

Abstract
We have developed composite hydrogels of chitosan (CS) and mesoporous silica nanoparticles (MSNs) in this study. The gelation rate, gel strength, drug delivery behavior and chondrocyte proliferation properties were investigated. The introduction of MSNs into CS accelerated the gelation process at body temperature and also increased the elastic modulus G′ from 1000 to 1800 Pa. When we used gentamicin (GS) and bovine serum albumin (BSA) as model small chemical drugs and biomacromolecules, respectively, the CS/MSN hydrogels released GS and BSA in a sustained manner simultaneously, but the CS hydrogels only showed sustained BSA release. Furthermore, in vitro chondrocyte culture showed that the CS/MSN composite hydrogels indeed performed much better in supporting chondrocyte growth and maintaining chondrocytic phenotype compared to the CS hydrogels. Therefore, the results suggest that the CS/MSN composite hydrogels can be potentially very useful for cartilage regeneration.
Keywords: hydrogels, chitosan, mesoporous silica nanoparticles, cartilage regeneration
1. Introduction
Cartilage regeneration is one of the most successful cases in tissue engineering research [1]. Many types of scaffolds with varied structures and morphologies, such as porous scaffolds, fibrous scaffolds, hydrogels and microcarriers [2–5], have been utilized in delivering chondrocytes and correlative molecules to repair cartilage defects. Since chondrocytes are of round or ellipsoidal morphologies in the natural cartilage matrix, which is crucial in maintaining phenotype, studies have demonstrated that hydrogels are helpful in maintaining the round or ellipsoidal cell shapes [6–8]. Moreover, the water-rich environment in hydrogels is beneficial in protecting cells, preventing the inactivation of drug molecules such as proteins and delivering nutrients as well [9, 10] It is worth noting that hydrogels are able to transform from the solution to gel form with fixed semi-solid shape and a certain mechanical strength under certain external physical or chemical stimulations. Therefore, these smart features endow the hydrogels with possibilities of repairing the defects of irregular shapes and minimizing the invasive damage/hurt to patients by using injectable scaffolds with any operation as well [11, 12].
Chitosan (CS), a naturally occurring polymer, has been widely studied as hydrogel material owing to its excellent biodegradability, biocompatibility, high hydrophilicity and other interesting chemical properties due to the presence of amino and hydroxyl groups [13]. Various external stimuli-responsive and injectable CS-based smart hydrogels have been prepared for biomedical application [14–17]. For example, Park et al [14] created an injectable CS composite hydrogel containing hyaluronic acid for cartilage repair, which increased the proliferation and deposition of the cartilaginous extracellular matrix by encapsulated chondrocytes. Nevertheless, one critical disadvantage of CS hydrogels is their low mechanical strength because of the high water content and a relatively loose three-dimensional (3D) network formed by linear polysaccharide molecules [18]. Another problem is that hydrophilic small drug molecules such as antibiotics will rapidly release from CS hydrogels, which is not satisfactory toward the in situ sustained chemotherapy required with drug-loaded implants, although CS hydrogel is an excellent carrier to release protein macromolecules slowly [19, 20]. Currently, design of nanoparticles/CS composite hydrogels is regarded as an effective strategy to overcome these disadvantages except from the polymer crosslinking method [21–24]. For example, Ramay et al [21] reported that the addition of hydroxyapatite nanoparticles into CS–alginate porous scaffolds improved their mechanical property. Gao et al [22] developed an injectable CS-based hydrogel containing hydroxyapatite nanoparticles for dexamethasone/rhBMP-2 delivery.
Mesoporous silica nanoparticles (MSNs) have recently brought new potential in nanomedicine due to their large surface area and pore volume, tunable mesopore size and biocompatibility [25–27]. The extensive mesoporosity and functionalized moieties on the pore surface make MSNs promising candidates to accomplish sustained and/or controlled drug delivery. For example, Zhu et al [28] demonstrated that ibuprofen-loaded MSNs exhibited sustained drug release behavior, and the drug release rate could be tuned by changing the pH environment. On the other hand, efforts have been made to introduce MSNs into a polymer matrix for drug delivery [29, 30]. For example, Ahmed et al [29] incorporated curcumin-loaded MSNs into curcumin-encapsulated porous CS scaffolds to obtain burst drug release from macroporous scaffolds and sustained slow drug release from mesoporous silica microspheres. Song et al [30] fabricated a dual drug-loaded poly(lactic-co-glycolic acid) (PLGA)/MSN electrospun composite mat, with the two model drugs (fluorescein and rhodamine B) releasing in separate and distinct release kinetics.
Therefore, evidence of the benefits of nanoparticles for the enhancement of the hydrogel strength and mesoporous structure of MSNs for the sustained/controlled release of small drug molecules motivated us to create CS/MSN composite hydrogels. We hypothesized that the introduction of MSNs into CS could enhance the gel mechanical strength through the strong intermolecular interaction between MSNs and the CS matrix; at the same time, MSNs are able to load small drug molecules and release them steadily over a long time period. To the best of our knowledge, there are no previous studies describing the introduction of MSNs into CS hydrogels for enhanced gel strength and sustained co-delivery of biomacromolecules and small chemotherapeutic drugs. In this study, the CS/MSN composite hydrogels were prepared, and the effects of MSNs on the gel structure, gel strength, drug delivery behavior, physicochemical and chondrocyte proliferation properties of the hydrogels were investigated.
2. Materials and methods
2.1. Materials
Tetraethoxysilane (TEOS, 98%), cetyltrimethylammonium bromide (CTAB, 99%) and glycerol 2-phosphate disodium salt hydrate (β-GP) were obtained from Sinopharm Chemical Reagent. Brij-56 (99%) and CS (Mw ∼ 2 × 105, degree of deacetylation ∼91%) were purchased from Sigma-Aldrich Company. Gentamicin (GS) powders were obtained from Nanyang Pukang Pharmaceutical and bovine serum albumin (BSA) from Shanghai Runcheng Reagent. All the chemicals were used directly without further purification.
2.2. Preparation of mesoporous silica nanoparticles
According to previously reported methods [31], MSNs were synthesized in an aqueous buffer solution of pH 7 from mixtures with the following molar ratios: Brij-56:CTAB:TEOS:H2O = 3.5:10:80:60 000. Briefly, CTAB and Brij-56 were dissolved in a buffer solution of 3.43 g KH2PO4 and 0.58 g NaOH under vigorous stirring and heating at 368 K. After the solution became homogeneous, TEOS was added dropwise. The reaction was maintained for 8 h. The products were centrifuged, washed with an excess amount of water and ethanol, and then dried under vacuum. As-synthesized materials were refluxed in a mixture solution of 250 ml ethanol and 2 ml hydrochloric acid (36–38%) at 353 K for 8 h to remove the surfactants, centrifuged, washed with ethanol and distilled water and dried in vacuum.
Transmission electron microscopy (TEM) images were obtained on a JEM-2010 electron microscope operating at an accelerating voltage of 200 kV. The size distribution of MSNs was measured by using a particle size analyzer (Zeta Plus, Brookhaven Instrument Corp., Holtsville, NY, USA).
2.3. Preparation of the chitosan/MSN composite hydrogels
In all, 100 mg of MSNs was added into 9 ml of acetic acid solution (0.1 M) under stirring and ultrasonicated to form a 1% w/v suspension; 200 mg of CS powder was then dissolved in the acidic suspension. The suspension was placed in an iced-water bath and subsequently 1 ml of β-GP (0.26 M) solution was added dropwise. Finally, the CS/MSN composite hydrogels (named CS/MSNs) were formed by placing the mixture in an oven heated to 37 °C. Pure CS hydrogels as control were also prepared following the same process but without MSNs.
The CS hydrogels and CS/MSN composite hydrogels were prefrozen at −20 °C for 12 h and then lyophilized for 48 h in a freeze drier. Lyophilized samples were characterized using field-emission scanning electron microscopy (JSM-6700F, JEOL) to assess the morphology and porous structure.
2.4. Rheological tests
The gelation temperature, gelation time and gel strength of both CS and CS/MSN hydrogels were measured using a Rheometer-SR5 rheometer (Rheometric Scientific) equipped with a parallel-plate sensor in oscillatory mode. The storage (elastic) modulus G′ and loss (viscous) modulus G″ versus temperature were measured at a plate gap of 0.1 mm and a frequency of 1.0 Hz. The temperature at the crossover point of G′ and G″ was defined as the gelation temperature. The gel processes were monitored by evaluating the gel rate when the temperature was settled at 37 °C.
2.5. Swelling measurements and in vitro enzymatic degradation
Lyophilized samples (M0) were immersed in ultrapure water at 37 °C. Swollen samples were taken out for certain time intervals. Excessive water on the surface was gently wiped with a filter paper and the samples were weighed (Me). The equilibrium swelling ratio (SR) was calculated as SR = (Me − M0)/M0.
The degradation of both CS and CS/MSNs hydrogels was studied in simulated body fluid (SBF) medium containing lysozyme at 37 °C. Hydrogels were weighed (W0) and put in dialysis bags (molecular weight cutoff: 3500), which were thoroughly washed with boiled ethanol and ethylenediaminetetraacetic acid (EDTA). Subsequently, the hydrogels in dialysis bags were incubated in SBF solution containing 4 mg ml−1 lysozyme for 18 d, and the hydrogel mass/SBF volume ratio was 10 mg ml−1. The samples were taken out at predetermined time intervals and washed in deionized water to remove ions absorbed on the surface. The hydrogels were freeze dried and the final weight was noted as Wt. The degradation was calculated using the equation %Degradation(t) = (W0 − Wt)/W0.
2.6. Loading and in vitro release of GS and BSA
Drug loading. In this study, GS and BSA were used as model small chemical drugs and biomacromolecules, respectively, to evaluate drug delivery behavior. GS drugs were initially loaded into the MSNs before hydrogel preparation. 0.1 g of MSNs was immersed in 10 ml of GS aqueous solution (30 mg ml−1) for 24 h under gentle stirring. After the centrifugation, the precipitates were carefully washed with distilled water and vacuum dried. The GS-loaded MSNs were dispersed into CS acidic solution and afterwards 1 mg of BSA and 1 ml of β-GP (0.26 M) were added in sequence to prepare the GS/BSA-loaded CS/MSN hydrogels. For CS hydrogels, GS and BSA drugs were simultaneously introduced before the addition of β-GP.
Drug release. Both GS/BSA-loaded hydrogels were cut into cubic pieces, each side of 8 mm length. The release test of every piece was carried out in 5 ml phosphate buffered saline (PBS) (pH = 7.4) solution. The medium was withdrawn at the predetermined time interval, and replaced with a fresh soaking medium heated to 37 °C. GS concentration in the medium was analyzed by measuring the maximum UV–Vis absorbance of the GS-o-phthaldialdehyde complex at 331 nm using a Shimadzu UV-3101PC spectroscope [32]. BSA concentration was determined via the Coomassie blue staining method [33].
2.7. Chondrocyte culture
Primary chondrocyte isolation and cell culture. Chondrocytes were isolated from the cartilage tissue of adult sheep ears under institutional guidelines. Briefly, the cartilage tissue was minced with scissors into pieces of about 1 mm3. The minced pieces were soaked in 40 ml Dulbecco’s modified Eagle’s medium (DMEM) containing 0.1% collagenase type II at 37 °C overnight and enzymatically digested. The isolated chondrocytes were centrifuged and suspended again in DMEM/10% fetal bovine serum (FBS) containing 100 IU ml−1 penicillin–streptomycin. The suspension was seeded into an 11 cm plastic tissue culture dish and incubated in a humidified atmosphere of 95% air and 5% CO2 at 37 °C. After a confluent cell layer was formed, the cells were detached using 0.25% trypsin in PBS and then were used for the experiment.
Quasi-quantitative analysis. CS, MSNs and β-GP powders were all sterilized under UV irradiation for 2 h before being dissolved or dispersed to form hydrogels. In all, 100 μl of pre-gel solution mixture was added into each well of a 48-well plate. 1 × 107 chondrocytes were then suspended in the solution as well. The plate was slightly shaken and put into the incubator to form gels. Another 250 μl of DMEM was added after complete gelation. The hydrogels were cultured in a 5% CO2 atmosphere. Chondrocyte proliferation, deoxyribonucleic acid (DNA) content and secreted glycosaminoglycan (GAG) content of the chondrocyte/hydrogels were measured after culturing for 4, 10 and 14 d.
Chondrocyte proliferation was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) assay. Briefly, the culture medium was sucked out. After being supplemented with 100 μl of MTT solution (0.5 mg ml−1), the cells were continually cultured for another 4 h. The hydrogel was smashed and 1 ml of dimethyl sulfoxide was added to dissolve the formed formazan pigment. The solution was centrifuged at 10 000 rpm, and the absorbance of 150 μl of upper solution at 490 nm was recorded by a microplate reader (Bio-TekELx 800) in a 96-well culture plate.
DNA content was determined using a DNA Quantization Kit, Fluorescence Assay (Hoechst 33258, Sigma). At each time point, the chondrocytes/hydrogels were taken out from the plate and were stored at −20 °C in an Eppendorf tube. After the experiment was finished, all the samples were frozen and thawed. This process was repeated several times. In all, 1 ml of 10 mg ml−1 papain solution (l-cysteine 0.005 M, EDTA 0.05 M, NaH2PO4–Na2HPO4 buffer solution, pH = 6.5) was added and the samples were placed in a water bath at 65 °C overnight to ensure the complete extraction of DNA. An amount of 100 μl of the above solution was mixed with 2 ml of 1 μg ml−1 bisbenzimide/fluorescent assay buffer solution and the fluorescent intensity of this mixture was measured rapidly on a fluorescence spectrometer (RF-5301C, Shimadzu). The excitation and emission wavelengths were 360 and 460 nm, respectively [34]. The concentration was found by referring to a calibration curve constructed from known concentrations of DNA solutions.
The amount of GAGs secreted in hydrogels was quantified by 1,9-dimethyl methylene blue (DMMB) examination. First, a DMMB working solution was prepared as follows: 1.9 mg DMMB, 0.304 g glycine and 0.237 g NaCl were dissolved in 90 ml of deionized water. The pH was adjusted to 3 using hydrochloride acid and the volume was finally made to 100 ml. Chondroitin sulfate was used instead to construct the GAGs-absorption calibration curve. Chondroitin sulfate solution with concentrations in the range from 10 to 100 μg ml−1 reacted to DMMB color developer reagent for 10 min. The resulting chelate precipitate was separated, washed with PBS and re-dissolved in guanidine hydrochloride solution containing 10% isopropanol (GuHCl, 4 M, pH = 6.8). The absorbances at 650 nm were recorded on a UV–Vis spectroscope (Shimadzu UV-3101PC). Similarly, GAG concentrations of the samples in papain solution were estimated by the same process.
3. Results and discussion
TEM images of the MSNs are shown in figure 1. It can be observed that the MSNs were monodispersed and a worm-like mesopore structure was formed on the MSNs. Particle size distribution of the MSNs was measured by dynamic light scattering (figure 1(C)). The hydrodynamic particle size of the MSNs was narrowly distributed at 78 nm, and the particle size was close to the TEM result, suggesting the good dispersity of MSNs. According to N2 sorption measurement, the Brunauer–Emmett–Teller surface area, pore volume and pore size were calculated to be 250 m2 g−1, 0.49 cm3 g−1 and 2.3 nm respectively, allowing small chemical drugs to diffuse into the mesoporous channels for high loading.
Figure 1.
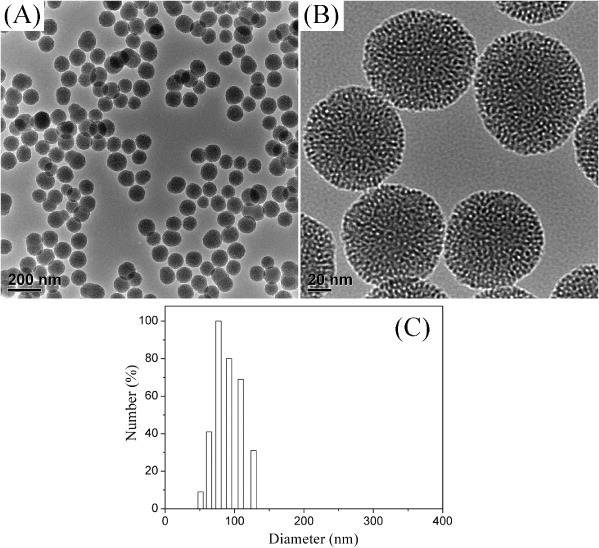
TEM images (A) and (B) and particle size distribution (C) of MSNs.
A hydrogel is a network of polymer chains in which water is the dispersion medium. Through lyophilization, both CS and CS/MSN hydrogels exhibited a 3D macroporous structure with completely inter-connected open pores as shown in figure 2, and the pore size varied from 200 to 500 μm, which was suitable for the encapsulation of chondrocytes. However, the lyophilized CS hydrogels showed cracks on the strut walls, indicating its quite brittle nature. In the case of the lyophilized CS/MSN hydrogels, the strut walls were intact. MSNs were dispersed homogeneously in the strut walls, resulting in much rougher surface morphology.
Figure 2.

SEM images of (A) and (C) the CS hydrogels and (B) and (D) the CS/MSN hydrogels after freeze drying for 48 h.
Rheology has been one of the most important methods employed to study the viscoelastic behavior of materials. Herein, the formation of CS/MSN composite hydrogels was studied using oscillatory rheometry at 1 Hz frequency and under low-strain conditions, which measures the storage modulus (G′) and loss modulus (G″) against the shear strain. Normally, the value of G′ when it reaches a plateau could indicate the completion of crosslinking and also serves as an indicator of stiffness. Furthermore, the gelation point, defined as the crossover of G′ and G″, is used to evaluate the gelation rate of hydrogels. In this study, as shown in figures 3(A) and (B), the changes of G′ and G″ values were investigated by slowly raising the water bath temperature from 25 °C at a rate of 0.5 °C min−1. At 25 °C, all the G′ values are about 10 Pa, which indicates their liquid states at the beginning. Along with the temperature increase, the G′ value slowly increases as well. Later on at a certain temperature, which is just the gelation point according to traditional definition, the G′ value increases rapidly to a high level and reaches a plateau gradually. At temperatures lower than the gelation point, the intermolecular forces in the CS solution are dominated by the hydrogen bonding between hydrophilic groups of CS and water molecules, and the hydrophobic interactions among the polymer chains prevailing at elevated temperatures, resulting in the formation of a long-range network—a gel. In figures 3(A) and (B), it can be seen that the gelation temperature was 37 °C for CS hydrogels in accordance with previous reports [35], whereas it decreased to 32 °C for the CS/MSN composite hydrogels. It suggested that the CS/MSN composite hydrogels exhibited a faster gelation rate compared to the CS hydrogels.
Figure 3.
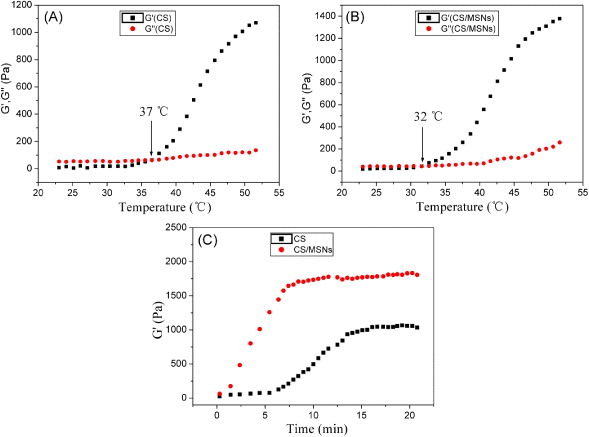
(A) and (B) Temperature dependences of the storage modulus G′ and loss modulus G″ showing the gelation transitions; (C) dependence of the storage modulus G′ value of the hydrogels on time at 37 °C to determine the gelation process.
As injectable tissue repair materials, the time duration of the whole gel process after injection under body temperature was another key parameter for CS-based thermal sensitive hydrogels. The time scan results at constant 37 °C are shown in figure 3(C). It took more than 5 min for the CS solution to start gelation, while the gelation of the CS/MSN solution took place immediately after being placed in an oven heated to 37 °C. Similarly, the duration to finish the whole gelation for the CS/MSN hydrogels was much earlier than that for the CS hydrogels. Importantly, the elastic modulus of the CS/MSN composite hydrogels became gradually stabilized at about 1800 Pa, which was significantly higher than the CS hydrogels (about 1000 Pa), indicating their higher gel strength than the CS hydrogels.
Therefore, it could be concluded that the introduction of MSNs into CS hydrogels has considerably enhanced mechanical and rheological performance. The enhancements can be mainly attributed to the abundant silanol groups on the particle surface. Together with the mesopores, the functional silanol groups could interact with and subsequently absorb a large number of CS molecules, which led to a dense CS chain meshwork all round the MSNs, which was consistent with previous studies on the interactions between silica and CS [36, 37]. At the same time, the steric stabilization effect from polymer molecules inhibited particle agglomerations so that phase separation was effectively prevented. When the temperature elevation stimulated the inter-entangling of high-density CS chains and furthermore their wrapping around MSN particles, gelation was induced in quite a short time period, resulting in the gel structure construction. Thus the composite system was more sensitive to environmental temperature variation, which was obviously advantageous for in vivo gelation after injection. On the other hand, the rigid structure of MSNs might also play an important role in strengthening the CS network.
Hydrogel swelling properties are related to the hydrogel network and will influence its mass transport characteristics. Figure 4(A) shows the SRs of both CS and CS/MSN hydrogels after 2 and 24 h of incubation. It can be seen that the SRs of both CS and CS/MSN hydrogels after 2 h water incubation were extremely close to those after 24 h of incubation, indicating that the whole swelling process reached equilibrium within only 2 h. However, the SR of the CS/MSN hydrogels was about 45.7%, which was remarkably higher than that of the CS hydrogels (about 12.7%). It might be that the rigid MSN particles might resist the external stress during the swelling process, resulting in the looser gel network. Also, the extensive mesoporosity in MSNs could be regarded as capillary tubes for water adsorption in the CS/MSN hydrogels. Figure 4(B) shows the enzymatic degradation of both CS and CS/MSN hydrogels in SBF solution containing lysozyme at 37 °C. The CS/MSN hydrogels exhibited slightly faster degradation compared to the CS hydrogels. It might be attributed to more absorbed lysozymes in the CS/MSN hydrogels during water transportation, because the CS/MSN hydrogels had higher SRs than the CS hydrogels.
Figure 4.
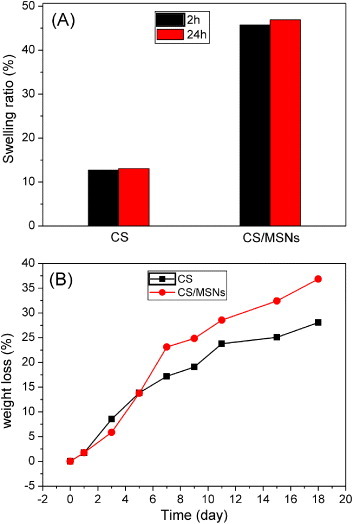
(A) SRs of the dried gels after immersing in water for 2 and 24 h; (B) weight losses of CS and CS/MSN hydrogels in SBF solution containing lysozymes as a function of immersion time.
CS chains in hydrogels have abundant positively charged amino groups, 3D interconnected macroporous structure and relatively good hydrophilicity; hence, CS-based gels are excellent candidates for the delivery of biomacromolecules, such as peptides, proteins, antigens, genes and so on. However, studies demonstrated that it was difficult to deliver most of the small hydrophilic drug molecules, for instance, antibiotics, in a sustained manner by CS hydrogels [38]. To overcome this drawback, the drug-loaded MSNs were introduced here into the CS network to form the drug-loaded CS/MSN hydrogels, which would be helpful in maintaining the small drug molecules for longer time release owing to their mesoporous structure. In this study, the GS loading capacity of MSNs was calculated to be approximately 311 mg g−1 using UV–Vis analysis. Moreover, 1 mg BSA was directly encapsulated into the hydrogel precursor solution. Therefore, the loading capacities of GS and BSA in the CS/MSN composite hydrogels were estimated to be 2.5 and 0.08 mg g−1, respectively. The release profiles of small drug molecules, GS, from the CS and CS/MSN hydrogels are shown in figure 5(A). The CS/MSN hydrogels exhibited much slower GS release than the CS hydrogels. The drug molecules were freed from the CS hydrogels within 24 h. However, the CS/MSN hydrogels were employed to load GS drugs in MSNs and exhibited good sustained drug release behavior. Only about 65% of the loaded drugs were released from the CS/MSN hydrogels in 7 d. Therefore, the MSNs played an important role in controlling the sustained release of small drug molecules.
Figure 5.
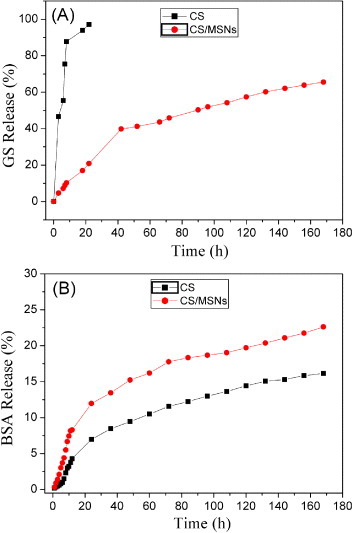
In vitro release of GS (A) and BSA (B) in PBS solution at 37 °C from both CS and CS/MSN hydrogels.
The release profiles of BSA molecules from CS and CS/MSN hydrogels are shown in figure 5(B). It can be seen that BSA releases from both CS and CS/MSN hydrogels were rather sustained, and about 16 and 22% of the loaded BSA molecules were released in 7 d, respectively. The effective confinement of BSA molecules in the CS hydrogels was attributed to the electrostatic attractions between BSA proteins and the CS long chains. Since MSNs with a negative surface could interact with CS, the electrostatic attraction forces between BSA and CS might be partially shielded in the CS/MSN hydrogels, suggesting that BSA molecules could diffuse more freely in the water microenvironment in the CS/MSN hydrogels. Also, the looser network of the CS/MSN hydrogels was beneficial for BSA diffusion. Therefore, the CS hydrogels could maintain macrobiomolecules for a longer time, but the CS/MSN hydrogels were capable of delivering both biomacromolecules and hydrophilic small drug molecules simultaneously in highly sustained patterns.
As an injectable hydrogel for cartilage regeneration, the hydrogel must have the ability to support chondrocyte proliferation and growth and maintain chondrocyte phenotype. The proliferation, growth and functional expression of chondrocytes encapsulated in the hydrogels were studied by MTT assay and the determination of the DNA and secreted GAGs content. Figure 6 shows chondrocyte proliferation in both CS and CS/MSN hydrogels measured by MTT assay. The chondrocyte viability increased when incubated with both CS and CS/MSN hydrogels in the time course of incubation, revealing good biocompatibility of both CS and CS/MSN hydrogels. Furthermore, the CS/MSN hydrogels had higher optical density values compared to the CS hydrogels, indicating that MSN introduction into the CS hydrogels was beneficial for chondrocyte proliferation. Previous studies indicate that MSNs are biocompatible and can easily be internalized into most normal and cancer cells without apparent deleterious effects on cellular growth, proliferation and differentiation [27]. However, Gordon et al demonstrated that silica nanoparticles (SNPs) in the CS/SNP hydrogels could elute and/or aggregate during a 12 d incubation at 37 °C [39]. Therefore, MSNs in the CS/MSN composite hydrogels might be eluted during the cell culture process, which would influence chondrocyte activities.
Figure 6.
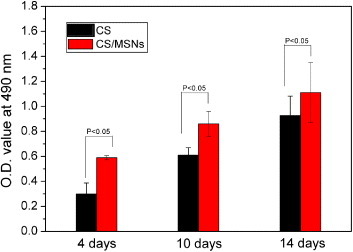
Chondrocyte proliferation after 4, 7 and 14 d of culture in both CS and CS/MSN hydrogels measured by MTT assay.
The DNA content in both CS and CS/MSN hydrogels increased along with the prolonged culture (figure 7(A)), demonstrating that the chondrocytes can normally proliferate and grow in both CS and CS/MSN hydrogels. However, the DNA amounts obtained in the CS/MSN hydrogels were always higher than those in the CS hydrogels (figure 7(A)). GAGs secreted by the chondrocytes are recognized as a signal of maintaining chondrocytic phenotype. As shown in figure 7(B), the amount of GAGs in the hydrogel/cell composites increased with the increase of culture time, and the CS/MSN hydrogels exhibited a higher amount of GAGs than the CS hydrogels, which were similar to those of the chondrocyte viability and DNA content. Therefore, MSN introduction into the CS hydrogels induced the promotion effect on chondrocyte proliferation, growth and chondrocyte phenotype.
Figure 7.
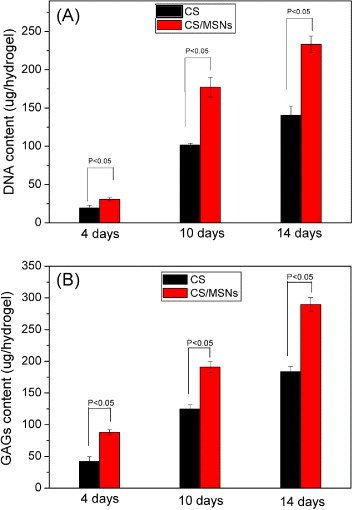
DNA content (A) and GAGs content (B) of chondrocytes in both CS and CS/MSN hydrogels after culturing for 4, 7 and 14 d.
A few studies demonstrated that the relatively loose structure and high gel strength of the hydrogels offered the cell more stable space to grow [40], thus stimulating the signaling and gene expression of correlative biomolecules. On the other hand, silicate ions (SiO44−) could be slowly released from MSNs in a physiological environment [41]. It has been reported that lower concentrations of silicate ions in the physiological environment were able to stimulate the activity of some metabolism correlated genes, which could regulate the cell division cycle and promote chondrocyte proliferation [42, 43]. Therefore, the CS/MSN composite hydrogels are much more advantageous in improving the inner environment favorable for chondrocyte proliferation and growth compared to the CS hydrogels.
4. Conclusion
In this study, MSNs have been introduced into injectable CS to form the CS/MSN composite hydrogels. The introduction of MSNs into CS accelerated the gelation rate of the hydrogels, and significantly enhanced the hydrogel strength. When using GS and BSA as examples of small chemotherapeutic drugs and biomacromolecules, the CS/MSN hydrogels delivered both GS and BSA simultaneously in a highly sustained release manner, because MSNs act as the sustained drug release carriers. The results of chondrocyte culture showed that the CS/MSN hydrogels were biocompatible, and chondrocyte proliferation and growth were significantly higher in the CS/MSN hydrogels than the CS hydrogels. In conclusion, the introduction of MSNs into CS hydrogels is a highly effective approach to fabricate composite hydrogels, which favor the sustained co-delivery of both biomacromolecules and small chemical drugs and chondrocyte proliferation and growth as well, and therefore is promising in the noninvasive therapy of cartilage regeneration.
Acknowledgments
This work was financially supported by the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning, National Natural Science Foundation of China (numbers 51102166, 51132009, 51072212, 51102259), Program for New Century Excellent Talent in University (number NCET-12-1053), Key Project of Chinese Ministry of Education (number 212055), Shanghai Pujiang Program (number 11PJ1407300), Shanghai Shuguang Project (number 12SG39) and Innovation Program of Shanghai Municipal Education Commission (number 12ZZ140).
References
- Chung C. and Burdick J A. Adv. Drug. Deliv. Rev. 2008;60:243. doi: 10.1016/j.addr.2007.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuernberger S, Cyran N, Albrecht C, Redl H, Vecsei V. and Marlovits S. Biomaterials. 2011;32:1032. doi: 10.1016/j.biomaterials.2010.08.100. [DOI] [PubMed] [Google Scholar]
- Alves da Silva M L, Martins A, Costa-Pinto A R, Costa P, Faria S. and Gomes M. Biomacromolecules. 2010;11:3228. doi: 10.1021/bm100476r. [DOI] [PubMed] [Google Scholar]
- Zheng L, Fan H S, Sun J, Chen X N, Wang G, Zhang L, Fan Y J. and Zhang X D. J. Biomed. Mater. Res. A. 2010;93:783. doi: 10.1002/jbm.a.32588. [DOI] [PubMed] [Google Scholar]
- Hong Y, Gong Y H, Gao C Y. and Shen J C. J. Biomed. Mater. Res. A. 2008;85:628. doi: 10.1002/jbm.a.31603. [DOI] [PubMed] [Google Scholar]
- Baker B M, Nathan A S, Gee A O. and Mauck R L. Biomaterials. 2010;31:6190. doi: 10.1016/j.biomaterials.2010.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullen S D, Autefage H, Callanan A, Gentleman E. and Stevens M M. Tissue Eng. A. 2012;18:2073. doi: 10.1089/ten.tea.2011.0606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W Q, Liu W L, Sun J, Dan X L, Wei D. and Fan H S. J. Bioact. Compat. Polym. 2012;27:327. doi: 10.1177/0883911512448692. [DOI] [Google Scholar]
- Hu X H, Ma L, Wang C C. and Gao C Y. Macromol. Biosci. 2009;9:1194. doi: 10.1002/mabi.200900275. [DOI] [PubMed] [Google Scholar]
- Zan J, Chen H H, Jiang G Q, Lin Y. and Ding F X. J. Appl. Polym. Sci. 2006;101:1892. doi: 10.1002/app.23613. [DOI] [Google Scholar]
- Nguyen M K. and Lee D S. Macromol. Biosci. 2010;10:563. doi: 10.1002/mabi.200900402. [DOI] [PubMed] [Google Scholar]
- Ashley A A. and Lakshmi S N. Biomed. Mater. 2012;7:4105. [Google Scholar]
- Di Martino A, Sittinger M. and Risbud M V. Biomaterials. 2005;30:5983. doi: 10.1016/j.biomaterials.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Park H, Choi B, Hu J. and Lee M. Acta Biomater. 2013;9:4779. doi: 10.1016/j.actbio.2012.08.033. [DOI] [PubMed] [Google Scholar]
- Nair L S, Starnes T, Ko J W. and Laurencin C T. Biomacromolecules. 2007;8:3779. doi: 10.1021/bm7006967. [DOI] [PubMed] [Google Scholar]
- Ahmadi R. and de Bruijn J D. J. Biomed. Mater. Res. A. 2008;86:824. doi: 10.1002/jbm.a.31676. [DOI] [PubMed] [Google Scholar]
- Guo B L. and Gao Q Y. Carbohyd. Res. 2007;16:2416. doi: 10.1016/j.carres.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Hoffman A S. Adv. Drug. Deliv. Rev. 2002;54:3. doi: 10.1016/S0169-409X(01)00239-3. [DOI] [PubMed] [Google Scholar]
- Bhattarai N, Ramay H R. and Gunn J. J. Control. Release. 2005;103:609. doi: 10.1016/j.jconrel.2004.12.019. [DOI] [PubMed] [Google Scholar]
- Vermonden T, Censi R. and Hennink W E. Chem. Rev. 2012;112:2853. doi: 10.1021/cr200157d. [DOI] [PubMed] [Google Scholar]
- Ramay H R, Li Z, Shum E. and Zhang M. J. Biomed. Nanotechnol. 2005;1:151. doi: 10.1166/jbn.2005.026. [DOI] [Google Scholar]
- Gao C, Cai Y, Kong X, Han G. and Yao J. Mater. Lett. 2013;93:312. doi: 10.1016/j.matlet.2012.11.106. [DOI] [Google Scholar]
- Gordon S, Teichmann E, Young K, Finnie K, Rades T. and Hook S. Eur. J. Pharm. Sci. 2010;4:360. doi: 10.1016/j.ejps.2010.07.004. [DOI] [PubMed] [Google Scholar]
- Peter M, Binulal N S, Soumya S, Nair S V, Furuike T, Tamura H. and Jayakumar R. Carbohyd. Polym. 2010;79:284. doi: 10.1016/j.carbpol.2009.08.001. [DOI] [Google Scholar]
- Trewyn B G, Giri S, Slowing I I. and Lin Victor S Y. Chem. Commun. 2007;31:3236. doi: 10.1039/b701744h. [DOI] [PubMed] [Google Scholar]
- Wu S H, Hung Y. and Mou C Y. Chem. Commun. 2011;47:9972. doi: 10.1039/c1cc11760b. [DOI] [PubMed] [Google Scholar]
- He Q. and Shi J. J. Mater. Chem. 2011;21:5845. doi: 10.1039/c0jm03851b. [DOI] [Google Scholar]
- Zhu Y, Shi J, Shen W, Chen H, Dong X. and Ruan M. Nanotechnology. 2005;16:2633. doi: 10.1088/0957-4484/16/11/027. [DOI] [Google Scholar]
- Ahmed A, Hearn J, Abdelmagid W. and Zhang H. J. Mater. Chem. 2012;22:25027. doi: 10.1039/c2jm35569h. [DOI] [Google Scholar]
- Song B, Wu C. and Chang J. Acta Biomater. 2012;8:1901. doi: 10.1016/j.actbio.2012.01.020. [DOI] [PubMed] [Google Scholar]
- He Q J, Cui X Z, Cui F M, Guo L M. and Shi J L. Micropor. Mesopor. Mater. 2009;117:609. doi: 10.1016/j.micromeso.2008.08.004. [DOI] [Google Scholar]
- Doadrio A L, Sousa E M B, Doadrio J C, Pariente J P, Izquierdo-Barba I. and Vallet-Regi M. J. Control. Release. 2004;97:125. doi: 10.1016/j.jconrel.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Brad M M. Anal. Biochem. 1976;72:248. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Teare J M, Islam R, Flanagan R, Gallagher S, Davies M G. and Grabau C. BioTechniques. 1997;22:1170. doi: 10.2144/97226pf02. [DOI] [PubMed] [Google Scholar]
- Chenite A, Chaput C. and Wang D. Biomaterials. 2000;21:2155. doi: 10.1016/S0142-9612(00)00116-2. [DOI] [PubMed] [Google Scholar]
- El-Barghouthi M, Eftaiha A, Rashi I, Al-Remawi M. and Badwan A. Drug Dev. Ind. Pharm. 2008;34:373. doi: 10.1080/03639040701657792. [DOI] [PubMed] [Google Scholar]
- Guo X, Zheng D. and Hu N. J. Phys. Chem. B. 2008;112:15513. doi: 10.1021/jp807452z. [DOI] [PubMed] [Google Scholar]
- Bhattarai N, Gunn J. and Zhang M Q. Adv. Drug. Deliv. Rev. 2010;62:83. doi: 10.1016/j.addr.2009.07.019. [DOI] [PubMed] [Google Scholar]
- Gordon S, Teichmann E, Young K, Finnie K, Rades T. and Hook S. Eur. J. Pharm. Sci. 2010;41:360. doi: 10.1016/j.ejps.2010.07.004. [DOI] [PubMed] [Google Scholar]
- Adelow C, Segura T, Hubbell J A. and Frey P. Biomaterials. 2008;29:314. doi: 10.1016/j.biomaterials.2007.09.036. [DOI] [PubMed] [Google Scholar]
- He Q, Shi J, Zhu M, Chen Y. and Chen F. Micropor. Mesopor. Mater. 2010;131:314. doi: 10.1016/j.micromeso.2010.01.009. [DOI] [Google Scholar]
- Shirosaki Y, Hayakawa S. and Osaka A. Key Eng. Mater. 2011;493–494:698. doi: 10.4028/www.scientific.net/KEM.493-494.698. [DOI] [Google Scholar]
- Ramaswamy Y, Wu C, Van Hummel A, Combes V, Grau G. and Zreiqat H. Biomaterials. 2008;29:4392. doi: 10.1016/j.biomaterials.2008.08.006. [DOI] [PubMed] [Google Scholar]