

Abstract
Background
Activins, members of the TGF-β superfamily, are key drivers of inflammation and are thought to play a significant role in ischemia-reperfusion injury (IRI), a process inherent to renal transplantation that negatively impacts early and late allograft function. Follistatin (FS) is a protein that binds activin and inhibits its activity. This study examined the response of activin A and B in mice after renal IRI and the effect of exogenous FS in modulating the severity of renal injury.
Methods
Mice were treated with recombinant FS288 or vehicle before renal IRI surgery. Activin A, B, and FS levels in the serum and kidney, and renal injury parameters were measured at 3, 6, and 24 hours after reperfusion.
Results
Serum and kidney activin B levels were increased within 6 hours postrenal IRI, accompanied by renal injury—increased serum creatinine, messenger (m)RNA expression of kidney injury molecule-1 (KIM-1) and neutrophil gelatinase-associated lipocalin (NGAL); endothelial activation—increased E-selectin mRNA; and systemic inflammation—increased serum levels of IL-6, monocyte chemotactic protein-1 and TNF-α. Further injury was potentiated by an upsurge in activin A by 24 hours, with further increases in serum creatinine, KIM-1 and NGAL mRNA expression. Follistatin treatment significantly reduced the level of serum activin B and subsequently blunted the increase in activin A. Renoprotection was evident with the attenuated rise in serum creatinine, KIM-1 and NGAL expression, tubular injury score, renal cell apoptosis, and serum IL-6 and monocyte chemotactic protein-1 levels.
Conclusions
We propose that activin B initiates and activin A potentiates renal injury after IRI. Follistatin treatment, through binding and neutralizing the actions of activin B and subsequently activin A, reduced renal IRI by minimizing endothelial cell activation and dampening the systemic inflammatory response. These data support the potential clinical application of FS treatment to limit IRI during renal transplantation.
The incidence and prevalence of end stage kidney disease is rising rapidly in Australia, a trend that is reflected worldwide.1 Although transplantation saves lives, only 6% of patients who receive dialysis in Australia were transplanted in 2011.2 The disparity between organ demand and supply has necessitated expanding the donor organ pool to include the use of organs from donors after circulatory death (DCD). In Australia, there were 120 DCD transplants in 2015 in contrast to 23 DCD transplants in 2008.3,4 However, organs from DCD are even more susceptible to ischemia-reperfusion injury (IRI), an unavoidable consequence of transplantation occurring when blood flow is interrupted for organ removal and reestablished after transplantation. Although the reestablishment of blood flow is essential to halt ongoing ischemic damage, reoxygenation is associated with an exacerbation of tissue injury and a profound inflammatory response. Ischemia-reperfusion injury adversely impacts early graft function increasing the risk of subsequent rejection episodes and eventual graft failure. Thus, approaches that attenuate renal IRI may improve short- and long-term graft function and survival.
Ischemia-reperfusion injury activates a cascade of proinflammatory and profibrotic events that involve the innate and adaptive immune response. Upregulation of adhesion molecules promotes recruitment of neutrophils, natural killer T cells, dendritic cells, monocytes and lymphocytes; complement is activated; and oxygen-free radicals and proinflammatory cytokines, including TNF-α, IL-1β, IL-6, and IL-18, are released.5-7
Activins are members of the TGF-β superfamily of growth and differentiation cytokines. Activins are disulphide-linked homodimers of 2 β-subunits. Activin A is a homodimer of the inhibin βA-subunits and activin B is a homodimer of the inhibin βB-subunits. The βB subunit has about 65% sequence homology with the βA-subunit.8 Although the proinflammatory actions of activin A are well established, there is increasing evidence that activin B acts as a slightly weaker activin agonist than activin A and may exert functionally distinct effects from those of activin A.9-11 Activin A is widely produced and distributed in various tissues although there is a poor relationship between messenger (m)RNA expression and protein content in mice, with the highest protein content is found in bone marrow-derived cells and the highest mRNA expression in the liver.12 Resting βB mRNA expression is highest in the gonads, placenta, pituitary, uterus, salivary gland, and the central nervous system.13 Activin A and likely activin B are produced by many different cell types, such as myeloid cells, granulocytes, some T-cell subsets, endothelial cells and smooth muscle cells, in response to inflammation, tissue repair, and fibrosis.13-16
Follistatin (FS), an activin-binding protein, is also synthesized in many tissues usually induced by activin stimulation including gonads, hypothalamus, pituitary, placenta, kidney and adrenal.17 Through alternative gene splicing, FS is produced in 2 main forms consisting of 288 (FS288) and 315 (FS315) amino acids.18,19 Follistatin binds activin A with high affinity (KD 50-900pM), which is comparable to the affinity of activin A for its receptor. Although the binding affinity of FS to activin B is 10-fold lower,20 it remains a potent antagonist. The activin-FS complex is internalized by endocytosis and cleared by the lysosomal degradation pathway.18,21 FS288 is more potent than FS315 in its ability to bind activin to the cell surfaces thereby accelerating its clearance.18
There have been limited studies examining the role of activin A and FS in murine models of IRI of the kidney, liver, and heart. Maeshima et al22 showed in a rat model of renal IRI that the mRNA expression of activin A was upregulated in renal tubular cells after ischemia, and that administration of recombinant FS288 reduced renal injury. In a rat model of hepatic IRI, Kanamoto et al23 demonstrated that treatment with FS reduced the expression of IL-6 and mRNA expression of activin A resulting in reduced liver enzymes. In a mouse model of myocardial IRI, Chen et al24 demonstrated that IRI stimulated the local production of activin A which damaged cardiomyocytes, and blocking activin A action by exogenous FS reduced this damage. However, the role of activin B in IRI has not been previously examined.
Our study documents the dynamic changes of both activin A and B levels in the serum and kidney tissue of mice in the first 24 hours after renal IRI. It adds to the limited data available regarding these proteins in renal IRI, which have been limited to the mRNA expression of the activin βA-subunit.22 We demonstrated for the first time that activin B as well as activin A are involved in renal IRI. In fact, the response of activin B was very rapid with a substantial increase within 6 hours of IRI, in contrast to the increase in activin A that was only detectable by 24 hours. Follistatin treatment blunted this response and reduced renal injury.
MATERIALS AND METHODS
Animals
Adult male C57BL/6 wild type mice aged 10 to 12 weeks were obtained from the Animal Resource Centre, Perth, Australia. Mice were housed in an approved animal facility within St. Vincent's Hospital Melbourne, and all experiments involving animals were approved by the Animal Ethics Committee of St. Vincent's Hospital Melbourne.
Renal IRI Model
Mice were anaesthetized with intraperitoneal ketamine and xylazine (16 and 8 mg/kg, respectively). Using a midline abdominal incision, a right nephrectomy was performed, and subsequently, the left renal pedicle was clamped with a microvascular clamp (Roboz, Rockville, MD) for 20 minutes. The clamp was removed after 20 minutes and the kidney was observed to confirm complete reperfusion. The abdominal wall was then sutured (Sofsilk Wax Coated Braided Silk 4-0). Mice were maintained at core body temperature of 37°C during the procedure and allowed to recover on a 37°C heat-pad for 2 to 3 hours. Mice were sacrificed at 3, 6, and 24 hours. Sham mice underwent right nephrectomy only. Baseline mice did not undergo any procedure.
Administration of FS or Vehicle
Human recombinant FS28824 was administered intravenously via the penile vein after the mice were anaesthetized and before the IRI surgery. The dose used was 0.4 μg/g body weight per mouse. Normal saline was used as vehicle. Baseline mice did not receive any treatment.
Protein Extraction From Kidney
Fresh frozen mouse kidneys were homogenized in phosphate-buffered saline containing protease inhibitor (EMD Millipore, San Diego, CA). After homogenization, samples were centrifuged at 14 000g at 4°C for 10 minutes. The supernatants were collected for protein measurement.
Immunoassays
Activin A was measured using a 2-site enzyme-linked immunosorbent assay (ELISA) (Oxford Bio-Innovations, Oxfordshire, UK) which measures total (free and FS-bound) activin A with no significant cross-reactivity with other forms of activin.25 Activin B was measured using an ELISA which measures total activin B with no significant cross-reactivity with activin A and other related proteins (Oxford Bio-Innovations).26 The average intraplate coefficient of variation (CV) was 5.4%, the interplate CV was 7.0% (n = 4 plates), and the detection limit was 0.01 ng/mL.
Follistatin concentrations were measured using a discontinuous radioimmunoassay which detects total FS using a reagent that dissociates the activin-follistatin complex.27 The samples were measured in a total of 5 assays, the average intra-assay CV was 10.1%, the inter-assay CV was 6.2%, and the limit of detection was 0.99 ng/mL.
Analysis of Renal Function
Renal function was assessed by measurement of serum creatinine at 3, 6, and 24 hours after renal IRI using a kinetic colorimetric assay based on the Jaffé method, analyzed on a Roche COBAS Integra 400 Plus analyzer.
RNA Extraction, Complementary DNA Synthesis, and Quantitative Real-Time Polymerase Chain Reaction
Total RNA from frozen mouse kidneys was isolated using the PureLink RNA mini kit (Life Technologies, Carlsbad, CA) according to the manufacturer's instructions. Total RNA quality and quantity were determined using the NanoDrop spectrophotometer (NanoDrop Technologies, Oxfordshire, UK). First-strand complementary DNA synthesis was performed by incubating 1.0 μg total RNA in a 50-μL reaction mix containing 0.5 μg Oligo (dT) primers and 1 μg random primers (both from Invitrogen, Carlsbad, CA) at 70°C for 10 minutes. A 50-μL reaction mix containing 10 mM dNTP, 300 U SuperScript III recombinant ribonuclease inhibitor, 60 U RNaseOUT recombinant ribonuclease inhibitor, 0.1 M DTT, and 5× first-strand buffer (all from Invitrogen) was then added. Reverse transcription was performed at 42°C for 1 hour and 70°C for 10 minutes. The complementary DNA was stored at −20°C. Real-time polymerase chain reaction was performed using the TaqMan Universal PCR Master Mix system according to the manufacturer's instructions (Applied Biosystem, Bedford, MA).
Analysis of Histological Changes
Kidneys were fixed in 10% formalin for at least 48 hours and embedded in paraffin wax at the Department of Anatomical Pathology, St. Vincent's Hospital Melbourne. Four-micron paraffin-embedded kidney sections mounted on Superfrost slides were stained with hematoxylin-eosin according to standard methods. Tissue sections were firstly dewaxed and rehydrated. Rehydrated sections were stained in filtered hematoxylin, counterstained with alcoholic eosin, dehydrated and cover-slipped. A score of 0 to 5 (0, normal; 1, <10% [minimal]; 2, 10-25% [mild]; 3, 26-50% [moderate]; 4, 51-75% [severe]; 5, >75% [very severe]) was determined without knowledge of the treatment group by assessing the percentage of renal tubular injury involvement (necrosis, cast formation, cell swelling, and dilatation) in 3 randomly selected corticomedullary fields from each upper, mid, and lower poles of the kidney.
Analysis of Apoptosis
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed using the QIA33 FragEL DNA Fragmentation Detection Kit (Calbiochem, Darmstadt, Germany) according to the manufacturer's instructions. Four-micron paraffin-embedded tissue sections were firstly dewaxed and rehydrated in 1× Tris-buffered saline. Sections were then permeabilized with Proteinase K, and endogenous peroxidase was blocked using 3% H2O2 diluted in 100% methanol. Subsequently, the apoptotic nuclei in tissues sections were labelled using the terminal deoxynucleotidyl transferase enzyme for 90 minutes and TUNEL staining was developed using the diaminobenzidine solution. Finally, TUNEL-stained tissue sections were counterstained with methyl green, dehydrated, and cover-slipped.
Measurements of Proinflammatory Cytokines
Serum was analyzed for proinflammatory cytokines using the BD cytometric bead array Mouse Inflammation Kit (BD Biosciences, San Diego, CA) as per the manufacturer's instructions.
Statistical Analyses
The Graphpad Prism 6 graphical and statistics package (Graph-Pad Software Inc., San Diego, CA) was used for presentation and analyses of the data. Results are expressed as mean ± standard error of the mean (SEM). Groups were compared using unpaired Student t test or Mann-Whitney U test as appropriate. A P value less than 0.05 was considered to be statistically significant.
RESULTS
Changes in Serum and Kidney Activin and FS Levels After Renal Ischemia and Reperfusion
Serum and kidney levels of activin A, activin B, and FS were measured 3, 6, and 24 hours after reperfusion. Compared with baseline (untreated) mice, sham-operated mice (ie right nephrectomy only) exhibited a modest increase in serum activin A at 24 hours (Figure 1A) and in serum activin B at 6 and 24 hours (Figure 1C), presumably representing an acute phase response to surgery. Neither activin A nor B was significantly increased in kidney tissue in sham mice during the 24 hours follow-up (Figures 1B, D). Both activins were increased after renal IRI however with different kinetics: serum and kidney activin A levels were unchanged at 3 and 6 hours, but were significantly increased by 24 hours (Figures 1A, B), whereas serum and kidney activin B levels increased earlier, at 6 hours and 3 hours, respectively (Figures 1C, D).
FIGURE 1.
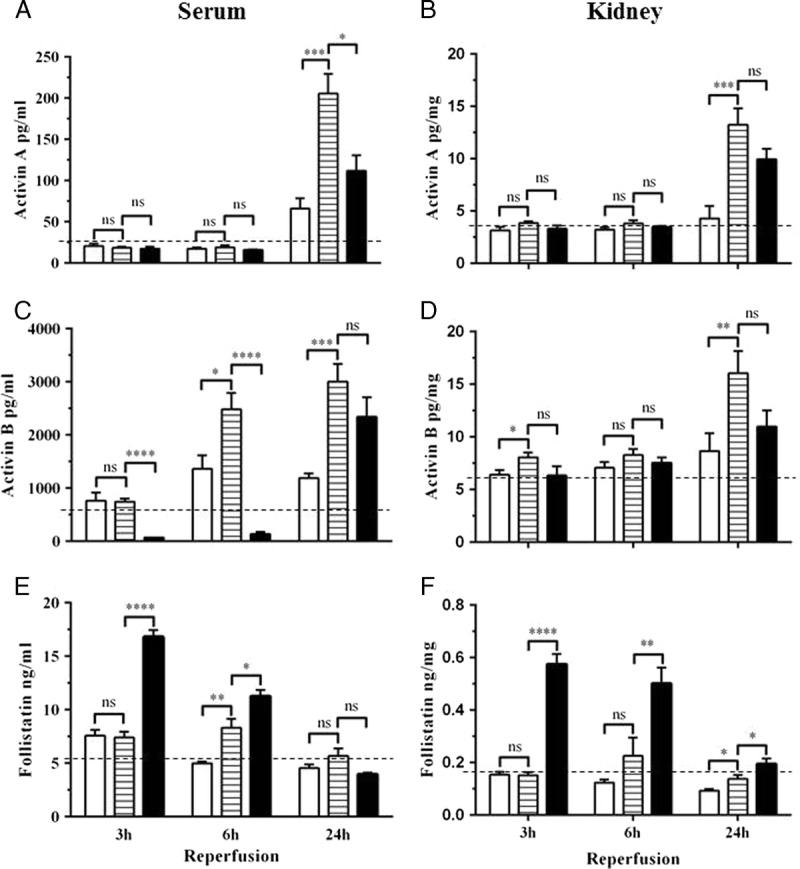
Serum and kidney tissue activin and FS levels after renal IRI. Protein levels of activin A, B and FS were determined within the serum (A, C, E) and kidney tissue (B, D, F) 3, 6 and 24 hours after reperfusion. Results expressed as mean ± SEM. * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001; ns = not significant. White bar, sham (n = 6-8); lined bar, vehicle-treated (n = 6-9); black bar, FS-treated (n = 6-7); dotted line, baseline (n = 4-7).
Endogenous serum FS levels were elevated at 6 hours in mice subjected to renal IRI compared to sham-operated mice, but returned to baseline at 24 hours (Figure 1E). In the kidney, endogenous FS remained close to baseline levels at all times (Figure 1F).
Effect of FS Treatment on Serum and Kidney Activin and FS Levels
After FS treatment, circulating FS levels were significantly increased at 3 and 6 hours before returning to baseline at 24 hours (Figure 1E). Treatment with FS reduced serum activin B at both 3 and 6 hours to below baseline levels, whereas activin B levels at 24 hours were similar to sham (Figure 1C). In contrast, FS treatment did not affect serum activin A levels at 3 and 6 hours, although a significant decrease was observed at 24 hours (Figure 1A).
Follistatin treatment also increased FS levels in the kidney at 3 hours and these remained elevated at 6 and 24 hours although decreasing over time (Figure 1F). Follistatin treatment had no significant effect on either activin A or B levels in the kidney at any time point (Figures 1B, D).
Effect of FS Treatment on Renal Injury After Ischemia-Reperfusion
In vehicle treated mice that were subjected to IRI, serum creatinine was elevated from 3 hours after reperfusion and reached a 5-fold increase by 24 hours, reflecting significant deterioration in renal function (Figure 2A). There was a concomitant increase in the mRNA expression of the biomarkers kidney injury molecule-1 (KIM-1) and neutrophil gelatinase-associated lipocalin (NGAL) at all time-points (Figures 2B, C). E-selectin, a marker of endothelial cell activation, was rapidly upregulated after IRI and persisted to 24 hours (Figure 2D). Histologically, kidneys harvested at 24 hours after IRI showed extensive tubular injury involving 50% to 75% of the renal cortex, cast formation, and overall disruption of the renal architecture (Figures 3A, B[ii]). Numerous TUNEL-positive apoptotic nuclei were observed (Figures 4A, B[ii]).
FIGURE 2.
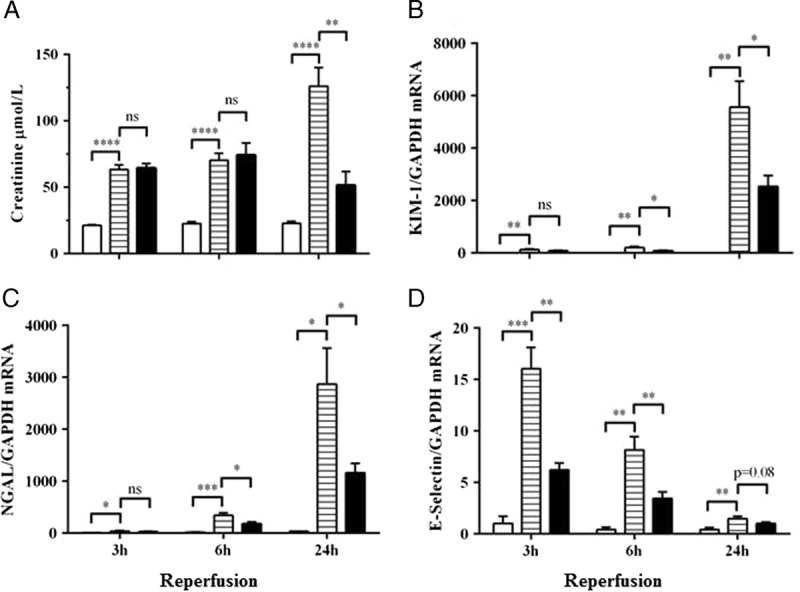
Follistatin treatment reduced renal injury and endothelial activation after IRI. Serum creatinine (A), and mRNA expression of KIM-1 (B), NGAL (C), and E-selectin (D) in kidney tissue at 3, 6 and 24 hours after renal IRI. Results expressed as mean ± SEM. * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001. White bar, sham (n = 6-8); lined bar, vehicle-treated (n = 7-9); black bar, FS-treated (n = 6-7).
FIGURE 3.
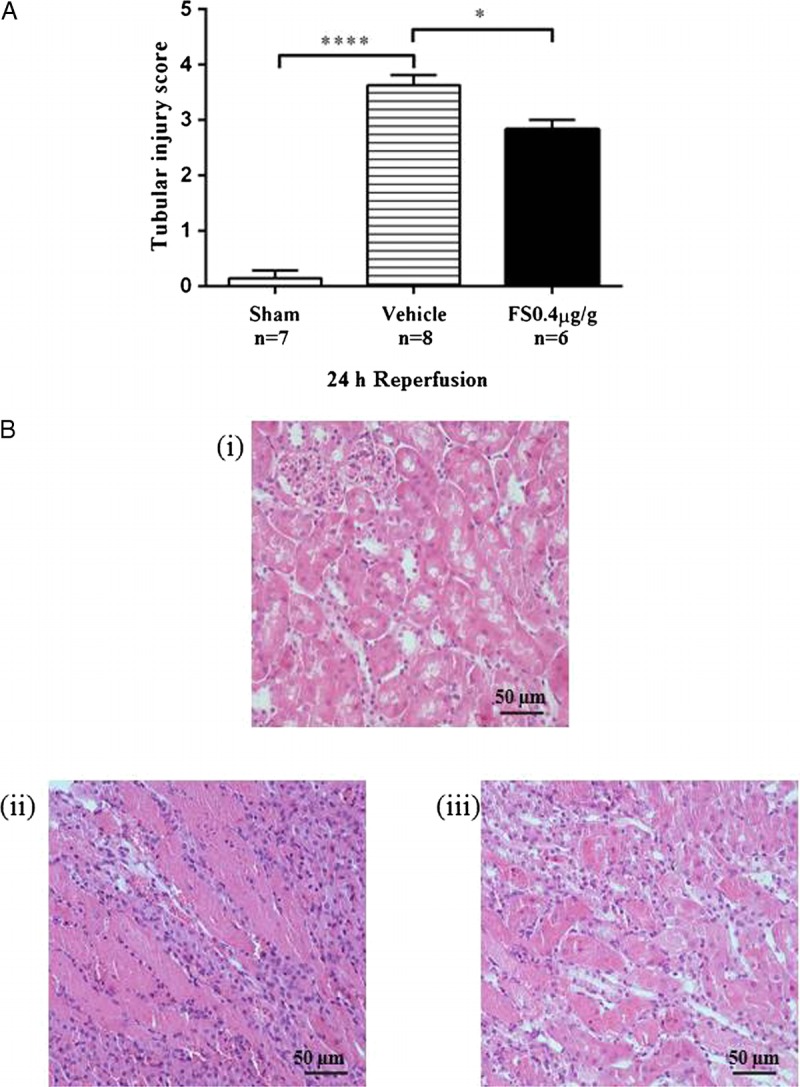
Renal morphology 24 hours after reperfusion. Semiquantitative scoring of tubular damage (A). Results expressed as mean ± SEM. * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001. Representative hematoxylin-eosin–stained kidney sections (B): (i) Kidney section from a baseline mouse showing normal histological morphology; (ii) Kidney section from a vehicle-treated mouse after renal IRI showing severe (50-75%) tubular injury. (iii) Kidney sections from a FS-treated mouse FS showing mild (26-50%) tubular injury.
FIGURE 4.
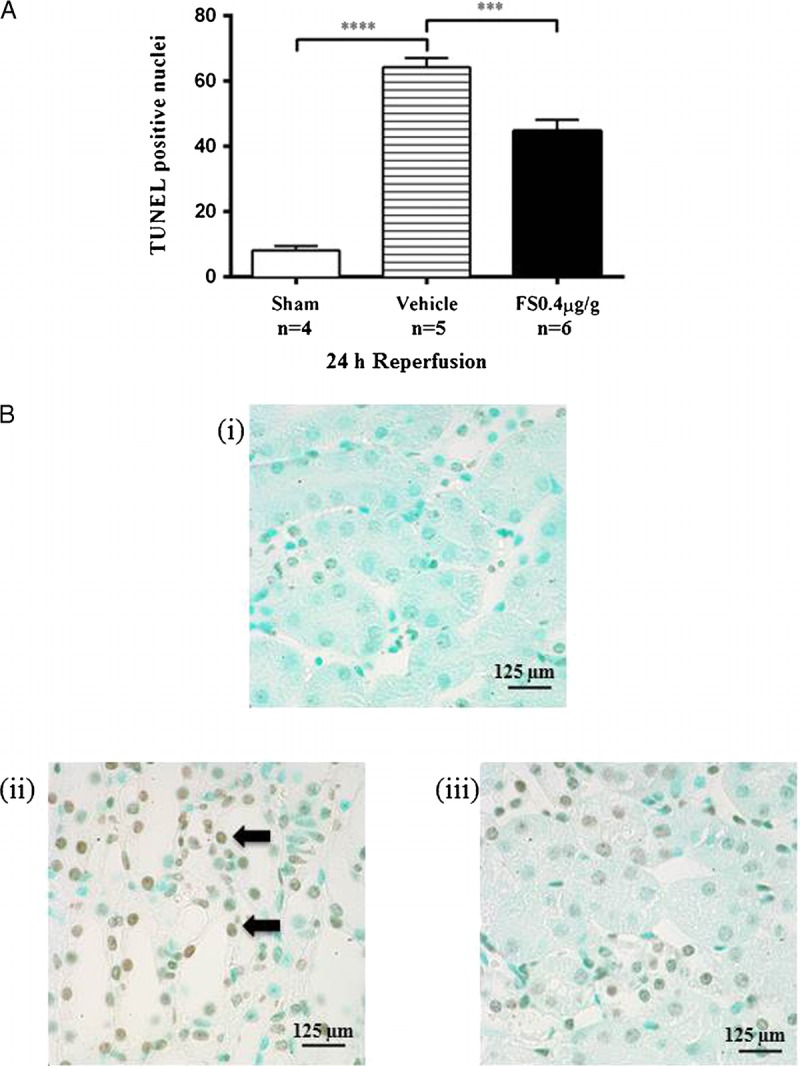
FS treatment reduced apoptosis 24 hours after renal IRI. Sections of renal tissue were analyzed by TUNEL-stain and the number of apoptotic nuclei per high power field (hpf) were counted (A). Results expressed as mean ± SEM. * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001. Representative sections of TUNEL-stained renal cortex at 24 hours (B): (i) Kidney section from a sham mouse showing no TUNEL-positive cells. (ii) Kidney section from a vehicle-treated mouse showing significant number of TUNEL-positive cells (black arrow). (iii) Kidney sections from a FS-treated mouse showing reduction in number of TUNEL-positive cells.
Treatment with FS reduced early injury and endothelial activation, noted as a reduction in mRNA expression of KIM-1, NGAL, and E-selectin (Figures 2B-D). There was no effect on serum creatinine at 3 and 6 hours after IRI but the late rise in serum creatinine between 6 and 24 hours was attenuated (Figure 2A). This was accompanied by reduced tubular injury (Figures 3A, B[iii]) and cellular apoptosis (Figures 4A, B[iii]).
Effect of FS Treatment on Proinflammatory Cytokine Expression
Ischemia-reperfusion injury initiates an inflammatory cascade, and expression of IL-6, monocyte chemotactic protein-1 (MCP-1) and TNF-α were elevated in vehicle treated mice subjected to IRI (Figures 5A-C). The level of IL-6 was significantly increased compared to sham mice up to 24 hours after IRI with the highest concentration seen at the earliest time point, 3 hours (Figure 6A). The increase in MCP-1 and TNF-α levels were evident from 6 hours and sustained through to 24 hours (Figures 6B, C). Follistatin treatment significantly reduced the concentrations of IL-6 and MCP-1, which were sustained to 24 hours (Figures 6A, B) but had little impact on TNF-α levels (Figure 6C).
FIGURE 5.
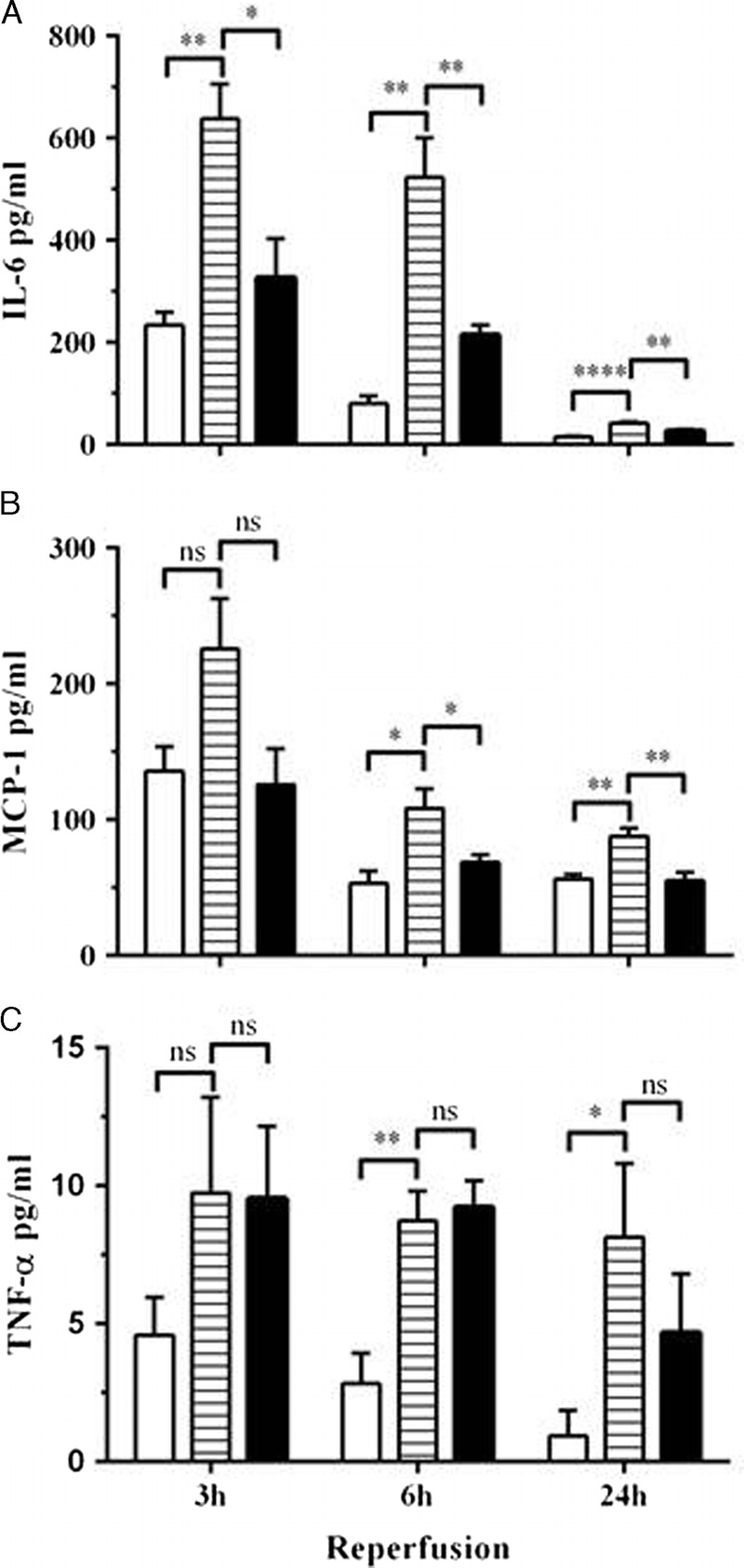
Expression of systemic proinflammatory cytokines after renal IRI. Serum IL-6 (A), MCP-1 (B), and TNF-α (C) were determined 3, 6, and 24 hours after reperfusion. Results expressed as mean ± SEM. * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001. White bar, sham (n = 4-5); lined bar, vehicle-treated (n = 4); black bar, FS-treated (n = 4-5).
FIGURE 6.
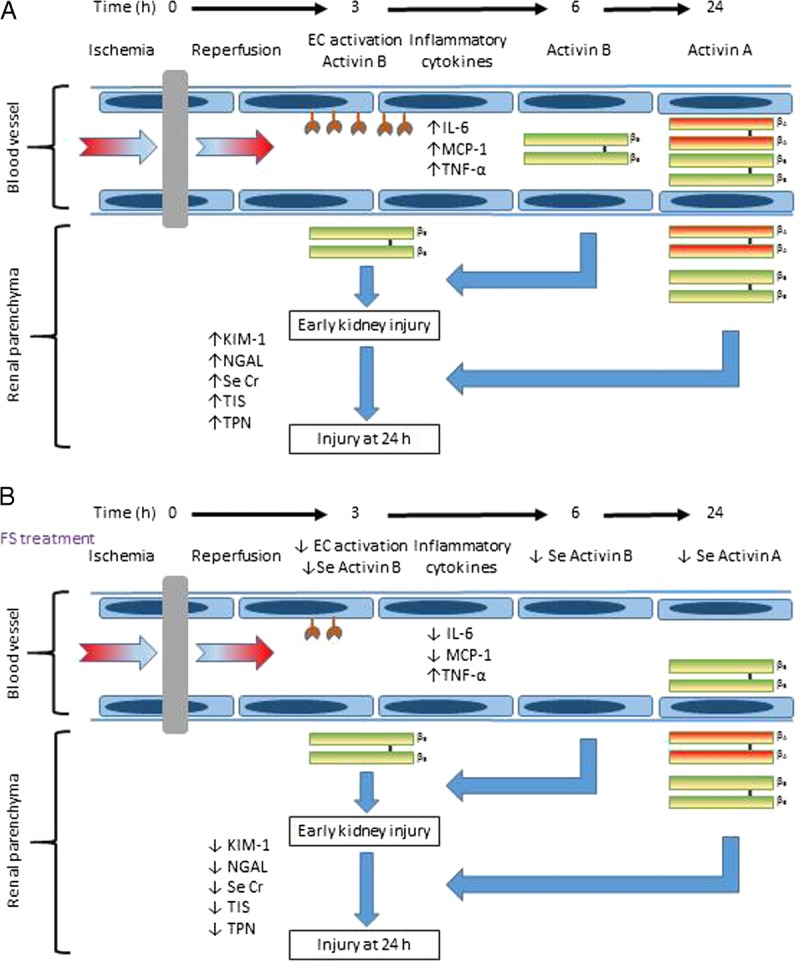
Proposed role of activins in renal IRI. The increase in activin B, which precedes that of activin A, initiates early renal injury by stimulating endothelial cell (EC) activation and pro-inflammatory cytokine expression. The later upregulation of activin A potentiates renal injury leading to further increases in KIM-1, NGAL, serum creatinine (Se Cr), tubular injury score (TIS) and cellular apoptosis (TPN, TUNEL positive nuclei) (A). FS treatment reduced Se activin B and subsequently activin A, with associated reduction in EC activation, serum IL-6 and MCP-1, KIM-1, NGAL, Se Cr, TIS and TPN (B).
DISCUSSION
Activin A and B play a key role in inflammation. There is increasing evidence that serum and tissue activin levels are increased in acute and chronic inflammatory conditions, such as septicemia, preeclampsia, asthma, inflammatory bowel disease, burns injuries, and rheumatoid arthritis.13,16 Studies in mice showed that activin A is rapidly increased in response to a lipopolysaccharide (LPS) challenge, and treatment with FS can halve the mortality in mice after a lethal LPS injection.28,29 Further, in patients with acute respiratory failure in intensive care units, the elevation of both activin A and B during the first 5 days in intensive care unit was associated with a marked increase in mortality up to 1 year.29
Maeshima et al22 showed in a rat model of renal IRI that mRNA expression of activin A was not detected in normal and sham-operated kidneys, but increased markedly after renal ischemia-reperfusion. Follistatin mRNA was abundantly expressed in normal kidneys but decreased significantly at 24 and 48 hours after IRI. In this study, we measured the levels of serum and kidney activin A and FS protein. We showed that activin A protein was present in the serum and kidney of baseline (normal) and sham-operated mice. This may represent bound activin A-FS complexes that are biologically inactive as the activin A ELISA measures total activin A and cannot differentiate between the free and bound forms of the protein. This finding of poor relationship between mRNA expression and protein content of activin A in mouse is consistent with that previously reported.12 We also showed that FS protein was present and measurable in normal kidneys, a finding consistent with the detection of abundant FS mRNA expression in normal kidneys but in contrast to Maeshima et al, we observed an increase in FS protein at 24 hours after IRI in the kidneys of the vehicle treated mice.
In addition to activin A, we also measured changes of activin B level in response to renal IRI, which has not been examined previously. We showed that the increase in activin B in the serum and kidney after renal IRI preceded that of activin A. These results contrast with those from an acute LPS inflammation mouse model, where serum activin A increased by 5- to 10-fold within 1 hour of administration of LPS12,28 and serum activin B increased several hours later, along with a secondary rise in activin A.30 Our findings suggest that the regulation of activin A and B levels in inflammation can differ depending on the stimulus.
Activin B may be an early mediator of injury after renal IRI which is later potentiated by activin A. We demonstrated that the early rise in serum and kidney activin B was associated with renal injury, characterized by an elevation in serum creatinine, and mRNA expression of KIM-1 and NGAL in the kidney. Renal tubules are the most susceptible region to IRI31 and both KIM-1 and NGAL are useful and sensitive early markers of renal tubular damage.32-34 Follistatin treatment prevented the rise in serum activin B level, and this was associated with a decrease in mRNA expression of KIM-1 and NGAL. We did not show an early reduction in serum creatinine, which may reflect the poor sensitivity of this marker for the detection of renal injury.33-35 In parallel, FS treatment also reduced systemic IL-6 and MCP-1, and mRNA expression of E-selectin, a cell adhesion molecule expressed on activated endothelial cells. Endothelial dysfunction is the result of initial ischemia and is well described in the pathogenesis of renal IRI.5,6,32,36 This finding suggests that FS treatment may confer early renal protection through minimizing endothelial cell activation and reducing activin B-mediated systemic inflammation.
Despite this early renal protection, injury propagated with evidence of further increases in serum creatinine and the mRNA expression of KIM-1 and NGAL. There was a concomitant significant upsurge in serum and kidney activin A in addition to the elevated activin B level, suggesting this late injurious effect was potentiated by activin A. Reducing the rise in serum activin A without significant reduction in serum activin B preserved renal function as illustrated by reduced serum creatinine, KIM-1 and NGAL expression, renal tubular injury score and renal cell apoptosis. This suggests that activin A may play a more potent role than activin B11,13,20 in renal IRI.
The renoprotective effect at 24 hours paralleled the extent of activin A reduction which was dependent on the FS treatment dose (our unpublished data).
Interestingly, FS treatment had no impact on activin levels within the kidney despite the demonstrable protective renal effect. It is well established that acute kidney injury ignites a systemic inflammatory response in addition to a local inflammatory response.37 We propose that the protective renal effect of FS was predominantly due to dampening of the systemic inflammatory response consequent to reduced serum activin B and A. Indeed, we showed that FS potently reduced systemic IL-6 and MCP-1 at all time-points, but had little effect on TNF-α which may have been the main source of activin A and B stimulation.11,28
In summary, these data provided novel information on the kinetics of activin A and B in response to renal IRI in mice. We demonstrated that the increase in activin B preceded that of activin A after renal IRI. We propose that IRI induces endothelial cell activation which augments serum activin B levels and promotes systemic inflammation. Together these trigger renal injury and increase the susceptibility of the renal tubules to further injury mediated by activin A (Figure 6A). We suggest that FS treatment binds circulating activin B and inhibits endothelial cell derived activin B release and systemic inflammation to reduce early injury. Follistatin also binds activin A. Thus, by 24 hours, after renal IRI, not only is there less activin A present in the serum, but also the renal tubules are less vulnerable to further injury having experienced less early injury (Figure 6B). These data add to the increasing literature on the role of activin in inflammatory conditions and point to potential clinical application in the transplantation process.
ACKNOWLEDGMENTS
The authors thank the Oxford Brookes University for providing reagents for measurement of activin A and B; staff at the Immunology Research Centre, St. Vincent’s Hospital Melbourne, for technical support and advice; staff at the Bioresources Centre for animal care; and Sylwia Glowacka (University of Melbourne) for assistance with COBAS analyses. The authors also thank the Victorian Government Operational Infrastructure Support Program.
Footnotes
Published online 6 June 2016.
This study was supported by Jacquot Research Entry Scholarship to D. Fang, and grants from the National Health and Medical Research Council of Australia (NHMRC 1048094) to D. M. de Kretser and K. M. Dwyer, and the CASS Foundation to D. M. de Kretser.
Prof David de Kretser is a director of Paranta Biosciences, a company developing follistatin as a therapeutic. The other authors declare no conflict of interest.
D.Y.P.F. participated in research design, performance of experiments, data analysis, preparation of the article, and intellectual input. B.L. participated in research design and performance of experiments. S.H. participated in the measurement of the activins and follistatin. D.M.d.K. participated in research design, preparation of manuscript, and intellectual input. P.J.C. participated in research design, preparation of manuscript, and intellectual input. K.M.D participated in research design, preparation of the article, and intellectual input.
REFERENCES
- 1.Cass A, Chadban SJ, Gallagher MP, et al. Kidney Health Australia. The Economic Impact of End-stage Kidney Disease in Australia—Projections to 2020. http://kidney.org.au/cms_uploads/docs/kha-economic-impact-of-eskd-in-australia-projections-2020.pdf. Published 2010. Accessed May 2, 2016.
- 2.McDonald S, Clayton P, Hurst K. Australia and New Zealand Dialysis and Transplant Registry The 35th Annual ANZDATA Report (2012). http://www.anzdata.org.au/v1/report_2012.html. Published 2013. Accessed May 2, 2016.
- 3.ANZOD Registry. 2008 Summary. http://www.anzdata.org.au/anzod/updates/anzodsummary.htm. Published 2009. Accessed May 2, 2016.
- 4.ANZOD Registry. Monthly Report on Deceased Organ Donation in Australia December 2015. http://www.anzdata.org.au/anzod/updates/anzod2015summary.pdf. Published 2016. Accessed May 2, 2016.
- 5.Jang HR, Ko GJ, Wasowska BA, et al. The interaction between ischemia-reperfusion and immune responses in the kidney. J Mol Med (Berl). 2009;87:859–864. [DOI] [PubMed] [Google Scholar]
- 6.Eltzschig HK, Eckle T. Ischemia and reperfusion—from mechanism to translation. Nature medicine. 2011;17(11):1391–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kinsey GR, Sharma R, Huang L, et al. Regulatory T cells suppress innate immunity in kidney ischemia-reperfusion injury. J Am Soc Nephrol. 2009;20(8):1744–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mason AJ, Niall HD, Seeburg PH. Structure of two human ovarian inhibins. Biochem Biophys Res Commun. 1986;135(3):957–964. [DOI] [PubMed] [Google Scholar]
- 9.Brown CW, Li L, Houston-Hawkins DE, et al. Activins are critical modulators of growth and survival. Mol Endocrinol. 2003;17(12):2404–2417. [DOI] [PubMed] [Google Scholar]
- 10.Thompson TB, Cook RW, Chapman SC, et al. Beta A versus beta B: is it merely a matter of expression? Mol Cell Endocrinol. 2004;225:9–17. [DOI] [PubMed] [Google Scholar]
- 11.Hedger MP, de Kretser DM. The activins and their binding protein, follistatin-Diagnostic and therapeutic targets in inflammatory disease and fibrosis. Cytokine Growth Factor Rev. 2013;24:285–95. [DOI] [PubMed] [Google Scholar]
- 12.Wu H, Chen Y, Winnall WR, et al. Acute regulation of activin A and its binding protein, follistatin, in serum and tissues following lipopolysaccharide treatment of adult male mice. Am J Physiol Regul Integr Comp Physiol. 2012;303:R665–R675. [DOI] [PubMed] [Google Scholar]
- 13.Hedger MP, Winnall WR, Phillips DJ, et al. The regulation and functions of activin and follistatin in inflammation and immunity. Vitam Horm. 2011;85:255–297. [DOI] [PubMed] [Google Scholar]
- 14.de Kretser DM, O'Hehir RE, Hardy CL, et al. The roles of activin A and its binding protein, follistatin, in inflammation and tissue repair. Mol Cell Endocrinol. 2012;359:101–106. [DOI] [PubMed] [Google Scholar]
- 15.Jones KL, de Kretser DM, Patella S, et al. Activin A and follistatin in systemic inflammation. Mol Cell Endocrinol. 2004;225:119–125. [DOI] [PubMed] [Google Scholar]
- 16.Werner S, Alzheimer C. Roles of activin in tissue repair, fibrosis, and inflammatory disease. Cytokine Growth Factor Rev. 2006;17:157–171. [DOI] [PubMed] [Google Scholar]
- 17.McConnell DS, Wang Q, Sluss PM, et al. A two-site chemiluminescent assay for activin-free follistatin reveals that most follistatin circulating in men and normal cycling women is in an activin-bound state. J Clin Endocrinol Metab. 1998;83:851–858. [DOI] [PubMed] [Google Scholar]
- 18.Hashimoto O, Nakamura T, Shoji H, et al. A novel role of follistatin, an activin-binding protein, in the inhibition of activin action in rat pituitary cells. Endocytotic degradation of activin and its acceleration by follistatin associated with cell-surface heparan sulfate. J Biol Chem. 1997;272:13835–13842. [DOI] [PubMed] [Google Scholar]
- 19.Shimasaki S, Koga M, Esch F, et al. Primary structure of the human follistatin precursor and its genomic organization. Proc Natl Acad Sci U S A. 1988;85:4218–4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schneyer A, Schoen A, Quigg A, et al. Differential binding and neutralization of activins A and B by follistatin and follistatin like-3 (FSTL-3/FSRP/FLRG). Endocrinology. 2003;144:1671–1674. [DOI] [PubMed] [Google Scholar]
- 21.Sugino H, Sugino K, Hashimoto O, et al. Follistatin and its role as an activin-binding protein. J Med Invest. 1997;44:1–14. [PubMed] [Google Scholar]
- 22.Maeshima A, Zhang YQ, Nojima Y, et al. Involvement of the activin-follistatin system in tubular regeneration after renal ischemia in rats. J Am Soc Nephrol. 2001;12:1685–1695. [DOI] [PubMed] [Google Scholar]
- 23.Kanamoto M, Shimada M, Morine Y, et al. Beneficial effects of follistatin in hepatic ischemia-reperfusion injuries in rats. Dig Dis Sci. 2011;56:1075–1081. [DOI] [PubMed] [Google Scholar]
- 24.Chen Y, Rothnie C, Spring D, et al. Regulation and actions of activin A and follistatin in myocardial ischaemia-reperfusion injury. Cytokine. 2014;69:255–262. [DOI] [PubMed] [Google Scholar]
- 25.Knight PG, Muttukrishna S, Groome NP. Development and application of a two-site enzyme immunoassay for the determination of ‘total’ activin-A concentrations in serum and follicular fluid. J Endocrinol. 1996;148:267–279. [DOI] [PubMed] [Google Scholar]
- 26.Ludlow H, Phillips DJ, Myers M, et al. A new ‘total’ activin B enzyme-linked immunosorbent assay (ELISA): development and validation for human samples. Clin Endocrinol. 2009;71(6):867–873. [DOI] [PubMed] [Google Scholar]
- 27.O'Connor AE, McFarlane JR, Hayward S, et al. Serum activin A and follistatin concentrations during human pregnancy: a cross-sectional and longitudinal study. Hum Reprod. 1999;14:827–832. [DOI] [PubMed] [Google Scholar]
- 28.Jones KL, Mansell A, Patella S, et al. Activin A is a critical component of the inflammatory response, and its binding protein, follistatin, reduces mortality in endotoxemia. Proc Natl Acad Sci U S A. 2007;104:16239–16244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Kretser D, Bensley J, Pettila V, et al. Serum activin A and B levels predict outcome in patients with acute respiratory failure: a prospective cohort study. Critical Care. 2013;17(5):R263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hedger MP, Phillips DJ, Mansell A, et al. Regulation of activins and the activin-binding protein, follistatin, by Toll-like receptor ligands in the adult mouse. Cytokine. 2014;70(1):46. [Google Scholar]
- 31.Sabbahy ME, Vaidya VS. Ischemic kidney injury and mechanisms of tissue repair. Wiley Interdiscip Rev Syst Biol Med. 2011;3(5):606–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. Clin Invest. 2011;121(11):4210–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han WK, Bailly V, Abichandani R, et al. Kidney Injury Molecule-1 (KIM-1): a novel biomarker for human renal proximal tubule injury. Kidney Int. 2002;62(1):237–244. [DOI] [PubMed] [Google Scholar]
- 34.Malyszko J, Malyszko JS, Mysliwiec M. Serum neutrophil gelatinase-associated lipocalin correlates with kidney function in renal allograft recipients. Clin Transplant. 2009;23. [DOI] [PubMed] [Google Scholar]
- 35.Star RA. Treatment of acute renal failure. Kidney Int. 1998;54(6):1817–1831. [DOI] [PubMed] [Google Scholar]
- 36.Khalid U, Pino-Chavez G, Nesargikar P, et al. Kidney ischaemia reperfusion injury in the rat: the EGTI scoring system as a valid and reliable tool for histological assessment. Journal of Histology and Histopathology. 2016;3(1):1. [Google Scholar]
- 37.Ratliff BB, Rabadi MM, Vasko R, et al. Messengers without borders: mediators of systemic inflammatory response in AKI. Clin J Am Soc Nephrol. 2013;24(4):529–536. [DOI] [PubMed] [Google Scholar]