

Abstract
Key points
Peripheral chemoreflex sensitization is a feature of renovascular hypertension.
Carotid sinus nerve denervation (CSD) has recently been shown to relieve hypertension and reduce sympathetic activity in other rat models of hypertension.
We show that CSD in renovascular hypertension halts further increases in blood pressure.
Possible mechanisms include improvements in baroreceptor reflex sensitivity and renal function, restoration of cardiac calcium signalling towards control levels, and reduced neural inflammation.
Our data suggest that the peripheral chemoreflex may be a viable therapeutic target for renovascular hypertension.
Abstract
The peripheral chemoreflex is known to be hyper‐responsive in both spontaneously hypertensive (SHR) and Goldblatt hypertensive (two kidney one clip; 2K1C) rats. We have previously shown that carotid sinus nerve denervation (CSD) reduces arterial blood pressure (ABP) in SHR. In the present study, we show that CSD ameliorates 2K1C hypertension and reveal the potential underlying mechanisms. Adult Wistar rats were instrumented to record ABP via telemetry, and then underwent CSD (n = 9) or sham CSD (n = 9) 5 weeks after renal artery clipping, in comparison with normal Wistar rats (n = 5). After 21 days, renal function was assessed, and tissue was collected to assess sympathetic postganglionic intracellular calcium transients ([Ca2+]i) and immune cell infiltrates. Hypertensive 2K1C rats showed a profound elevation in ABP (Wistar: 98 ± 4 mmHg vs. 2K1C: 147 ± 8 mmHg; P < 0.001), coupled with impairments in renal function and baroreflex sensitivity, increased neuroinflammatory markers and enhanced [Ca2+]I in stellate neurons (P < 0.05). CSD reduced ABP in 2K1C+CSD rats and prevented the further progressive increase in ABP seen in 2K1C+sham CSD rats, with a between‐group difference of 14 ± 2 mmHg by week 3 (P < 0.01), which was accompanied by improvements in both baroreflex control and spectral indicators of cardiac sympatho‐vagal balance. Furthermore, CSD improved protein and albuminuria, decreased [Ca2+]i evoked responses from stellate neurons, and also reduced indicators of brainstem inflammation. In summary, CSD in 2K1C rats reduces the hypertensive burden and improves renal function. This may be mediated by improvements in autonomic balance, functional remodelling of post‐ganglionic neurons and reduced inflammation. Our results suggest that the peripheral chemoreflex may be considered as a potential therapeutic target for controlling renovascular hypertension.
Keywords: carotid sinus denervation, chemoreceptor reflex, renovascular hypertension
Key points
Peripheral chemoreflex sensitization is a feature of renovascular hypertension.
Carotid sinus nerve denervation (CSD) has recently been shown to relieve hypertension and reduce sympathetic activity in other rat models of hypertension.
We show that CSD in renovascular hypertension halts further increases in blood pressure.
Possible mechanisms include improvements in baroreceptor reflex sensitivity and renal function, restoration of cardiac calcium signalling towards control levels, and reduced neural inflammation.
Our data suggest that the peripheral chemoreflex may be a viable therapeutic target for renovascular hypertension.
Abbreviations
- 2K1C
two kidney one clip hypertension
- ABP
arterial blood pressure
- Ang II
angiotensin II
- AT1R
angiotensin type 1 receptor
- CSD
carotid sinus denervation
- HR
heart rate
- FACS
fluorescence‐activated cell sorting
- HF
high frequency
- LF
low frequency
- MAP
mean arterial pressure
- SBP
systolic blood pressure
- SHR
spontaneously hypertensive rat
Introduction
Clinical renovascular hypertension occurs when the renal artery is narrowed, predominantly as a result of atherosclerosis (70–90% of cases) or less commonly as a result of dysplasia. It usually involves the ostium and proximal third of the main renal artery and the peri‐renal aorta (Safian & Textor, 2001; Lopez‐Novoa et al. 2011). Renovascular hypertension affects ∼6% of the elderly population, regardless of race or sex (Hansen et al. 2002; Piecha et al. 2012; Weber, 2014). For many years, the Goldblatt or two kidney‐one clip (2K1C) experimental model has been used to study renovascular hypertension (Goldblatt et al. 1934) and has been adopted in the present study.
In both 2K1C model and human patients, renal artery stenosis is associated with a decrease in renal perfusion pressure, activation of the renin–angiotensin system, suppression of the baroreceptor reflex and increased sympathetic nerve activity (Johansson et al. 1999; Oliveira‐Sales et al. 2014). Angiotensin II (Ang II) has been shown to drive increases in reactive oxygen species production and inflammatory markers in renovascular hypertension (Ruiz‐Ortega et al. 2006), and inhibition of these pathways can mitigate the increase in arterial pressure in the 2K1C model (Bivol et al. 2008). Previous studies in renovascular hypertensive rats have found evidence of elevated inflammatory markers in the heart (Nicoletti et al. 1996) and kidneys (Bivol et al. 2008; Cheng et al. 2009). We have previously shown that carotid sinus nerve denervation (CSD) in spontaneously hypertensive rats (SHR) reduces T‐cell infiltration in the aorta and brainstem (McBryde et al. 2013), although it is currently unknown whether a similar benefit might be seen in renovascular hypertension.
The carotid body has attracted considerable interest as a potential new treatment target for sympathetically‐mediated cardiovascular disease (Paton et al. 2013 a; Paton et al. 2013 b). We have recently shown that removal of peripheral chemoreceptor afferents by CSD ameliorates hypertension in the SHR (Abdala et al. 2012), in conjunction with an improvement in cardiac baroreceptor reflex gain and a profound (∼50%) reduction in renal sympathetic activity (McBryde et al. 2013). In other conditions where the renin–angiotensin system is activated, Ang II has been shown to facilitate the release of noradrenaline from sympathetic nerves (Maruyama et al. 2000; Fabiani et al. 2001). Given our recent finding that peripheral chemoreceptor reflex sensitivity to stimulation sodium cyanide is greatly increased in 2K1C rats (Oliveira‐Sales et al. 2016), we hypothesized that CSD would alleviate hypertension and improve renal function in experimental renovascular hypertension. We further hypothesized that the mechanisms underlying the responses to CSD may involve improvements in baroreceptor reflex function, reductions in inflammation and sympathetic post‐ganglionic remodelling.
Methods
Ethical approval
Procedures were carried out according to the United Kingdom Home Office Guidelines Scientific Procedures Act of 1986. Rats were housed individually, given normal rat chow and drinking water ad libitum, and maintained under a 14:10 h light/dark cycle. Animals were divided into three experimental groups: (1) two kidney one clip sham carotid sinus denervated (2K1C+sham CSD; n = 9); (2) two kidney one clip carotid sinus denervated (2K1C+CSD; n = 9); and (3) age‐matched normotensive Wistar controls (Wistar; n = 5). All surgeries were conducted under aseptic conditions, with animals anaesthetized with ketamine (60 mg kg−1; Vetalar, Zoetis, London, UK) and medetomidine (250 μg kg−1; Elanco Animal Health, Basingstoke, UK) via i.m. injection. The level of anaesthesia was checked frequently by testing limb withdrawal reflexes. Body temperature was regulated using a feedback‐controlled heating pad (Harvard Apparatus, Cambridge, UK). Postoperatively, non‐steroidal anti‐inflammatory pain relief was given (0.004 ml/100 g of Metacam; Boehringer Ingelheim, Germany) for 3 days following surgery. At the completion of the experimental protocol, animals were anaesthetized with isofluorane, then killed with an i.p. injection of sodium pentobarbital (100 mg kg−1).
Goldblatt model of hypertension (2K1C)
Male Wistar rats (150–180 g) were anaesthetized and the left renal artery accessed via a retroperitoneal incision. A silver clip with an internal width of 0.2 mm was used to partially obstruct the artery, as described previously (Oliveira‐Sales et al. 2014). On completion of the surgery, anaesthesia was reversed with a s.c. injection of atipamezole (1 mg kg−1, Antisedan; Zoetis) and the animals were returned to a warm recovery box.
CSD
Bilateral CSD was performed 5 weeks after renal artery clipping, using a surgical approach as described previously (Abdala et al. 2012; McBryde et al. 2013). Briefly, the carotid sinus was accessed via a ventral midline neck incision, the carotid sinus nerve branches removed, and the adventitia of the bifurcation and the internal carotid artery carefully stripped. Sham‐operated rats underwent the same surgical procedures, although the carotid sinus nerves were left intact.
Experimental design
Four weeks after the implantation of renal clips, rats were instrumented to record systemic arterial pressure via wireless telemetry (Data Sciences International Ltd, St Paul, MN, USA) and a catheter was inserted into the femoral vein and externalized between the scapulae, as described previously (Waki et al. 2006; McBryde et al. 2013). After 5 days of recovery, a baseline period was recorded. CSD or sham CSD was performed 5 weeks after renal artery clipping. Renal function was tested in each group 21 days after CSD (2K1C+CSD) or sham surgery (2K1C+sham CSD) and in aged‐matched control animals (Wistar). Chemoreflex sensitivity was assessed before and after CSD or sham CSD, with an i.v. bolus infusion of sodium cyanide (120 μg kg−1, i.v.).
Cardiac baroreflex sensitivity analysis
Bradycardic and tachycardic reflex responses produced by bolus infusion of phenylephrine (0.1 mg ml−1, i.v.) and sodium nitroprusside (0.1 mg ml−1, i.v.) (Sigma‐Aldrich Co., Poole, UK) were measured to generate baroreflex function curves, as reported previously (Abdala et al. 2012; Lincevicius et al. 2015). Values of matching systolic blood pressure (SBP) variations with reflex heart rate (HR) responses were plotted separately for each vasoactive drug to create linear regression curves for each group, and their slopes were compared to evaluate changes in baroreflex sensitivity (beats min–1 mmHg–1).
Renal function
Renal function was studied as described previously (McBryde et al. 2013; Pijacka et al. 2015). Briefly, animals were housed in metabolic cages with free access to food and water for 24 h. Blood was collected from the tail vein, and was kept on ice until centrifugation. Plasma and urine creatinine were measured by the improved method of Jaffe (1886) with the commercially available QuantiCrom Creatinine Assay Kit (DICT‐500; Universal Biologicals, Cambridge, UK). Creatinine clearance was estimated as: [urinary creatinine (μmol l–1) × urine volume produced in 24 h (ml)]/[plasma creatinine (μmol l–1) × 1440 (min)] (Cornock et al. 2010). Urinary albumin was measured by the improved bromocresol green method with the commercially available Albumin Assay (Randox Laboratories Ltd, Crumlin, UK) and total protein was measured by the method of Lowry et al. (1951) with the DC Protein Assay Kit (Bio‐Rad Laboratories Ltd, Hemel Hempstead, UK).
Primary cultures of dissociated sympathetic neurons
Sympathetic neurons were isolated using a previously reported method (Shanks et al. 2013). Following death, the stellate and mesenteric ganglia were removed from rats and isolated enzymatically with collagenase and trypsin. Experiments were performed 2–3 days after plating. In addition to the 2K1C+sham CSD and 2K1C+CSD groups described above, neurons were also cultured from separate 2K1C (n = 8) and normotensive Wistar (n = 8) groups.
Measurement of free intracellular calcium concentration
[Ca2+]i was determined in single neurons using Fura‐2 acetoxymethyl ester (Fura‐2/AM; 2 μmol l–1) as described previously (Li et al. 2012). Loaded neurons were imaged with a QICLICK digital CCD camera (Photometrics, Tucson, AZ, USA) connected to an OptoLED fluorescence imaging system (Cairn Research Ltd, Faversham, UK), housed on an inverted microscope (Nikon, Tokyo, Japan) equipped with a 40×, oil‐immersion objective. The cover slip containing the neurons was placed into a temperature‐controlled (36 ± 0.5°C), gravity fed, perfusion chamber (volume: 100 μl), perfused with Tyrode solution at a flow rate of 2 ml min−1. The evoked [Ca2+]i transient was evaluated by 30 s of exposure to 50 mmol l−1 KCl (with equimolar reduction in NaCl) in the Tyrode solution. Fura‐2AM was excited alternately at 355 and 380 nm and the emitted at 510 nm. Fluorescence excitation ratios were calculated.
Fluorescence‐activated cell sorting (FACS) analysis of cellular inflammation
Analyses of T cells and macrophages in tissue homogenates of the aorta and brainstem were performed as described previously (McBryde et al. 2013) using FACS. To analyse leukocytes in the aorta and brain, tissue was digested using collagenase type IX (125 U ml−1), collagenase type IS (450 U ml−1) and hyaluronidase IS (60 U ml−1) dissolved in 20 mm Hepes‐PBS buffer for 30 min at 37° C, with constant agitation. The dissolved tissue was then passed through a 70 μm sterile filter (Falcon; BD Biosciences, San Jose, CA, USA), yielding a single cell suspension. An additional step was applied for brain tissue using a 30%/70% Percoll gradient to separate out the mononuclear cell layer. All cells were then washed twice with FACS buffer (0.5% BSA in PBS) then counted, stained and analysed using multicolour flow cytometry. The antibodies (BD Biosciences) used for staining were V450 anti‐CD45, anti‐CD3 PerCP‐eFluor® 710 and anti‐macrophage markers. After immunostaining, cells were re‐suspended in FACS buffer and analysed immediately on a LSR‐II flow cytometer with DIVA software (BD Biosciences). Data were analysed with FlowJo software (Tree Star Inc., Ashland, OR, USA) and an initial gate was applied to exclude cell debris from the analysis. CD45 positive cells were identified as leukocytes within the tissue cell suspension and T cells and macrophages were identified with the above antibodies.
Data acquisition and analysis
Blood pressure telemetry data were acquired and analysed using purpose‐written scripts in Spike2 (CED Ltd, Cambridge, UK). HR, respiratory rate and spectral parameters, including spontaneous cardiac baroreflex gain, were obtained from the arterial waveform data, as described previously (McBryde et al. 2013). Briefly, power spectral density was computed using purpose‐written scripts in Spike2. The frequencies calculated in normalized units were: <0.27 Hz [very low frequency (LF)], 0.27–0.75 Hz (LF) and 0.75–3.3 Hz [high frequency (HF)]. The ratio of the LF to the HF component was used as an indicator of cardiac sympatho‐parasympathetic balance. Spontaneous baroreflex gain was computed and the respiratory rate was inferred from the peaks of respiratory modulation of the systolic pressure frequency spectrum. Averaged data were expressed as the daily average mean or as the within‐animal change from baseline.
Statistical analysis
Data were analysed using Prism, version 6.05 (GraphPad Software Inc., San Diego, CA, USA). Once a normal distribution was confirmed, between groups statistical differences were analysed using a one‐way ANOVA, with Newman‐Keuls post hoc comparisons. Within‐group analyses were performed as the comparison between baseline (days −5 to 0) and week 3 (days 14–21) by repeated measures ANOVA and the Holm–Sidek multiple comparisons test. The type of post hoc analysis performed is indicated as appropriate. P < 0.05 was considered statistically significant. All results are reported as the mean ± SEM.
Results
Cardiovascular responses to CSD
Prior to CSD/sham surgeries, baseline mean arterial pressure (MAP) was significantly elevated after 5 weeks of renal artery clipping in both 2K1C groups compared to normotensive control rats (Wistar: 98 ± 4 mmHg vs. 2K1C+CSD: 142 ± 8 mmHg and 2K1C+sham CSD: 147 ± 8 mmHg; P < 0.001) and there was no significant difference in the level of hypertension between the two 2K1C rat groups. At this time, rats were either sham‐operated or underwent CSD. In the 3 weeks following sham CSD surgery, arterial pressure continued to increase gradually, reaching a level 8 ± 2 mmHg above baseline (P < 0.01). By contrast, 2K1C+CSD animals showed a decrease in MAP by day 21 (−6 ± 2 mmHg; P = 0.02). Thus, a between‐group comparison found that MAP was 14 ± 2 mmHg lower in 2K1C+CSD vs. 2K1C+sham CSD animals (P < 0.01) (Fig. 1). The day/night difference was similar between Wistar and 2K1C rats (6.4 ± 0.8 vs. 3.8 ± 2 mmHg; P = 0.28) and was not changed after CSD (4.6 ± 2 vs. 3.7 ± 1.6 mmHg; P = 0.83).
Figure 1. Cardiovascular response to CSD or sham surgery (sham CSD) 5 weeks after induction of 2K1C renovascular hypertension .
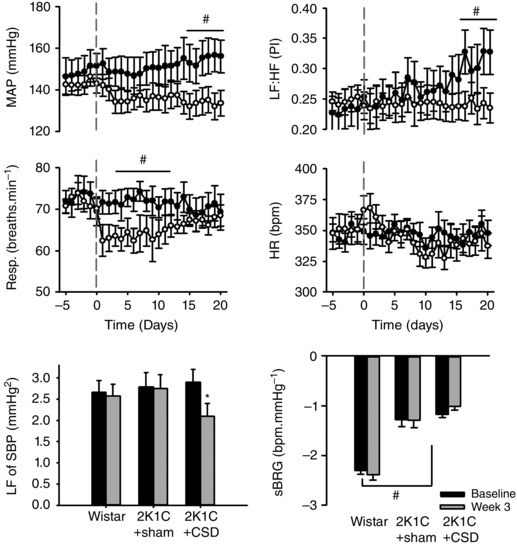
Top: temporal responses in MAP, respiratory frequency (Resp.), HR and heart rate variability (LF:HF) responses 5 days before and 21 days after CSD or sham CSD surgery (grey dashed vertical line; n = 9 per group). Bottom: average responses of baseline (days −5 to 0) vs. week 3 (days 14–21) in Wistar, 2K1C+sham CSD and 2K1C+CSD rats for LF power of systolic blood pressure (LF of SBP) and spontaneous baroreflex gain (sBRG). Differences were evaluated using one‐way ANOVA with Holm–Sidek post hoc comparisons; # P < 0.05 between‐groups comparison; * P < 0.05 repeated measures within‐subject comparison.
The respiration rate was not significantly different at baseline between groups (75 ± 1 vs. 76 ± 2 breaths min−1) but fell by 9 ± 1breaths min−1 at around day 5 in 2K1C+CSD but not 2K1C+sham CSD rats (P < 0.01). Respiration recovered to baseline levels by day 8, with no significant difference from either baseline, or between groups, for the remainder of the experiment (Fig. 1).
Baseline heart rate was not significantly different between 2K1C and normotensive rats (Wistar: 345 ± 7 beats min–1 vs. 2K1C+sham CSD: 346 ± 7 beats min–1 and 2K1C+CSD: 352 ± 8 beats min–1). There were no statistically significant changes in heart rate after CSD.
Spontaneous baroreflex gain was significantly reduced in 2K1C hypertension compared to Wistar rats (−1.3 ± 0.13 vs. −2.3 ± 0.08 beats min–1 mmHg−1) but did not significantly differ within or between groups following CSD or sham CSD surgery (Fig. 1). The LF:HF ratio of pulse interval was significantly lower in both 2K1C groups compared to Wistar control rats (0.23 ± 0.02 and 0.25 ± 0.03 vs. 0.39 ± 0.04; P > 0.01). Over the 21 day recording window, the LF:HF ratio increased in 2K1C+sham CSD rats (0.23 ± 0.02 to 0.32 ± 0.03; P < 0.05) but not 2K1C+CSD rats (0.25 ± 0.02 to 0.25 ± 0.03; P = 0.69), leaving a significant between groups difference by week 3 (P < 0.05) (Fig. 1). This indicates that CSD may prevent further deterioration in cardiac sympatho‐vagal balance in 2K1C rats. The LF component of SBP decreased from baseline by day 21 in 2K1C+CSD rats (2.9 ± 0.3 to 2.1 ± 0.3 mmHg2; P < 0.05) (Fig. 1).
Chemoreflex and baroreflex responses to CSD
The success of CSD was confirmed by 2K1C+CSD rats showing little or no arterial pressure response and no bradycardia response (ΔHR: 2K1C+sham CSD −191.8 ± 30.5; 2K1C+CSD −4.4 ± 3.6; Wistar −182.9 ± 48.0) to transient sodium cyanide activation of the chemoreflex compared to the powerful pressor response seen in sham CSD rats (P < 0.001) (Fig. 2 A). 2K1C+CSD rats also exhibited an improved tachycardic response to induced falls in blood pressure compared to 2K1C+sham CSD animals (P < 0.05) (Fig. 2 B). However, the bradycardic response to induced increases in blood pressure was not significantly different between 2K1C+CSD and 2K1C+sham CSD rats (P = 0.1) (Fig. 2 B).
Figure 2. Assessment of chemoreflex and baroreflex sensitivity in 2K1C+sham CSD and 2K1C+CSD rats .
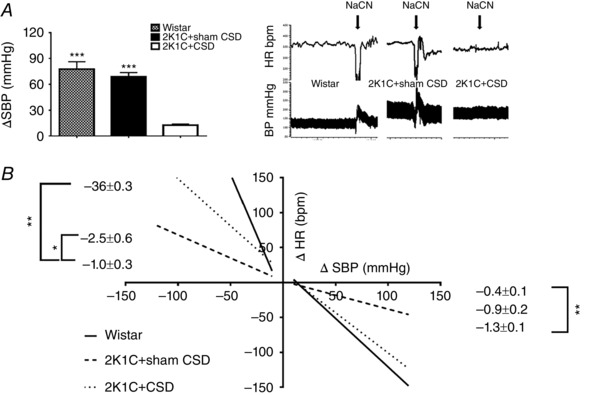
A, chemoreflex‐induced increases in SBP by bolus infusion of sodium cyanide (NaCN; 120 μg kg−1, i.v.) were abolished after CSD; P < 0.001. B, cardiac baroreceptor reflex curves produced with i.v. infusions of vasoactive drugs (sodium nitroprusside and phenylephrine) showed an increase in sensitivity during sodium nitroprusside infusion in 2K1C+CSD vs. 2K1C+sham CSD rats (P < 0.05). Note that the responses in the 2K1C+CSD occurred despite a loss of carotid sinus baroreceptor inputs and were mediated by aortic baroreceptors. Data were analysed by one‐way ANOVA with Newman–Keuls multiple comparisons post hoc test (n = 4). * P < 0.05, ** P < 0.01 and *** P < 0.001. Data are presented as the mean ± SEM.
Intracellular free calcium transients in sympathetic neurons
During depolarization with high K+, neurons cultured from the stellate ganglia of 2K1C rats showed a greater increase in [Ca2+]i (2.71 ± 0.26, n = 10) compared to those from Wistar animals (2.08 ± 0.13, n = 20, P < 0.05). This heightened [Ca2+]i response evoked was decreased by 24% in 2K1C+CSD rats (P < 0.05) (Fig. 3 C and D). By contrast, no significant differences in [Ca2+]i were observed for neurons cultured from the mesenteric ganglia (Fig. 3 E) of Wistar, 2K1C or 2K1C+CSD rats.
Figure 3. Intracellular free calcium transients in sympathetic neurons from the 2K1C+sham CSD rats, 2K1C+CSD rats and age‐matched controls (Wistar) .
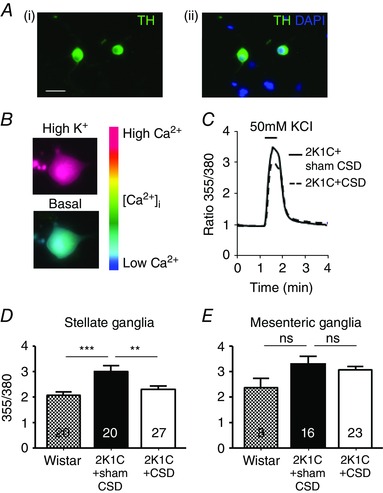
A, (i) all neurons imaged were sympathetic as indicated by tyrosine hydroxylase immunopositivity (green) and (ii) were localized in relation to other nuclei stained in blue by 4′,6‐diamidino‐2‐phenylindole. B, pseudocolour‐coded ratio images of Fura‐2‐loaded neurons were obtained by conventional fluorescence microscopy. Ca2+ concentrations were colour‐coded with a basal Ca2+ concentration in blue and a high Ca2+ concentration in red. C, example recording from a stellate neuron exposed to 50 mmol l−1 of KCl (30 s) depolarization, resulting in a rise in intracellular free calcium concentration ([Ca2+]i). Note that CSD attenuated this response. D and E, quantitative data showing the difference in the peak evoked [Ca2+]i between Wistar and Goldblatt hypertensive rats with CSD (2K1C+CSD) or sham CSD (2K1C+sham CSD), obtained from stellate ganglia (D) and mesenteric ganglia (E) neurons collected and cultured from >3 rats per group. Numbers within bars refers to the number of neurons tested, with each response per neuron representing one K exposure. ** P < 0.01 and *** P < 0.001; one‐way ANOVA with Holm–Sidek post hoc comparisons test.
Inflammatory changes in the aorta and brainstem
When compared with 2K1C+Sham CSD rats, no differences were observed in the percentage of vascular aortic immune cell infiltrates (CD45+, CD3+ cells and macrophages) in 2K1C+CSD rats. However, the percentage of macrophages in brainstem homogenates was significantly reduced in 2K1C+CSD rats (P < 0.05) (Fig. 4).
Figure 4. FACS data showing immune cell infiltration of vascular aortic and brainstem tissue in the 2K1C+sham CSD rats, 2K1C+CSD rats and age‐matched control Wistars .
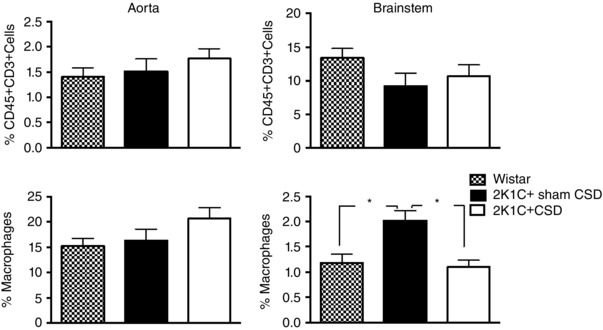
CSD reduced macrophage infiltration in the brainstem but had no effect on CD45 and CD3 expressing T cells. One‐way ANOVA with Newman–Keuls multiple comparisons test. * P < 0.05. Data are presented as the mean ± SEM.
Renal function after CSD
2K1C hypertension was associated with increased plasma creatinine, decreased creatinine clearance, elevated albuminuria and proteinuria and increases in both water intake and urine production (P < 0.05 Wistar vs. sham CSD) (Fig. 5). CSD did not change plasma creatinine or creatinine clearance but did normalize urinary protein and albumin levels (P < 0.05 CSD vs. Wistar and sham CSD) (Fig. 5). Following CSD, water intake and urine production tended to decrease slightly to an intermediate level not significantly different from either Wistar or sham CSD groups (Fig. 5).
Figure 5. Effect of CSD or sham surgery on renal function .
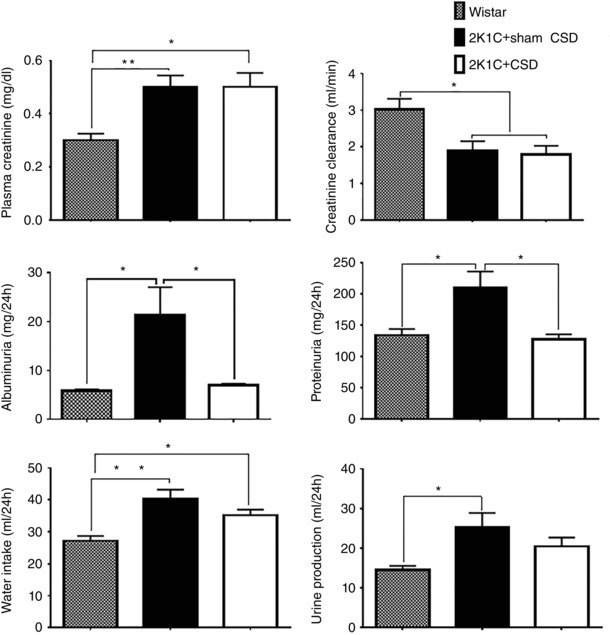
Renal function was assessed at 21 days after CSD (n = 10) or sham CSD (n = 14) in 2K1C hypertensive rats, as well as in age‐matched Wistar controls (n = 11). 2K1C hypertension was associated with multiple impairments in renal function compared to Wistar controls. CSD normalized proteinuria and albuminuria (P < 0.05). Data were analysed by one‐way ANOVA with Newman–Keuls multiple comparisons tests. * P < 0.05 and ** P < 0.01. Data are presented as the mean ± SEM.
Discussion
The results of the present study are the first ever demonstration indictating that renovascular hypertension can be ameliorated by CSD in the 2K1C model. We found that resecting carotid sinus nerves bilaterally 5 weeks after renal artery clipping reduced arterial pressure, thereby preventing the further increases in arterial pressure seen in sham‐operated animals where the carotid nerves were left intact. Our observed reduction in the hypertensive burden of 2K1C rats was accompanied by some improvements in baroreflex control and renal function, a restoration of stellate calcium signalling, and a reduction in neural inflammatory markers. The present study is an important extension of our previous work demonstrating the efficacy of CSD in another experimental model of hypertension (Abdala et al. 2012; McBryde et al. 2013), which identified the carotid bodies as a potential therapeutic target for treating refractory or essential hypertension in human patients (Paton et al. 2013 b; Ratcliffe et al. 2014). The results of the present study suggest that the carotid body may be a viable therapeutic target for renovascular hypertension.
We observed a significant impairment in renal function assessed after 8 weeks of renal artery clipping, with reduced creatinine clearance, and increased plasma creatinine, albuminuria and proteinuria, suggesting a breakdown in the filtration barrier. Carotid sinus nerve resection at 5 weeks post clipping was observed to normalize albuminuria and proteinuria but not other indicators of renal function (Fig. 5). Although a presumed decrease in renal sympathetic drive may be involved, we suggest that this may also have occurred secondary to the decrease in arterial pressure, which could itself reduce the excessive pressure in glomerular capillaries and also reduce leakage from the intact kidney. The lack of improvement in other indicators of renal function such as creatinine clearance may be explained by an already prolonged exposure to protein in the filtrate, which can cause inflammation and scarring in the renal tubules, with subsequent nephron loss, and an ensuing fall in renal filtration rate that is more difficult to rescue (Abbate et al. 2006). Thus, we conclude that changes in renal function are probably do not comprise the primary mechanism for the anti‐hypertensive effect of CSD in renovascular hypertension.
Using both direct and indirect measures, our group has previously suggested that 2K1C hypertension is associated with an increase sympathetic drive, which precedes the increase in arterial pressure (Oliveira‐Sales et al. 2014; Campos et al. 2015; Oliveira‐Sales et al. 2016). Consistent with earlier work (Oliveira‐Sales et al. 2014; Campos et al. 2015; Oliveira‐Sales et al. 2016), the results of the present study show that 2K1C hypertension is associated with impaired cardiac baroreflex sensitivity and impaired cardiac sympatho‐vagal balance (Fig. 2 B), which has previously been reported to contribute to the development and maintenance of essential hypertension (Abdala et al. 2012; McBryde et al. 2013). Interestingly, neither the bradycardia response to an increase in arterial pressure, nor the net spontaneous baroreflex gain was observed to change with CSD. Importantly, despite the complete bilateral resection of the carotid sinus nerves and the removal of carotid baroreceptor input, no impairment in baroreflex function was observed. This result may initially be counterintuitive, although the aortic baroreceptors are known to play a dominant role in mediating cardiac baroreflex in the rat (Pickering et al. 2008). Furthermore, the result is consistent with our previous work where CSD was found to improve baroreflex gain in the SHR model (Abdala et al. 2012). We suggest that the improvements in baroreceptor reflex function may be a result of central remodelling following the removal of the antagonism of peripheral chemoreceptor overactivity.
Analysis of the calcium transients in post‐ganglionic mesenteric and stellate ganglia revealed an increase in stellate [Ca2+]i transients in 2K1C rats. These results indicate that CSD may be causing remodelling of post‐ganglionic sympathetic neuronal function. This is consistent with previous results reported in the SHR, which also showed that, at the level of the end organ, abnormalities in post‐ganglionic intracellular calcium signalling (Li et al. 2013) may result in the enhanced exocytosis and impaired re‐uptake of noradrenaline from sympathetic neuronal terminals (Li et al. 2012; Shanks et al. 2013). We have shown previously that activation of the peripheral chemoreflex can evoke powerful increases in inferior cardiac sympathetic activity (Boscan et al. 2001). Interestingly, although we observed attenuation of stellate [Ca2+]i transients with CSD in the present study, we did not observe any corresponding reduction in heart rate after CSD. We suggest that any functional effects of cardiac post‐ganglionic sympathetic remodelling are probbaly iono‐ or dromotrophic rather than chronotrophic; this is an area that warrants further investigation. It is unclear whether our observed effects of CSD on calcium signalling are a result of the direct resetting of the arterial chemoreceptor cardiac sympathetic axis, or may have occurred secondary to the differences in arterial blood pressure itself. Although the within‐subject fall in blood pressure after CSD was modest, hypertension in the sham‐operated group continued to increase with time, such that, at the time tissue was removed for testing, the between group difference was ∼14 mmHg. Our finding that calcium signalling in the mesenteric ganglia was not affected by CSD should be taken with the caveat that our tissue collection protocol was unable to discriminate between the clipped and unclipped kidneys. Earlier work has shown that renal denervation can mitigate hypertension and reduce sympathetic tone in the 2K1C rat, although only when the nerves from the clipped kidney are ablated (Katholi et al. 1982). Subsequent work has shown a similar fall in arterial pressure after selective renal afferent denervation by dorsal rhizotomy but, again, no effect is seen with a contralateral denervation (Wyss et al. 1986). This suggests that the clipped and unclipped renal nerves probably play quite different roles in the development and maintenance of renovascular hypertension. Thus, it is perhaps not surprising that our ‘mixed approach’ did not yield any significant results, and any future work should attempt to clarify these differences.
CSD was associated with reduced brainstem macrophage infiltration compared to sham‐operated animals, consistent with our previously findings in the SHR (McBryde et al. 2013). We suggest that there are several possible explanations for this observation. First, the reduction in inflammation may simply occur secondary to the reduction in the prevailing level of pressure. Alternatively, given that the brainstem is a key region responsible for the regulation of arterial blood pressure and sympathetic activity, if the removal of carotid body input itself reduces brainstem inflammation, this may contribute causally to the reduction in arterial pressure. In favour of the latter, it has been reported recently that the generation of the sympathetic vasomotor tone and maintenance of hypertension in 2K1C rats is mediated by increases in reactive oxygen species production in the rostroventrolateral medulla (Oliveira‐Sales et al. 2010). We speculate that an increase in brainstem reactive oxygen species may contribute to brainstem inflammation, or vice versa. Alternatively, brainstem inflammation may be triggered via an afferent feedback mechanism from the ischaemic kidney, and/or high circulating levels of Ang II (Marvar et al. 2010; Oliveira‐Sales et al. 2014). Further characterization of the inflammatory cells following CSD is required to determine the potential neuro‐immune modulation in the 2K1C model of renovascular hypertension, and we recognize the importance of assessing whether CSD altered the inflammatory state of the kidney in future studies.
Both renovascular and essential hypertension are associated with sympathetic overactivity (Johansson et al. 1999; Carthy, 2014; Grassi et al. 2014). Recent evidence has demonstrated that the carotid bodies contribute to increased sympathetic drive in several models of cardiovascular disease (McBryde et al. 2013; Marcus et al. 2014 a). In the present study conducted in rats with renovascular hypertension, CSD improved indicators of cardiac sympatho‐vagal balance compared to sham‐operated animals. Although the commonly‐used (Kuwahara et al. 1994; Waki et al. 2006) indirect methods used in the present study to assess autonomic tone are not universally accepted (Billman, 2013), our results are consistent with both our present finding that CSD reduced [Ca2+]i in stellate cells from 2K1C rats and our previous finding that CSD reduces renal sympathetic nerve activity in another model of hypertension (McBryde et al. 2013). Thus, we suggest that there is a strong likelihood that our observed fall in arterial pressure after the removal of peripheral chemoreceptor input is the result, at least in part, of a reduction in sympathetic drive. Although the purpose of the present study was to examine the impact of CSD on established renovascular hypertension, it would be interesting to investigate whether CSD can prevent the onset of renovascular hypertension, as we have previously shown to be the case in the pre‐hypertensive young SHR (Abdala et al. 2012). We predict that, although CSD probbaly does not fully prevent renovascular hypertension, given that both chemoreflex sensitivity and sympathetic outflow appear to pre‐date the increase in blood pressure in 2K1C hypertension, the pre‐emptive removal of the carotid bodies may limit the degree of hypertension.
Following CSD, we observed a transient fall in respiratory rate, which returned to baseline rates within 5 days. We have previously observed a similar response after CSD in SHR but not Wistar rats (Abdala et al. 2012; McBryde et al. 2013). We interpret this result to indicate that there is tonic drive from the peripheral chemoreceptors in both the SH and 2K1C rat, the interruption of which is responsible for the temporary dip in the rate of breathing until compensatory central remodelling occurs. This is a further indicator of abnormal peripheral chemoreceptor function in the 2K1C hypertensive rat.
In terms of possible mechanisms linking renal stenosis to carotid body function, we speculate that these effects may be mediated in glomus cells by Ang II via type 1 angiotensin II receptors (AT1R), which have been shown to increase carotid body activity (Allen, 1998). In pathological conditions where Ang II production is chronically increased, carotid body chemoreceptor sensitivity to hypoxia and/or cyanide is enhanced, and has been linked to AT1R up‐regulation (Li & Schultz, 2006; Li et al. 2006; Marcus et al. 2010; Peng et al. 2011; Oliveira‐Sales et al. 2016). Because 2K1C hypertension is known to be Ang II‐dependent, it is possible that increased Ang II production could contribute to the excitation of the glomus cells in this model. Furthermore, the actions of Ang II on the AT1R have also been shown to drive inflammation (Chabrashvili et al. 2003) and facilitate the release of noradrenaline from sympathetic nerves (Maruyama et al. 2000; Fabiani et al. 2001), which is consistent with the findings of the present study. The increased sympathetic tone seen in renovascular hypertension has also recently been linked to an up‐regulation of AT1R in the rostroventrolateral medulla (Lincevicius et al. 2015). Thus, it is possible that brainstem inflammation and sympathetic overactivity in renovascular hypertension may be driven by elevated ANG II signalling. Healy et al. (1989) found that sino‐aortic denervation was associated with a decrease in brainstem AT1‐R in normal Wistar rats. This suggests that interrupting the neural reflex arcs controlling arterial pressure can directly impact brainstem Ang II signalling, although no attempt was made to resolve chemoreceptor vs. baroreceptor pathways. We speculate that CSD may act, at least in part, via reducing central AT1 receptors to reduce brainstem inflammation. This putative relationship between Ang II/AT1‐R signalling and carotid body function in renovascular hypertension should be examined in future studies.
Perspectives
The results reported in the present study support the concept that the carotid body should be considered as a potential therapeutic target in renovascular hypertension. This extends our previously published results showing benefits in an animal model of neurogenic hypertension (Abdala et al. 2012; McBryde et al. 2013) and adds to the growing body of evidence suggesting that the carotid body may be a suitable target in a range of cardiovascular diseases characterized by parallel changes in peripheral chemoreceptor function and autonomic imbalance (Del Rio et al. 2013; McBryde et al. 2013; Niewinski et al. 2013; Paton et al. 2013 a; Paton et al. 2013 b; Marcus et al. 2014 b; Del Rio et al. 2015; Moraes et al. 2015). Recent studies have evaluated the feasibility of both surgical resection (Narkiewicz et al. 2016) and a novel pharmacological compound (Pijacka et al. 2016) to selectively target the carotid body in resistant hypertension. Notably, renal artery stenosis remains difficult to identify and treat clinically. Renal function studies show that only 5–10% of patients with renal artery stenosis exhibit increased plasma creatinine, yet most have high arterial pressure (Safian & Textor, 2001); thus, many renovascular pathology patients may be initially misdiagnosed with essential hypertension. Because removing chemoreflex drive is effective in animal models of both neurogenic and renovascular hypertension, we suggest that the the carotid body presents a viable therapeutic target independent of these types of hypertension. This, together with an improved selective targeting of the carotid body (perhaps via pharmacological modulation), awaits further testing.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
JFRP was responsible for acquisition of funding, administrative support, study conception and experimental design, and data interpretation. FDM, WP, LW and APA designed the experiments, as well as collected, analysed and interpreted data for the main study, performed at the University of Bristol. PJM was responsible for the inflammatory markers component during a fellowship at the University of Bristol. GSL, DL and DJP were responsible for the calcium imaging study performed at Oxford University, using tissue collected from Bristol. All authors contributed to the writing of this study. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by funding from the British Heart Foundation (RG/12/6/29670), Royal Society, NIH (1R01AT008632‐01), European Commission Research Executive Agency Marie Curie International Incoming Fellowship (P.J.M) (MC‐IIF – 276147); NIH R00 HL107675‐03 (PJM); American Heart Association 15CSA24340001 (PJM) and a gift from CiBiem NC1 to JFRP.
Acknowledgements
We acknowledge the assistance of Dr Elizabeth Oliviera‐Sales in establishing the 2K1C model of hypertension.
Linked articles This article is highlighted by a Perspective by Marcus. To read this Perspective, visit http://dx.doi.org/10.1113/JP273323.
This is an Editor's Choice article from the 1 November 2016 issue.
References
- Abbate M, Zoja C & Remuzzi G (2006). How does proteinuria cause progressive renal damage? J Am Soc Nephrol 17, 2974–2984. [DOI] [PubMed] [Google Scholar]
- Abdala AP, McBryde FD, Marina N, Hendy EB, Engelman ZJ, Fudim M, Sobotka PA, Gourine AV & Paton JF (2012). Hypertension is critically dependent on the carotid body input in the spontaneously hypertensive rat. J Physiol 590, 4269–4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen A (1998). Angiotensin AT1 receptor‐mediated excitation of rat carotid body chemoreceptor afferent activity. J Physiol 510, 773–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billman GE (2013). The LF/HF ratio does not accurately measure cardiac sympatho‐vagal balance. Front Physiol 4, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bivol LM, Berge RK & Iversen BM (2008). Tetradecylthioacetic acid prevents the inflammatory response in two‐kidney, one‐clip hypertension. Am J Physiol Regul Integr Comp Physiol 294, R438–R447. [DOI] [PubMed] [Google Scholar]
- Boscan P, Allen AM & Paton JF (2001). Baroreflex inhibition of cardiac sympathetic outflow is attenuated by angiotensin II in the nucleus of the solitary tract. Neuroscience 103, 153–160. [DOI] [PubMed] [Google Scholar]
- Campos RR, Oliveira‐Sales EB, Nishi EE, Paton JF & Bergamaschi CT (2015). Mechanisms of renal sympathetic activation in renovascular hypertension. Exp Physiol 100, 496–501. [DOI] [PubMed] [Google Scholar]
- Carthy ER (2014). Autonomic dysfunction in essential hypertension: A systematic review. Ann Med Surg 3, 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabrashvili T, Kitiyakara C, Blau J, Karber A, Aslam S, Welch WJ & Wilcox CS (2003). Effects of ANG II type 1 and 2 receptors on oxidative stress, renal NADPH oxidase, and SOD expression. Am J Physiol Regul Integr Comp Physiol 285, R117–R124. [DOI] [PubMed] [Google Scholar]
- Cheng J, Zhou W, Warner GM, Knudsen BE, Garovic VD, Gray CE, Lerman LO, Platt JL, Romero JC, Textor SC, Nath KA & Grande JP (2009). Temporal analysis of signaling pathways activated in a murine model of two‐kidney, one‐clip hypertension. Am J Physiol Renal Physiol 297, F1055–F1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornock R, Langley‐Evans SC, Mobasheri A & McMullen S (2010). The impact of maternal protein restriction during rat pregnancy upon renal expression of angiotensin receptors and vasopressin‐related aquaporins. Reprod Biol Endocrinol 8, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Rio R, Andrade DC, Marcus NJ & Schultz HD (2015). Selective carotid body ablation in experimental heart failure: a new therapeutic tool to improve cardiorespiratory control. Exp Physiol 100, 136–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Rio R, Marcus NJ & Schultz HD (2013). Carotid chemoreceptor ablation improves survival in heart failure: rescuing autonomic control of cardiorespiratory function. J Am Coll Cardiol 62, 2422–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiani ME, Sourial M, Thomas WG, Johnston CI, Johnston CI & Frauman AG (2001). Angiotensin II enhances noradrenaline release from sympathetic nerves of the rat prostate via a novel angiotensin receptor: implications for the pathophysiology of benign prostatic hyperplasia. J Endocrinol 171, 97–108. [DOI] [PubMed] [Google Scholar]
- Goldblatt H, Lynch J, Hanzal RF & Summerville W (1934). Studies on Experimental Hypertension 1. The production of persistent elevation of systlic blood pressure by means of renal ischemia. J Exp Med 59, 347–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi G, Seravalle G, Brambilla G, Pini C, Alimento M, Facchetti R, Spaziani D, Cuspidi C & Mancia G (2014). Marked sympathetic activation and baroreflex dysfunction in true resistant hypertension. Int J Cardiol 177, 1020–1025. [DOI] [PubMed] [Google Scholar]
- Hansen KJ, Edwards MS, Craven TE, Cherr GS, Jackson SA, Appel RG, Burke GL & Dean RH (2002). Prevalence of renovascular disease in the elderly: a population‐based study. J Vasc Surg 36, 443–451. [DOI] [PubMed] [Google Scholar]
- Healy DP, Rettig R, Nguyen T & Printz MP (1989). Quantitative autoradiography of angiotensin II receptors in the rat solitary‐vagal area: effects of nodose ganglionectomy or sinoaortic denervation. Brain Res 484, 1–12. [DOI] [PubMed] [Google Scholar]
- Jaffe M (1886). Ueber den Niederschlag welchen Pikrinsäure in normalen Harn erzeugt und über eine neue reaction des Kreatinins. Z Physiol Chem 10, 391–400. [Google Scholar]
- Johansson M, Elam M, Rundqvist B, Eisenhofer G, Herlitz H, Lambert G & Friberg P (1999). Increased sympathetic nerve activity in renovascular hypertension. Circulation 99, 2537–2542. [DOI] [PubMed] [Google Scholar]
- Katholi RE, Whitlow PL, Winternitz SR & Oparil S (1982). Importance of the renal nerves in established two‐kidney, one clip Goldblatt hypertension. Hypertension 4, 166–174. [PubMed] [Google Scholar]
- Kuwahara M, Yayou K, Ishii K, Hashimoto S, Tsubone H & Sugano S (1994). Power spectral analysis of heart rate variability as a new method for assessing autonomic activity in the rat. J Electrocardiol 27, 333–337. [DOI] [PubMed] [Google Scholar]
- Li D, Lee CW, Buckler K, Parekh A, Herring N & Paterson DJ (2012). Abnormal intracellular calcium homeostasis in sympathetic neurons from young prehypertensive rats. Hypertension 59, 642–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Nikiforova N, Lu CJ, Wannop K, McMenamin M, Lee CW, Buckler KJ & Paterson DJ (2013). Targeted neuronal nitric oxide synthase transgene delivery into stellate neurons reverses impaired intracellular calcium transients in prehypertensive rats. Hypertension 61, 202–207. [DOI] [PubMed] [Google Scholar]
- Li YL & Schultz HD (2006). Enhanced sensitivity of Kv channels to hypoxia in the rabbit carotid body in heart failure: role of angiotensin II. J Physiol 575, 215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YL, Xia XH, Zheng H, Gao L, Li YF, Liu D, Patel KP, Wang W & Schultz HD (2006). Angiotensin II enhances carotid body chemoreflex control of sympathetic outflow in chronic heart failure rabbits. Cardiovasc Res 71, 129–138. [DOI] [PubMed] [Google Scholar]
- Lincevicius GS, Shimoura CG, Nishi EE, Perry JC, Casarini DE, Gomes GN, Bergamaschi CT & Campos RR (2015). Aldosterone Contributes to Sympathoexcitation in Renovascular Hypertension. Am J Hypertens 28, 1083–1090. [DOI] [PubMed] [Google Scholar]
- Lopez‐Novoa JM, Rodriguez‐Pena AB, Ortiz A, Martinez‐Salgado C & Lopez Hernandez FJ (2011). Etiopathology of chronic tubular, glomerular and renovascular nephropathies: clinical implications. J Transl Med 9, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL & Randall RJ (1951). Protein measurement with the Folin phenol reagent. J Biol Chem 193, 265–275. [PubMed] [Google Scholar]
- Marcus JA, Pothineni A, Marcus CZ & Bisognano JD (2014. a). The role of obesity and obstructive sleep apnea in the pathogenesis and treatment of resistant hypertension. Curr Hypertens Rep 16, 411. [DOI] [PubMed] [Google Scholar]
- Marcus NJ, Del Rio R & Schultz HD (2014. b). Central role of carotid body chemoreceptors in disordered breathing and cardiorenal dysfunction in chronic heart failure. Front Physiol 5, 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus NJ, Li YL, Bird CE, Schultz HD & Morgan BJ (2010). Chronic intermittent hypoxia augments chemoreflex control of sympathetic activity: role of the angiotensin II type 1 receptor. Respir Physiol Neurobiol 171, 36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama R, Hatta E, Yasuda K, Smith NC & Levi R (2000). Angiotensin‐converting enzyme‐independent angiotensin formation in a human model of myocardial ischemia: modulation of norepinephrine release by angiotensin type 1 and angiotensin type 2 receptors. J Pharmacol Exp Ther 294, 248–254. [PubMed] [Google Scholar]
- Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C, Gordon FJ & Harrison DG (2010). Central and peripheral mechanisms of T‐lymphocyte activation and vascular inflammation produced by angiotensin II‐induced hypertension. Circ Res 107, 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBryde FD, Abdala AP, Hendy EB, Pijacka W, Marvar P, Moraes DJ, Sobotka PA & Paton JF (2013). The carotid body as a putative therapeutic target for the treatment of neurogenic hypertension. Nat Commun 4, 2395. [DOI] [PubMed] [Google Scholar]
- Moraes DJ, Machado BH & Paton JF (2015). Carotid body overactivity induces respiratory neurone channelopathy contributing to neurogenic hypertension. J Physiol 593, 3055–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narkiewicz K, Ratcliffe L, Hart EC, Briant LJB, Chrostowska M, Wolf J, Szyndler A, Hering D, Abdala AP, Manghat N, Burchell A, Durant C, Lobo M, Sobotka PA, Nk Patel, Leiter J, Engelman ZJ, Nightengale A & Paton JFR (2016). Unilateral carotid body resection in resistant hypertension: a safety and feasibility trial. J Am Coll Cardiol 1, 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoletti A, Heudes D, Mandet C, Hinglais N, Bariety J & Michel JB (1996). Inflammatory cells and myocardial fibrosis: spatial and temporal distribution in renovascular hypertensive rats. Cardiovasc Res 32, 1096–1107. [DOI] [PubMed] [Google Scholar]
- Niewinski P, Janczak D, Rucinski A, Jazwiec P, Sobotka PA, Engelman ZJ, Fudim M, Tubek S, Jankowska EA, Banasiak W, Hart EC, Paton JF & Ponikowski P (2013). Carotid body removal for treatment of chronic systolic heart failure. Int J Cardiol 168, 2506–2509. [DOI] [PubMed] [Google Scholar]
- Oliveira‐Sales EB, Colombari DS, Davisson RL, Kasparov S, Hirata AE, Campos RR & Paton JF (2010). Kidney‐induced hypertension depends on superoxide signaling in the rostral ventrolateral medulla. Hypertension 56, 290–296. [DOI] [PubMed] [Google Scholar]
- Oliveira‐Sales EB, Colombari E, Abdala AP, Campos RR & Paton JF (2016). Sympathetic overactivity occurs before hypertension in the two‐kidney, one‐clip model. Exp Physiol 101, 67–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira‐Sales EB, Toward MA, Campos RR & Paton JF (2014). Revealing the role of the autonomic nervous system in the development and maintenance of Goldblatt hypertension in rats. Auton Neurosci 183, 23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton JF, Ratcliffe L, Hering D, Wolf J, Sobotka PA & Narkiewicz K (2013. a). Revelations about carotid body function through its pathological role in resistant hypertension. Curr Hypertens Rep 15, 273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton JF, Sobotka PA, Fudim M, Engelman ZJ, Hart EC, McBryde FD, Abdala AP, Marina N, Gourine AV, Lobo M, Patel N, Burchell A, Ratcliffe L & Nightingale A (2013. b). The carotid body as a therapeutic target for the treatment of sympathetically mediated diseases. Hypertension 61, 5–13. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Raghuraman G, Khan SA, Kumar GK & Prabhakar NR (2011). Angiotensin II evokes sensory long‐term facilitation of the carotid body via NADPH oxidase. J Appl Physiol (1985) 111, 964–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering AE, Simms AE & Paton JF (2008). Dominant role of aortic baroreceptors in the cardiac baroreflex of the rat in situ. Auton Neurosci 142, 32–39. [DOI] [PubMed] [Google Scholar]
- Piecha G, Wiecek A & Januszewicz A (2012). Epidemiology and optimal management in patients with renal artery stenosis. J Nephrol 25, 872–878. [DOI] [PubMed] [Google Scholar]
- Pijacka W, Clifford B, Tilburgs C, Joles JA, Langley‐Evans S & McMullen S (2015). Protective role of female gender in programmed accelerated renal aging in the rat. Physiol Rep 3, e12342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijacka W, Moraes DJ, Ratcliffe L, Nightengale A, Hart EC, da Silva MP, Machado BH, McBryde FD, Abdala AP, Ford AP & Paton JFR (2016). Purinergic receptors in the carotid body as a new drug target for controlling hypertension. Nat Med 22, 1151–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliffe LE, Pijacka W, McBryde FD, Abdala AP, Moraes DJ, Sobotka PA, Hart EC, Narkiewicz K, Nightingale AK & Paton JF (2014). CrossTalk opposing view: Which technique for controlling resistant hypertension? Carotid chemoreceptor denervation/modulation. J Physiol 592, 3941–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz‐Ortega M, Esteban V, Ruperez M, Sanchez‐Lopez E, Rodriguez‐Vita J, Carvajal G & Egido J (2006). Renal and vascular hypertension‐induced inflammation: role of angiotensin II. Curr Opin Nephrol Hypertens 15, 159–166. [DOI] [PubMed] [Google Scholar]
- Safian RD & Textor SC (2001). Renal‐artery stenosis. N Engl J Med 344, 431–442. [DOI] [PubMed] [Google Scholar]
- Shanks J, Mane S, Ryan R & Paterson DJ (2013). Ganglion‐specific impairment of the norepinephrine transporter in the hypertensive rat. Hypertension 61, 187–193. [DOI] [PubMed] [Google Scholar]
- Waki H, Katahira K, Polson JW, Kasparov S, Murphy D & Paton JF (2006). Automation of analysis of cardiovascular autonomic function from chronic measurements of arterial pressure in conscious rats. Exp Physiol 91, 201–213. [DOI] [PubMed] [Google Scholar]
- Weber MA (2014). The evolving clinical management of hypertension. J Clin Hypertens (Greenwich) 16, 917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss JM, Aboukarsh N & Oparil S (1986). Sensory denervation of the kidney attenuates renovascular hypertension in the rat. Am J Physiol Heart Circ Physiol 250, H82–H86. [DOI] [PubMed] [Google Scholar]