by measuring differential gene regulation in treated ECs, from three separate donors, using real-time PCR. Briefly, total RNA extraction was performed with TRIzol Reagent (Invitrogen), according to the manufacturer's instructions. Reverse transcription was performed with 2 µg total RNA using the First-Strand cDNA Synthesis Kit (Invitrogen). 50 ng of cDNA was used for the qPCR reaction. Quantitative PCR was performed in duplicates, using the iCycler (Bio-Rad Laboratories, Munich, Germany) with specific TaqMan Gene Expression assays (Applied Biosystems). The reactions were incubated in a 96-well optical plate at 50°C for 2 min (UDG incubation) and 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 60 sec.
Active caspases
Levels of active caspase-3/7 in treated ECs were assayed using the commercially available SensoLyte Homogeneous AnaRed Caspase-3/7 Assay Kit (AnaSpec Inc., Fremont, CA), according to manufacturer's instructions. Briefly ECs were seeded at a density of 6 × 103 cells/ well in 96-well black cluster plates and grown to 60% confluence. Various SF-CMs were added to cultured cells and incubated at 37°C under normoxic or hypoxic conditions. At 16 h and 24 h of treatment, caspase 3/7 substrate solution was added to the cells, and incubated in the dark for 1 min with gentle shaking. Fluorescence intensity was measured at 635 nm using a fluorescence microplate reader.
Western blot analysis
To determine pathway effectors of the survival response, sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis was performed. Briefly, subconfluent EC cultures were washed twice and serum starved for 12 h in medium containing 0.5% serum, followed by treatment with fractionated or unfractionated conditioned media. Treated ECs were rinsed twice with ice-cold PBS, scraped, and pelleted. Lysis was conducted in the presence of protease and phosphatase inhibitor cocktail tablets (Roche) and the protein concentration was measured with Coomassie Plus Reagent (Pierce). Equal amounts of proteins were loaded on 12% gels, electrophoresed, and transferred to polyvinylidene difluoride membranes (BioRad). Membranes were blocked for 1 h at room temperature with 5% nonfat dried milk (BioRad) in PBS/ 0.05% Tween-20, followed by overnight incubations with primary antibodies against total AKT (rabbit polyclonal, 1:1000 dilution), phospho-AKT (Ser473) (mouse monoclonal, 1:1000 dilution), total ERK-1/2 (rabbit polyclonal, 1:1000 dilution), phospho-ERK (mouse monoclonal, 1:1000 dilution), total p38 MAPK (rabbit polyclonal, 1:1000 dilution), and Phospho-p38 MAPK (Thr180/Tyr182) (mouse monoclonal, 1:2000 dilution). Post-washing, the membranes were incubated with IRDYE labeled secondary antibodies (LI-COR) for 1 h at room temperature and scanned using the Odyssey Infrared Imaging System (LI-COR Biosciences).
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This study was supported in part by the Stichting Erasmus Heelkundig Kankeronderzoek (SEHK) and the EORTC Melanoma Group.
References
- 1.Eggermont AM, Spatz A, Robert C. Cutaneous melanoma. Lancet 2014; 383:816-27; PMID:24054424; http://dx.doi.org/ 10.1016/S0140-6736(13)60802-8 [DOI] [PubMed] [Google Scholar]
- 2.Cummins DL, Cummins JM, Pantle H, Silverman MA, Leonard AL, Chanmugam A. Cutaneous malignant melanoma. Mayo Clinic proceedings 2006; 81:500-7; PMID:16610570; http://dx.doi.org/ 10.4065/81.4.500 [DOI] [PubMed] [Google Scholar]
- 3.Hamid O, Boasberg PD, Rosenthal K, O'Day SJ. Systemic treatment of metastatic melanoma: new approaches. J Surg Oncol 2011; 104:425-9; PMID:21858838; http://dx.doi.org/ 10.1002/jso.22034 [DOI] [PubMed] [Google Scholar]
- 4.Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, Buzaid AC, Cochran AJ, Coit DG, Ding S et al.. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol 2009; 27:6199-206; PMID:19917835; http://dx.doi.org/ 10.1200/JCO.2009.23.4799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schadendorf D, Fisher DE, Garbe C, Gershenwald JE, Grob J-J, Halpern A, Herlyn M, Marchetti MA, McArthur G, Ribas A et al.. Melanoma. Nat Rev Dis Primers. 2015; 1:15003; PMID:27188223; http://dx.doi.org/21639808 10.1038/nrdp.2015.3 [DOI] [PubMed] [Google Scholar]
- 6.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M et al.. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011; 364:2507-16; PMID:21639808; http://dx.doi.org/ 10.1056/NEJMoa1103782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L et al.. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 2015; 372:30-9; PMID:25399551; http://dx.doi.org/ 10.1056/NEJMoa1412690 [DOI] [PubMed] [Google Scholar]
- 8.Carvajal RD, Antonescu CR, Wolchok JD, Chapman PB, Roman RA, Teitcher J, Panageas KS, Busam KJ, Chmielowski B, Lutzky J et al.. KIT as a therapeutic target in metastatic melanoma. JAMA 2011; 305:2327-34; PMID:21642685; http://dx.doi.org/ 10.1001/jama.2011.746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC et al.. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711-23; PMID:20525992; http://dx.doi.org/ 10.1056/NEJMoa1003466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E et al.. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015; 372:320-30; PMID:25399552; http://dx.doi.org/ 10.1056/NEJMoa1412082 [DOI] [PubMed] [Google Scholar]
- 11.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M et al.. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med 2015; 372:2521-32; PMID:25891173; http://dx.doi.org/ 10.1056/NEJMoa1503093 [DOI] [PubMed] [Google Scholar]
- 12.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P et al.. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med 2015; 373:23-34; PMID:26027431; http://dx.doi.org/ 10.1056/NEJMoa1504030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Basu B, Biswas S, Wrigley J, Sirohi B, Corrie P. Angiogenesis in cutaneous malignant melanoma and potential therapeutic strategies. Expert Rev Anticancer Ther 2009; 9:1583-98; PMID:19895243; http://dx.doi.org/ 10.1586/era.09.135 [DOI] [PubMed] [Google Scholar]
- 14.Mahabeleshwar GH, Byzova TV. Angiogenesis in melanoma. Semin Oncol 2007; 34:555-65; PMID:18083379; http://dx.doi.org/ 10.1053/j.seminoncol.2007.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kashani-Sabet M, Sagebiel RW, Ferreira CM, Nosrati M, Miller JR 3rd. Tumor vascularity in the prognostic assessment of primary cutaneous melanoma. J Clin Oncol 2002; 20:1826-31; PMID:11919240; http://dx.doi.org/ 10.1200/JCO.2002.07.082 [DOI] [PubMed] [Google Scholar]
- 16.Mehnert JM, McCarthy MM, Jilaveanu L, Flaherty KT, Aziz S, Camp RL, Rimm DL, Kluger HM. Quantitative expression of VEGF, VEGF-R1, VEGF-R2, and VEGF-R3 in melanoma tissue microarrays. Hum Pathol 2010; 41:375-84; PMID:20004943; http://dx.doi.org/ 10.1016/j.humpath.2009.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reed JA, McNutt NS, Albino AP. Differential expression of basic fibroblast growth factor (bFGF) in melanocytic lesions demonstrated by in situ hybridization. Implications for tumor progression. Am J Pathol 1994; 144:329-36; PMID:8311116 [PMC free article] [PubMed] [Google Scholar]
- 18.Nurnberg W, Tobias D, Otto F, Henz BM, Schadendorf D. Expression of interleukin-8 detected by in situ hybridization correlates with worse prognosis in primary cutaneous melanoma. J Pathol 1999; 189:546-51; PMID:10629556; http://dx.doi.org/ [DOI] [PubMed] [Google Scholar]
- 19.Tas F, Duranyildiz D, Oguz H, Camlica H, Yasasever V, Topuz E. Circulating serum levels of angiogenic factors and vascular endothelial growth factor receptors 1 and 2 in melanoma patients. Melanoma Res 2006; 16:405-11; PMID:17013089; http://dx.doi.org/ 10.1097/01.cmr.0000222598.27438.82 [DOI] [PubMed] [Google Scholar]
- 20.Ugurel S, Rappl G, Tilgen W, Reinhold U. Increased serum concentration of angiogenic factors in malignant melanoma patients correlates with tumor progression and survival. J Clin Oncol 2001; 19:577-83; PMID:11208853 [DOI] [PubMed] [Google Scholar]
- 21.Mouawad R, Spano JP, Comperat E, Capron F, Khayat D. Tumoural expression and circulating level of VEGFR-3 (Flt-4) in metastatic melanoma patients: correlation with clinical parameters and outcome. Eur J Cancer 2009; 45:1407-14; PMID:19157860; http://dx.doi.org/ 10.1016/j.ejca.2008.12.015 [DOI] [PubMed] [Google Scholar]
- 22.Zaki KA, Basu B, Corrie P. The role of angiogenesis inhibitors in the management of melanoma. Curr Top Med Chem 2012; 12:32-49; PMID:22196268; http://dx.doi.org/ 10.2174/156802612798919240 [DOI] [PubMed] [Google Scholar]
- 23.Nikolaou V, Stratigos A, Bafaloukos D, Katsambas A. Antiangiogenic and antiapoptotic treatment in advanced melanoma. Clin Dermatol 2013; 31:257-63; PMID:23608445; http://dx.doi.org/ 10.1016/j.clindermatol.2012.08.018 [DOI] [PubMed] [Google Scholar]
- 24.Emmett MS, Dewing D, Pritchard-Jones RO. Angiogenesis and melanoma - from basic science to clinical trials. Am J Cancer Res 2011; 1:852-68; PMID:22016833 [PMC free article] [PubMed] [Google Scholar]
- 25.Liao D, Johnson RS. Hypoxia: a key regulator of angiogenesis in cancer. Cancer Metastasis Rev 2007; 26:281-90; PMID:17603752; http://dx.doi.org/ 10.1007/s10555-007-9066-y [DOI] [PubMed] [Google Scholar]
- 26.Rofstad EK, Rasmussen H, Galappathi K, Mathiesen B, Nilsen K, Graff BA. Hypoxia promotes lymph node metastasis in human melanoma xenografts by up-regulating the urokinase-type plasminogen activator receptor. Cancer Res 2002; 62:1847-53; PMID:11912164 [PubMed] [Google Scholar]
- 27.Rofstad EK, Halsor EF. Hypoxia-associated spontaneous pulmonary metastasis in human melanoma xenografts: involvement of microvascular hot spots induced in hypoxic foci by interleukin 8. Br J Cancer 2002; 86:301-8; PMID:11870523; http://dx.doi.org/ 10.1038/sj.bjc.6600052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsushita H, Morishita R, Nata T, Aoki M, Nakagami H, Taniyama Y, Yamamoto K, Higaki J, Yasufumi K, Ogihara T. Hypoxia-induced endothelial apoptosis through nuclear factor-kappaB (NF-kappaB)-mediated bcl-2 suppression: in vivo evidence of the importance of NF-kappaB in endothelial cell regulation. Circ Res 2000; 86:974-81; PMID:10807870; http://dx.doi.org/ 10.1161/01.RES.86.9.974 [DOI] [PubMed] [Google Scholar]
- 29.Westphal JR, van't Hullenaar RG, van der Laak JA, Cornelissen IM, Schalkwijk LJ, van Muijen GN, Wesseling P, de Wilde PC, Ruiter DJ, de Waal RM. Vascular density in melanoma xenografts correlates with vascular permeability factor expression but not with metastatic potential. Br J Cancer 1997; 76:561-70; PMID:9303353; http://dx.doi.org/ 10.1038/bjc.1997.427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Muijen GN, Cornelissen LM, Jansen CF, Figdor CG, Johnson JP, Brocker EB, Ruiter DJ. Antigen expression of metastasizing and non-metastasizing human melanoma cells xenografted into nude mice. Clin Exp Metastasis 1991; 9:259-72; PMID:2060184; http://dx.doi.org/ 10.1007/BF01753729 [DOI] [PubMed] [Google Scholar]
- 31.Westphal JR, Van't Hullenaar R, Peek R, Willems RW, Crickard K, Crickard U, Askaa J, Clemmensen I, Ruiter DJ, De Waal RM. Angiogenic balance in human melanoma: expression of VEGF, bFGF, IL-8, PDGF and angiostatin in relation to vascular density of xenografts in vivo. Int J Cancer 2000; 86:768-76; PMID:10842189; http://dx.doi.org/ 10.1002/(SICI)1097-0215(20000615)86:6%3c768::AID-IJC3%3e3.0.CO;2-E [DOI] [PubMed] [Google Scholar]
- 32.Ezhilarasan R, Mohanam I, Govindarajan K, Mohanam S. Glioma cells suppress hypoxia-induced endothelial cell apoptosis and promote the angiogenic process. Int J Oncol 2007; 30:701-7; PMID:17273772; http://dx.doi.org/ 10.3892/ijo.30.3.701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lazar-Molnar E, Hegyesi H, Toth S, Falus A. Autocrine and paracrine regulation by cytokines and growth factors in melanoma. Cytokine 2000; 12:547-54; PMID:10843728; http://dx.doi.org/ 10.1006/cyto.1999.0614 [DOI] [PubMed] [Google Scholar]
- 34.Streit M, Detmar M. Angiogenesis, lymphangiogenesis, and melanoma metastasis. Oncogene 2003; 22:3172-9; PMID:12789293; http://dx.doi.org/ 10.1038/sj.onc.1206457 [DOI] [PubMed] [Google Scholar]
- 35.Potgens AJ, Westphal HR, de Waal RM, Ruiter DJ. The role of vascular permeability factor and basic fibroblast growth factor in tumor angiogenesis. Biol Chem Hoppe Seyler 1995; 376:57-70; PMID:7540844 [DOI] [PubMed] [Google Scholar]
- 36.Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F et al.. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006; 10:515-27; PMID:17157791; http://dx.doi.org/ 10.1016/j.ccr.2006.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deer EL, Gonzalez-Hernandez J, Coursen JD, Shea JE, Ngatia J, Scaife CL, Firpo MA, Mulvihill SJ. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010; 39:425-35; PMID:20418756; http://dx.doi.org/ 10.1097/MPA.0b013e3181c15963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shalini S, Dorstyn L, Dawar S, Kumar S. Old, new and emerging functions of caspases. Cell Death Differ 2015; 22:526-39; PMID:25526085; http://dx.doi.org/ 10.1038/cdd.2014.216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Becker D. Antisense targeting of basic fibroblast growth factor and fibroblast growth factor receptor-1 in human melanomas blocks intratumoral angiogenesis and tumor growth. Nat Med 1997; 3:887-93; PMID:9256280; http://dx.doi.org/ 10.1038/nm0897-887 [DOI] [PubMed] [Google Scholar]
- 40.Graeven U, Rodeck U, Karpinski S, Jost M, Philippou S, Schmiegel W. Modulation of angiogenesis and tumorigenicity of human melanocytic cells by vascular endothelial growth factor and basic fibroblast growth factor. Cancer Res 2001; 61:7282-90; PMID:11585767 [PubMed] [Google Scholar]
- 41.Benjamin LE, Golijanin D, Itin A, Pode D, Keshet E. Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J Clin Invest 1999; 103:159-65; PMID:9916127; http://dx.doi.org/ 10.1172/JCI5028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benjamin LE, Keshet E. Conditional switching of vascular endothelial growth factor (VEGF) expression in tumors: induction of endothelial cell shedding and regression of hemangioblastoma-like vessels by VEGF withdrawal. Proc Natl Acad Sci U S A 1997; 94:8761-6; PMID:9238051; http://dx.doi.org/ 10.1073/pnas.94.16.8761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Munoz-Chapuli R, Quesada AR, Angel Medina M. Angiogenesis and signal transduction in endothelial cells. Cell Mol Life Sci 2004; 61:2224-43; PMID:15338053; http://dx.doi.org/ 10.1007/s00018-004-4070-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anfuso CD, Giurdanella G, Motta C, Muriana S, Lupo G, Ragusa N, Alberghina M. PKCalpha-MAPK/ERK-phospholipase A2 signaling is required for human melanoma-enhanced brain endothelial cell proliferation and motility. Microvasc Res 2009; 78:338-57; PMID:19747926; http://dx.doi.org/ 10.1016/j.mvr.2009.09.001 [DOI] [PubMed] [Google Scholar]
- 45.Liu W, Davis DW, Ramirez K, McConkey DJ, Ellis LM. Endothelial cell apoptosis is inhibited by a soluble factor secreted by human colon cancer cells. Int J Cancer 2001; 92:26-30; PMID:11279602; http://dx.doi.org/ 10.1002/1097-0215(200102)9999:9999%3c::AID-IJC1151%3e3.0.CO;2-T [DOI] [PubMed] [Google Scholar]
- 46.Sviderskaya EV, Gray-Schopfer VC, Hill SP, Smit NP, Evans-Whipp TJ, Bond J, Hill L, Bataille V, Peters G, Kipling D et al.. p16/cyclin-dependent kinase inhibitor 2A deficiency in human melanocyte senescence, apoptosis, and immortalization: possible implications for melanoma progression. J Natl Cancer Inst 2003; 95:723-32; PMID:12759390; http://dx.doi.org/ 10.1093/jnci/95.10.723 [DOI] [PubMed] [Google Scholar]
- 47.Jaffe EA, Nachman RL, Becker CG, Minick CR. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J Clin Invest 1973; 52:2745-56; PMID:4355998; http://dx.doi.org/ 10.1172/JCI107470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crop MJ, Baan CC, Korevaar SS, Ijzermans JN, Pescatori M, Stubbs AP, van Ijcken WF, Dahlke MH, Eggenhofer E, Weimar W et al.. Inflammatory conditions affect gene expression and function of human adipose tissue-derived mesenchymal stem cells. Clin Exp Immunol 2010; 162:474-86; PMID:20846162; http://dx.doi.org/ 10.1111/j.1365-2249.2010.04256.x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.