

Abstract
Helicobacter and Campylobacter species are Gram-negative microaerophilic host-associated heterotrophic bacteria that invade the digestive tract of humans and animals. Campylobacter jejuni is the major worldwide cause of foodborne gastroenteritis in humans, while Helicobacter pylori is ubiquitous in over half of the world's population causing gastric and duodenal ulcers. The colonisation of the gastrointestinal system by Helicobacter and Campylobacter relies on numerous cellular defences to sense the host environment and respond to adverse conditions, including those imposed by the host immunity. An important antimicrobial tool of the mammalian innate immune system is the generation of harmful oxidative and nitrosative stresses to which pathogens are exposed during phagocytosis. This review summarises the regulators, detoxifying enzymes and subversion mechanisms of Helicobacter and Campylobacter that ultimately promote the successful infection of humans.
Keywords: Helicobacter, Campylobacter, pathogen, innate immunity, bacterial defences
This review summarises the regulators, detoxifying enzymes and subversion mechanisms of Helicobacter and Campylobacter that ultimately promote the successful infection of humans.
Graphical Abstract Figure.
This review summarises the regulators, detoxifying enzymes and subversion mechanisms of Helicobacter and Campylobacter that ultimately promote the successful infection of humans.
INTRODUCTION
Helicobacter and Campylobacter species are Gram-negative microaerophilic host-associated heterotrophic bacteria that belong to the ε-proteobacteria phylum (Nachamkin, Szymanski and Blaser 2008; Sutton and Mitchell 2010). Although Helicobacter species were first classified as members of the Campylobacter genus, a separated Helicobacter genus was established in 1989 and now comprises over 30 species with more than 150 genome sequences currently available (HelicoBase; www.Helicobacter.um.edu.my). Helicobacter and Campylobacter grow optimally at temperatures ranging from 37°C to 42°C under conditions of reduced oxygen concentration. Morphologically, the two species are similar, both being spiral shaped and highly motile bacteria (Altekruse et al. 1999; Hofreuter 2014).
Helicobacter species have been isolated from the gastric mucosa, faeces, saliva and dental plaques of infected people, as well as from the upper gastrointestinal tract and liver of some birds and mammals (Menard, Pere-Vedrenne et al. 2014; Flahou et al. 2015). The best-studied species is Helicobacter pylori, which colonises the gastric mucous layer in close contact with epithelial cells and is ubiquitous in over half of the world's population (Correa and Piazuelo 2011). Helicobacter pylori establishes lifelong infections that can persist asymptomatically over decades due to several mechanisms that allow the bacterium to evade immune clearance (Algood and Cover 2006; Muller, Oertli and Arnold 2011; Monack 2013). However, in many cases infection is also linked to gastroduodenal pathologies such as gastric and duodenal ulcers. Helicobacter pylori is a highly genetically diverse species with geographically related virulence (Salama et al. 2000; Ali et al. 2015). Strains that express specific gene products, namely those encoding the vacuolating cytotoxin (vacA), and cytotoxin-associated gene A and L (cagA and cagL), have been proposed to have an augmented risk for malignancy including gastric cancer (Polk and Peek 2010; Yamaoka 2010; Salama, Hartung and Muller 2013; Hardbower, Peek and Wilson 2014; Kaakoush et al. 2015). Helicobacter pylori may also play a role in other diseases such as thrombocytopenic purpura, sideropenic anaemia and cardiovascular disease (Bohr et al. 2007; Yamaoka 2010).
The human stomach is a hostile environment that exposes H. pylori to strong acidity, high oxygen tension, and fluctuations of nutrient availability and osmolarity. In this niche, H. pylori is submitted to nitric oxide (NO) from the acid-induced chemical decomposition of dietary and salivary nitrites present in the gastric lumen (Iijima et al. 2003). Moreover, H. pylori triggers immune defence cells that produce toxic compounds such as superoxide, nitric oxide and their derivatives. Therefore, H. pylori colonisation success relies on several detoxifier systems, one of the most important being the nickel-dependent urease (ureAB) that hydrolyses urea to ammonia and neutralises the stomach's acidity. This enzyme, which is expressed in amounts that represent ∼10% of the total H. pylori cellular protein content, has also been proposed to play a role in NO stress adaptation (Bauerfeind et al. 1997; Kuwahara et al. 2000).
In general, H. pylori infections can be successfully treated with a combination of drugs, which consists of two or three antibiotics (metronidazole, clarithromycin, amoxicillin and tetracycline) in addition to an acid-suppressive drug (a proton pump inhibitor, e.g. omeprazole). Nevertheless, the emergence of metronidazole-resistant H. pylori strains is currently one of the major causes of H. pylori treatment failure (Kanizaj and Kunac 2014).
In the recent years, the number and type of known Helicobacter species has expanded along with the number of sequenced genomes, and includes species such as H. canadensis, H. bilis, H. heilmannii, H. canis, H. macacae, H. fennelliae, H. cetorum and H. suis. Although first isolated from animals such as wild birds (H. valdiviensis), Bengal tigers and Australian marsupials, these so-called non-H. pylori species have been also found in humans (H. heilmannii, H. cinaedi, H. pullorum, H. bizzozeronii), and associated with a range of human diseases in children and adults including chronic gastritis (e.g. H. suis and H. felis), hepatobiliary malignancies, Crohn's disease and sepsis (H. cinaedi, H. bilis and H. canis) (Goldman and Mitchell 2010; Rossi and Hanninen 2012; Segura-Lopez, Guitron-Cantu and Torres 2015).
The Campylobacter genus includes at least 15 different species (e.g. Campylobacter fetus, C. coli, C. jejuni, C. sputorum) which are found in a wide range of niches from environmental habitats to humans and animals (Debruyne, Gevers and Vandamme 2008). Campylobacter jejuni is the more studied species and is a foodborne bacterial pathogen that colonises the gastrointestinal tract of a multitude of hosts including birds, chicken, turkeys, swine, cattle, sheep and humans (Altekruse et al. 1999; Humphrey, O'Brien and Madsen 2007). Colonisation of birds and chicken is typically commensal as these hosts are asymptomatic; however, these animals can act as vehicles for transmission of the bacteria to disease susceptible hosts. In humans, C. jejuni is pathogenic and infection typically results in gastroenteritis due to ingestion of contaminated food products or liquids. The predominant sources of infection in humans include intake of contaminated chicken, unpasteurised milk or contaminated water (Young, Davis and Dirita 2007) with doses as low as 500 organisms leading to the onset of illness (Robinson 1981). The development of gastroenteritis occurs within 2–5 days of C. jejuni ingestion and symptoms manifest in the form of bloody diarrhoea, abdominal pain, fever, vomiting and/or dizziness (Robinson 1981; Black et al. 1988). The illness is usually resolved within 2–10 days without the use of antibiotics and without complications. However, C. jejuni infection can result in the development of the rare neuromuscular disease Guillian–Barré syndrome, which occurs at a frequency of about 1 per 1000 infected people (Nachamkin 2002).
Campylobacter jejuni is equipped with numerous cellular defences to facilitate transit through the low pH of the stomach and exposure to bile salts in the duodenum prior to colonisation of target intestinal segments. The colon, caecum and to a lesser extent the ileum display the highest levels of bacterial colonisation (Humphrey, O'Brien and Madsen 2007). It is thought that the corkscrew-like architecture and high motility of the bacterial cells aid in the penetration of the viscous mucus layers that protect the underlying epithelial cells of the intestine (Ferrero and Lee 1988). Once C. jejuni transits through the mucus layer, the bacteria adhere to and invade the intestinal epithelium resulting in gastroenteritis (Humphrey, O'Brien and Madsen 2007). Flagellar (FlaAB), chemotaxis (CheY), adhesion and surface proteins (Peb1a, CadF, JlpA), and invasion antigens (CiaBCI) all play an important role in the adherence and invasion of C. jejuni into the intestinal epithelium (Masanta et al. 2013). Furthermore, C. jejuni produces and secretes a cytolethal distending toxin (CtdABC) that causes DNA damage, cell cycle arrest and cell death (Whitehouse et al. 1998). Subsequently, the host tissues mount an inflammatory response to protect against C. jejuni invasion; however, such a response results in host cell damage, bleeding, decreased water absorption in the lumen and consequently diarrhoea (Masanta et al. 2013).
In both developed and developing nations, Campylobacter is one of the principal bacteria associated with gastroenteritis disorders. In developed nations, young adults, especially young men show the greatest prevalence of C. jejuni infection, while in developing countries young children are most commonly affected by campylobacteriosis (Coker et al. 2002; Cooke 2010). Globally, there are approximately 400–500 million cases of C. jejuni infection worldwide (Ruiz-Palacios 2007). In the European Union, Campylobacter is the leading cause of bacterial-induced gastroenteritis in humans with approximately 214 779 reported cases in 2013 (EFSA 2015). Similar findings have been reported in Canada, where data from 2012 indicated 10 174 cases of campylobacteriosis, making it the main bacterial enteric disease (PHAC 2012). In the USA, 6486 cases of campylobacteriosis were reported in 2014, accounting for the second leading cause of foodborne illness behind salmonellosis with 7452 reported cases (Crim et al. 2015). The number of infected people in the European Union, Canada and USA is thought to be significantly underreported as the total number of cases is estimated to range from 2–20, 145 000 and 2 million for the European Union, Canada and USA, respectively (EFSA 2010; Thomas et al. 2013; CDC 2014).
Invasion by pathogens activates the host immune system that produces a myriad of antimicrobial compounds that microbes have to resist (Poole 2012). The next paragraphs describe the nature of the chemicals that expose Helicobacter and Campylobacter to oxidative and nitrosative stresses and their protective systems.
REACTIVE OXYGEN AND NITROGEN SPECIES: TOOLS OF THE INNATE IMMUNE SYSTEM
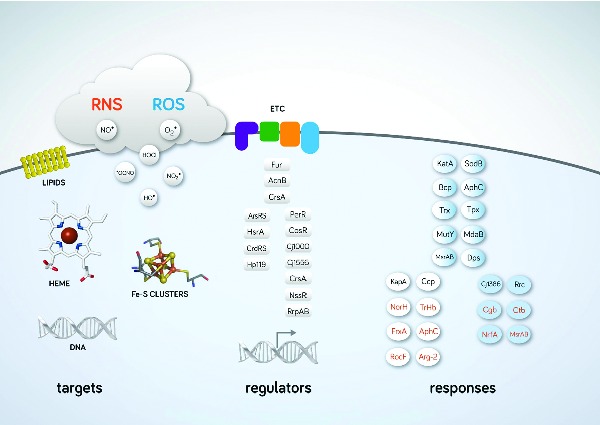
The mammalian innate immune system uses macrophages, neutrophils and inflammatory monocytes to phagocytise and expose microbes to harmful compounds such as reactive oxygen species (ROS) and reactive nitrogen species (RNS) (Fig. 1) (Fang 2004).
Figure 1.

Biochemical pathways of oxidative and nitrosative species production within host or bacterial cells. Molecular oxygen is reduced to superoxide within innate immune cells by NADPH oxidase or produced inadvertently by complexes of the respiratory electron transport chain. Within neutrophils and inflammatory monocytes, H2O2 formed by dismutation of O2•− is converted to toxic HOCl by myeloperoxidase. Generation of the powerful oxidising species, the hydroxyl radical, is produced by the Fenton chemistry within the host and/or bacterial cells. Macrophages produce nitric oxide via NO synthase, which can interact with superoxide to produce peroxynitrite. Autoxidation of NO produces N2O3 and subsequent downstream reactions generate nitrogen dioxide radicals. The generation of ROS and RNS plays an important role in antibacterial defence by damaging important Campylobacter and Helicobacter cellular molecules.
An important antibacterial weapon present in the plasma membrane of innate immune cells is the multisubunit complex NADPH oxidase that reduces O2 to produce superoxide anions (O2•−) (Slauch 2011; Hurst 2012). The recognition and engulfment of bacteria by macrophages induces the expression of the mammalian NADPH oxidase that results in a marked increase of O2 consumption and generation of a superoxide burst (DeLeo et al. 1999; Pan et al. 2009). Further dismutation of O2•− produces a second ROS, hydrogen peroxide (H2O2), which can freely diffuse across membranes and into the phagocytosed bacterial cells (Fig. 1). Neutrophils and inflammatory monocytes utilise the enzyme myeloperoxidase to catalyse the formation of additional bactericidal compounds (Fig. 1), such as HOCl, BrO− or IO−, by oxidation of Cl−, Br− or I− anions using the O2•− or H2O2 derived from NADPH oxidase (Klebanoff et al. 2013).
Immune cells of infected mammals express another key enzyme, the inducible nitric oxide (NO) synthase (iNOS) that produces NO from L-arginine (Bogdan 2015). NO has a complex chemistry (Fig. 1) and itself reacts rapidly with metals (nitrosylation reaction), such as Fe2+ in haem and iron-sulphur proteins, and combines with superoxide to generate peroxynitrite (ONOO−) that is a strong oxidant by itself. Furthermore, in the presence of O2 and other strong oxidants, NO loses one electron forming the nitrosonium ion (NO+), which reacts with nucleophilic groups such as amines and thiolates generating the later S-nitrosothiols RSNO (nitrosation reaction). Intracellularly, S-nitrosothiols are also formed by translocation of a NO+ group between two sulphur atoms (transnitrosation reaction) (Pacher, Beckman and Liaudet 2007).
Several works have shown that ROS and RNS, released by macrophages activated by pathogen-associated molecular patterns such as lipopolysaccharide (LPS), contribute to bacterial eradication, due to their ability to damage DNA, lipids, amino acid residues and metal centres that lead to protein inactivation (Pacher, Beckman and Liaudet 2007).
Helicobacter pylori and Campylobacter jejuni set off a host inflammatory response upon invasion of the gastric epithelium and intestinal epithelium, respectively (Hu et al. 2006; Borlace et al. 2012; Fehlings et al. 2012; Bhattacharyya et al. 2014). Consequently, these pathogens elicit the formation of ROS and RNS by increasing the expression of NADPH oxidase and iNOS in immune cells. Helicobacter pylori LPS has been shown to activate the transcription of NADPH oxidase in guinea pig gastric mucosal cells (Kawahara et al. 2005) and H. pylori-infected patients presented augmented expression of NADPH oxidase and high ROS levels (Augusto et al. 2007; Rokutan et al. 2008; Elfvin et al. 2014). Moreover, H. pylori causes upregulation of iNOS in gastric epithelial cells, neutrophils and macrophages, as judged by the higher systemic and intraluminal levels of stable NO-derived metabolites (nitrite and nitrate) measured in H. pylori-infected patients (Shapiro and Hotchkiss 1996; Wilson et al. 1996; Cherdantseva et al. 2014). Additionally, nitric oxide synthase (iNOS) deleted mice infected with H. pylori had increased mortality compared with infected wild-type mice (Nam et al. 2004). Campylobacter jejuni has also been reported to induce the formation of ROS generated by epithelial NADPH oxidases, which were described to impair bacterial capsule formation and modify bacterial signal transduction (Corcionivoschi et al. 2012). Infection of murine macrophages by C. jejuni highly induced the expression of iNOS, and macrophages lacking iNOS exhibited increased C. jejuni viability, particularly after 24 h of infection (Iovine et al. 2008; Tarantino et al. 2009).
The bacterial intracellular ROS and RNS targets are multiple, and the degree of damage depends on the concentration and time of exposure. In vitro studies of Helicobacter and Campylobacter under stress conditions have been utilising several sources of ROS and RNS. For ROS generation, oxygen exposure, H2O2 and other peroxides have been the chosen sources; for RNS, the majority of the studies have been performed with NO donors such as S-nitrosoglutathione (GSNO), sodium nitroprusside (SNP) and NONOates. However, antimicrobial assays that utilise different NO donors are difficult to compare due to the different NO chemistry associated with each compound. Indeed, a recent study of H. pullorum exposed to three different sources of NO, namely spermine-NONOate, DPTA-NONOate and GSNO, showed that their effects differ, with GNSO being the less effective one. Interestingly, the higher viability impairment was observed for H. pullorum stressed at the initial growth phase, an effect that was independent of the nitrosative stress's source (Parente et al. 2016).
One of the primary targets of O2•− and H2O2 are iron-sulphur (Fe-S)-containing enzymes (e.g. the respiratory dehydrogenases and hydratases), which results in the release of iron ions (Imlay 2008). Work done in the bacterial prototype Escherichia coli has shown that the univalent O2•− oxidation of solvent exposed [4Fe-4S] 2+/1+ clusters generates the catalytically inactive [3Fe-4S] 1+/0 cluster that is formed by release of one Fe2+ ion. Similarly, the divalent oxidation of the [4Fe-4S]2+/1+ group by H2O2, also results in the generation of an inactive [3Fe-4S]1+/0 cluster (Jang and Imlay 2007).
Besides enzyme damage, the generated free ferrous ions undergo the Fenton reaction in which the Fe2+ is oxidised by H2O2 to produce Fe3+ and the hydroxyl radical (•OH), which acts at diffusion limiting rates and is a powerful oxidant of most biological molecules (Imlay 2003, 2008).
Proteins containing mononuclear iron centres may be also damaged by H2O2 via the Fenton chemistry (Anjem and Imlay 2012). Furthermore, the generation of a •OH radical can result in further irreversible damage by reacting with the local polypeptide (Anjem and Imlay 2012). Superoxide may also exert deleterious effects on these proteins via loss of the Fe2+ ions and subsequent enzymatic inactivation (Gu and Imlay 2013). Although the inactive enzyme can undergo remetallation by the cell to restore function, mismetallation may occur (Gu and Imlay 2013). Mononuclear Fe2+-containing enzymes that have been shown to be sensitive to ROS damage include epimerases, dehydrogenases, deformylases and deaminases (Sobota and Imlay 2011; Anjem and Imlay 2012).
Bacterial metalloproteins with key metabolic functions are also impaired by NO due to direct binding to metal centres, such as Fe-S (e.g, aconitase of the citric acid cycle) and haem-iron (e.g. respiratory oxygen reductases) (Sarti et al. 2003). Furthermore, toxic GSNO is generated inside cells by the univalent oxidation of NO to the nitrosonium ion (NO+), promoted by free intracellular or protein-bound metals, and its interaction with glutathione (Bowman, McLean et al. 2011, Broniowska and Hogg 2012, Broniowska, Diers and Hogg 2013). NO is also an indirect DNA-damaging agent as autooxidation to N2O3 can result in deamination of DNA bases. Additionally, the peroxynitrite produced by reaction with NO also promotes nitration of the DNA bases.
Both Campylobacter and Helicobacter species encode [4Fe-4S]2+/1+-containing enzymes that have important functions in energy metabolism and are known targets of oxidative and nitrosative damage (Velayudhan et al. 2004). These enzymes include aconitase B (AcnB), serine dehydratase (SdaA), pyruvate: acceptor oxidoreductase (Por) and 2-oxoglutarate: acceptor oxidoreductase (Oor). In C. jejuni, exposure to oxygen was shown to cause inactivation of the [4Fe-4S]2+/1+ clusters with decrease of the enzymatic activity and cell viability (Kendall, Barrero-Tobon et al. 2014). In Campylobacter jejuni and H. pylori the peptide deformylase (def) is also a target for ROS inactivation.
Thus, the successful colonisation of Helicobacter and Campylobacter spp. critically depends on their protective mechanisms against ROS and RNS, which are described in the following sections.
OXIDATIVE STRESS DEFENCES OF HELICOBACTER PYLORI AND CAMPYLOBACTER JEJUNI
To combat the deleterious effects of ROS and inactivation of key cellular proteins, pathogens express inducible detoxification enzymes that convert harmful reactive oxygen intermediates into less harmful or inert products, and have cellular mechanisms to repair ROS-induced macromolecule damage. The next paragraphs describe the ROS protective systems so far identified in H. pylori and C. jejuni (Fig. 1, Tables 1 and 2).
Table 1.
Summary of Helicobacter systems against phagocytes’ oxidative and nitrosative burst. Protein, gene name and function are listed.
| Protein | Gene | Function | References |
|---|---|---|---|
| Oxidative response | |||
| Superoxide dismutase SodB | hp0389 | Superoxide detoxification | Pesci and Pickett (1994); Seyler et al. (2001) |
| Catalase KatA | hp0875 | H2O2 detoxification | Harris et al. (2002); Harris and Hazell, (2003); Hong et al. (2007); Mehta et al. (2007) |
| Catalase-associated protein KapA | hp0874 | KatA chaperone | Harris et al. (2003) |
| Alkyl hydroperoxidase AhpC | hp1563 | Peroxides detoxification | Baker et al. (2001); Olczak et al. (2002, 2003); Charoenlap et al. (2012) |
| Thiol peroxidase Tpx | hp0390 | Peroxides detoxification | Comtois et al. (2003); Olczak et al. (2003) |
| Bacterioferritin comigratory protein BCP | hp0136 | H2O2 and organic peroxide detoxification | Comtois et al. (2003); Wang et al. (2005b) |
| Thioredoxins Trx1, Trx2 | hp1458, hp0824 | Oxidative stress | Windle et al. (2000); Comtois et al. (2003); McGee et al. (2006) |
| NADPH-specific quinone oxidoreductase, MdaB | hp0630 | Reduction of soluble quinones | Wang and Maier (2004); Hong et al. (2008) |
| Neutrophil-activating protein NapA | hp0243 | Oxidative stress | Cooksley et al. (2003); Wang et al. (2006) |
| Cytochrome c peroxidase Ccp | hp1461 | Oxidative stress | |
| Methionine sulfoxide reductase MsrAB | hp0224 | Reduction of oxidised methionine | Alamuri and Maier (2004, 2006); Mahawar et al. (2011); Benoit et al. (2013); Kuhns et al. (2013) |
| DNA glycolase MutY | hp0142 | DNA-repairing protein | Eutsey et al. (2007) |
| Nitrosative response | |||
| Nitric oxide reductase NorH | hp0013 | NO detoxification | Justino et al. (2012) |
| Haemoglobin-like (H. pullorum) | hpmg_00979 hpmg_00954 | NO detoxification | Saraiva et al. (unpublished) |
| Nitroreductase FrxA | hpB8_844 | GSNO reduction | Justino et al. (2014) |
| Alkyl hydroperoxidase AhpC | hp1563 | Peroxynitrite reduction | Bryk et al. (2000) |
| Regulators | |||
| Ferric uptake regulator Fur | hp1027 | Iron homeostasis | Danielli et al. (2006); Miles et al. (2010) |
| CrdRS two-component regulator | hp1364 | Nitrosative stress | Hung et al. (2015) |
| hp1365 | |||
| Aconitase B AcnB | hp0779 | Oxidative stress | Austin and Maier (2013) |
| Carbon storage regulator CrsA | hp1442 | Oxidative stress | Fields and Thompson (2008) |
| ArsRS two-component regulator | hp0165 | Oxidative stress | Loh et al. (2010) |
| hp0166 | |||
| Homeostatic global regulator HsrA | hp1043 | Oxidative stress | Olekhnovich et al. (2014) |
| Histone-like protein | hp0835 | Oxidative stress | Wang and Maier (2015) |
Table 2.
Summary of Campylobacter systems against phagocytes’ oxidative and nitrosative burst. Protein, gene name and function are listed.
| Protein | Gene | Function | References |
|---|---|---|---|
| Oxidative response | |||
| Superoxide dismutase SodB | cj0169 | Superoxide detoxification | Pesci et al. (1994); Purdy and Park (1994); Palyada et al. (2009); Novik et al. (2010) |
| Catalase KatA | cj1385 | H2O2 detoxification | Day et al. (2000); Palyada et al. (2009) |
| Catalase biogenesis protein Cj1386 | cj1386 | Trafficking haem to KatA | Flint et al. (2012); Flint and Stintzi (2015) |
| Alkyl hydroperoxide reductase AhpC | cj0344 | Peroxides detoxification | Baillon et al. (1999); Palyada et al. (2009) |
| Thiol peroxidase Tpx | cj0779 | H2O2 detoxification | Atack et al. (2008) |
| Bacterioferritin comigratory protein BCP | cj0271 | H2O2 and organic peroxide detoxification | Atack et al. (2008) |
| DNA-binding protein Dps | cj01534c | Sequestration of Fe ions | Ishikawa et al. (2003); Theoret et al. (2011, 2012); Huergo et al. (2013) |
| Desulforubrerythrin DRbr | cj0012c | H2O2 detoxification | Pinto et al. (2011); Flint et al. (2014) |
| Methionine sulfoxide reductase MsrA | cj0637c | Reduction of oxidised methionine (S-MetSO) | Atack and Kelly (2008) |
| Methionine sulfoxide reductase MsrB | cj1112c | Reduction of oxidised methionine (R-MetSO) | Atack and Kelly (2008) |
| Nitrosative response | |||
| Single domain haemoglobin Cgb | cj1586 | NO detoxification | Elvers et al. (2004); Tinajero-Trejo et al. (2013) |
| Truncated haemoglobin Ctb | cj0465c | NO detoxification | Smith et al. (2011) |
| Nitrite reductase NrfA | cj1357c | Nitrite reduction and NO detoxification | Pittman et al. (2007) |
| Regulators | |||
| Peroxide regulator PerR | cj0322 | Oxidative stress | Palyada et al. (2009); Butcher et al. (2015) |
| Ferric uptake regulator Fur | cj0400 | Iron homeostasis | Palyada et al. (2009); Butcher et al. (2015) |
| Aconitase B AcnB | cj0835c | Oxidative stress | Flint et al. (2014) |
| Campylobacter oxidative stress regulator CosR | cj0355c | Oxidative stress | Garenaux et al. (2008); Hwang et al. (2011, 2012) |
| LysR-type regulator Cj1000 | cj1000 | Oxidative stress | Dufour et al. (2013) |
| Regulator of response to peroxide RrpA | cj1546 | Oxidative stress | Gundogdu et al. (2015) |
| Regulator of response to peroxide RrpB | cj1556 | Oxidative stress | Gundogdu et al. (2011) |
Superoxide dismutase SodB
As mentioned above, NADPH oxidases produce superoxide. The major bacterial detoxifiers of superoxide are the superoxide dismutase (Sod) enzymes that convert O2•− into O2 and H2O2. Bacterial pathogens may contain up to four different forms of superoxide dismutase enzymes, SodA, SodB, SodC and SodN, which contain manganese, iron, copper-zinc and nickel, respectively, or superoxide reductases (Sheng et al. 2014).
Unlike other enteric pathogens such as Escherichia coli, Shigella and Salmonella, H. pylori encodes only one cytoplasmic Fe-superoxide dismutase (SodB). The protein is a dimer consisting of two identical subunits and displays 53% and 48% amino acid sequence identity to the structurally characterised SodB enzymes of E. coli and Pseudomonas ovalis, respectively (Ringe et al. 1983; Lah et al. 1995; Esposito et al. 2008; Sheng et al. 2014). At certain stages of H. pylori growth, SodB seems to be localised on the cell surface and even attached to the flagellar sheath, although no export sequence has been identified in its amino acid sequence (Spiegelhalder et al. 1993). However, H. pylori SodB has a C-terminus extension, which is absent in the E. coli and P. ovalis SodB structures (Esposito et al. 2008) but present in C. jejuni SodB (Pesci, Cottle and Pickett 1994), that has been proposed to play a role in SodB cell surface location in H. pylori. Alternatively, the serine and/or tyrosine residues located in the C-terminal tail may serve as phosphorylation sites as phosphoproteome studies showed that C. jejuni and H. pylori SodB are major phosphoproteins (Voisin et al. 2007). However, the role of these post-translational modifications on SodB function remains to be elucidated. Interestingly, other H. pylori cytoplasmic proteins, such as urease and heat shock protein B, have also been detected on the bacterial cell surface. Since these proteins were found to be incorporated onto the cell surface following bacterial lysis, their localisation has been considered to be the result of spontaneous bacterial cell autolysis that occurs during the late growth phase (Phadnis et al. 1996). Additional studies have argued that specific mechanisms (such as a type III secretion apparatus or ABC transporters) may be responsible for the secretion of the cytoplasmic proteins to the cell surface (Vanet and Labigne 1998). Despite these studies, the molecular mechanism behind the SodB surface localisation remains unclear.
The H. pylori sodB mutant exhibits impaired growth under typical laboratory microaerobic culturing conditions (12% O2 partial pressure), atmospheric O2 concentrations (Seyler, Olson and Maier 2001) and in the presence of H2O2 (Seyler, Olson and Maier 2001; Palyada et al. 2009). Transcriptome studies of H. pylori-infected macrophages revealed significant upregulation of sodB (Singh et al. 2012). The importance of H. pylori SodB for ROS survival during in vivo colonisation has also been demonstrated in murine gastric colonisation assays, in which the ΔsodB mutant failed to colonise the majority of mice contrary to the wild-type strain (Seyler, Olson and Maier 2001, Stent, Every and Sutton 2012).
Campylobacter jejuni expresses a single cytoplasmic Fe-superoxide dismutase SodB (Pesci and Pickett 1994; Pesci, Cottle and Pickett 1994; Parkhill et al. 2000). Deletion of sodB resulted in hypersensitivity towards O2•−, H2O2 and organic peroxides (Palyada et al. 2009). The higher H2O2 sensitivity of the ΔsodB mutant has been related to the augmented levels of endogenous superoxide and free ferrous ions, which arise from damaged Fe-S clusters, and sustain the Fenton chemistry. A similar mechanism is also proposed to occur in the presence of organic peroxides, as deletion of sodB resulted in the production of alkoxyl radicals and lipid peroxidation (Palyada et al. 2009). Investigation of the role of SodB in cellular pathogenesis showed that the ΔsodB mutant in C. jejuni 81–176 has a much lower survival (12-fold decrease) following invasion of human cervix carcinoma INT407 cells relative to the wild-type strain (Pesci et al. 1994). Experiments on epithelium cell adhesion, invasion and survival further revealed that the ΔsodB mutant has decreased adhesion and intracellular survival but is not defective for the invasion process itself (Novik, Hofreuter and Galan 2010). Additionally, the C. jejuni SodB has been shown to support in vivo colonisation of chicks with the ΔsodB mutant being significantly impaired in colonisation relative to the wild-type strain (Pesci et al. 1994).
Catalase (KatA) and catalase accessory and biogenesis proteins (KapA and Cj1386)
Dismutation of O2•− by SodB yields H2O2, which must be detoxified by additional cellular antioxidant enzymes to limit the Fenton chemistry and prevent macromolecule damage within the bacterial cell. Several enzymes exist within H. pylori and C. jejuni to keep the H2O2 levels from reaching lethal concentrations, such as catalases (Day et al. 2000; Harris et al. 2002; Palyada et al. 2009) and peroxiredoxins (see Section Peroxiredoxins: AhpC, Tpx and BCP).
Catalases have been extensively studied over the past 100 years and play a critical role in allowing organisms to survive within micro- and aerobic environments. Catalases are grouped into three main classes including monofunctional catalases, bifunctional catalase-peroxidases and non-haem-containing catalases (Zamocky et al. 2012). Bacterial species containing all three classes of catalase enzymes have been identified (Robbe-Saule et al. 2001). Furthermore, in Gram-negative bacteria these enzymes are usually not restricted to the cytoplasmic space and can be exported to the periplasm (Brunder, Schmidt and Karch 1996, Harris and Hazell 2003). At high concentrations of H2O2, catalases are the major detoxifiers of H2O2 (Alfonso-Prieto et al. 2009), do not display saturation even at millimolar H2O2 concentrations (Imlay 2008) and are stable under a variety of physiological conditions (Chelikani, Fita and Loewen 2004).
Helicobacter pylori and C. jejuni express one small subunit monofunctional tetrameric catalase, KatA, consisting of four identical subunits each containing a haem b prosthetic group (Grant and Park 1995; Odenbreit, Wieland and Haas 1996; Zamocky et al. 2012). The haem catalyses a two-step oxidation–reduction reaction to dismutate two molecules of H2O2 into O2 and two molecules of H2O (Alfonso-Prieto et al. 2009). Helicobacter pylori KatA was shown to be enzymatically active in both the cytoplasmic and periplasmic compartments (Harris and Hazell 2003). Deletion of katA in H. pylori and C. jejuni abolished catalase activity and resulted in hypersensitivity of the mutant strains to H2O2 when compared with the parental strain (Day et al. 2000; Harris et al. 2002; Bingham-Ramos and Hendrixson 2008; Palyada et al. 2009). Moreover, C. jejuni ΔkatA was attenuated for macrophage survival (Day et al. 2000). Long-term survival assays revealed that the wild-type strain remains viable 72 h post-infection, while the ΔkatA mutant loses viability (Day et al. 2000; Basu, Czinn and Blanchard 2004). In support of the role of ROS-mediated bacterial killing in macrophages, the use of an NADPH oxidase inhibitor facilitated survival of the ΔkatA mutant within macrophages (Day et al. 2000).
Despite the large amount of knowledge on catalase structure and biochemical properties, the study of the catalase chaperone proteins is still in its infancy. Indeed, Helicobacter and Campylobacter are among the few bacterial organisms in which catalase chaperons have been identified, namely KapA and Cj1386.
In H. pylori, the catalase-associated KapA does not exhibit catalase activity and thus is not expected to be directly involved in H2O2 detoxification. However, KapA has been proposed to promote in the translocation of KatA to the periplasmic space and therefore to play a role in hydrogen peroxide defence (Harris et al. 2002; Harris and Hazell 2003). The presence of a twin-arginine translocation motif in the KapA protein sequence (Harris and Hazell 2003) as well as the predicted KatA-KapA protein–protein interaction (Rain et al. 2001) supports the role of KapA in KatA translocation into the periplasmic space. In agreement, deletion of kapA significantly lowered the catalase activity detected in the periplasm but not in the cytoplasm (Harris and Hazell 2003). The importance of KatA and KapA in the resistance to ROS in vivo has been highlighted in murine gastric mucosa colonisation assays (Harris et al. 2003). Initial colonisation of the mice was not found to be attenuated in either ΔkatA or ΔkapA deletion mutants relative to the wild-type strain. However, long-term colonisation assays revealed a fitness defect in both the ΔkatA and ΔkapA deletion mutants relative to the wild-type (Harris et al. 2003).
Recent studies in C. jejuni have also identified a novel gene, cj1386, encoded directly downstream from katA, which functions in H2O2 defence (Flint, Sun and Stintzi 2012). Characterisation of the Cj1386 protein revealed that it binds haem b at a 1:1 ratio and that it interacts with KatA (Flint and Stintzi 2015). Similarly to H. pylori KapA, Cj1386 did not show catalase activity, but was found to indirectly contribute towards optimal catalase activity within cells (Flint, Sun and Stintzi 2012). Indeed, immunoprecipitation of KatA from a C. jejuni Δcj1386 mutant strain contained a significantly lower amount of haem b bound to KatA, suggesting a role for Cj1386 in haem trafficking to KatA (Flint, Sun and Stintzi 2012). In vivo, KatA and Cj1386 proteins provide important oxidative stress protection as assessed in a neonate piglet Campylobacter pathogenesis model, in which ΔkatA and Δcj1386 mutant strains were outcompeted by the wild-type C. jejuni for colonisation of the gastrointestinal tract (Flint, Sun and Stintzi 2012). Furthermore, the ΔkatA and Δcj1386 mutant strains were significantly attenuated for colonisation of chick ceca relative to the parental strain (Palyada et al. 2009; Flint, Sun and Stintzi 2012).
Peroxiredoxins: AhpC, Tpx and BCP
Although bacteria use catalases as major detoxifiers of H2O2, other peroxide detoxification enzymes also contribute to limit oxidative damage. These enzymes include alkyl hydroperoxide reductase (AhpC), thiol peroxidase (Tpx) and bacterioferritin comigratory protein (BCP). The peroxiredoxin enzymes can be classified according to their mechanism into 1-Cys and 2-Cys peroxiredoxins; both classes reduce H2O2 to H2O through an active site cysteine that is oxidised to sulfenic acid. In the 2-Cys subtype, the first conserved cysteine residue is oxidised by H2O2 to sulfenic acid and subsequently forms a disulphide bridge by condensing with the second conserved cysteine residue. Both 1-Cys and 2-Cys peroxidase enzymes are regenerated to their original reduced state by NADH-dependent oxidoreductases (Wood et al. 2003; Flohe et al. 2011; Nelson et al. 2011).
Helicobacter pylori encodes an AhpC that detoxifies a wide range of peroxides including H2O2, t-butyl, cumene, ethyl and linoleic acid hydroperoxides (Baker et al. 2001). In contrast to H. pylori wild-type, strains lacking AhpC displayed growth inhibition and high sensitivity to oxygen under normal microaerobic growth conditions (12% O2 partial pressure) (Olczak, Olson and Maier 2002). The ΔahpC mutant was also found to have hypersensitivity to the superoxide radical producing compound, paraquat (Olczak, Olson and Maier 2002). Moreover, DNA mutation frequency assays done with the ΔahpC mutant showed that deletion of ahpC resulted in a significant 20-fold increase in the frequency of rifampicin-resistant colonies when compared with the wild-type strain, demonstrating the protective role of AhpC against DNA oxidative damage (Olczak, Olson and Maier 2002). In vivo, ahpC-deficient H. pylori was completely attenuated for colonisation of the murine stomach (Olczak et al. 2003). Interestingly, in H. hepaticus the deletion of ahpC generated a strain with increased resistance to H2O2, which was linked to a higher KatA protein abundance (Hong, Wang and Maier 2007).
AhpC also shelters C. jejuni from oxidative stress. Stationary-phase C. jejuni ΔahpC mutants had increased sensitivity when exposed to atmospheric O2 conditions and cumene hydroperoxide (Baillon et al. 1999; Palyada et al. 2009). Although Baillon et al. (1999) found no significant difference in sensitivity of the ΔahpC mutant towards H2O2, Palyada et al. (2009) observed the opposite. However, the absence of a hypersensitive phenotype to H2O2 in the ΔahpC mutant may be due to compensation by other H2O2 detoxification enzymes such as KatA, as seen in H. hepaticus. In vivo, the C. jejuni ahpC mutant had lower viability in colonisation of chicks relative to the wild-type strain (Palyada et al. 2009).
Helicobacter pylori expresses two thioredoxin enzymes (Trx1 and Trx2) and a Trx reductase (TrxR). The TrxA1/TrxR enzymes form a reductase system for the AhpC peroxiredoxin (Baker et al. 2001). Additionally, Trx1 acts as a chaperone that assists in the reactivation of denatured arginase, which is inhibited by most reactive oxygen and nitrogen intermediates (Comtois, Gidley and Kelly 2003; McGee et al. 2006). Cells harbouring single and double knockout mutations of trx1 and trx2 have increased sensitivity not only to oxygen, H2O2, cumene hydroperoxide and paraquat, but also to SNP and GSNO (Comtois, Gidley and Kelly 2003; McGee et al. 2006). Kuhns, Wang and Maier (2015) showed that the two thioredoxins are important in protecting H. pylori from oxidative stress, with single mutants having a higher abundance of lipid peroxides and suffering more DNA damage and protein carbonylation than the parental strain.
Tpx and BCP belong to the 2-Cys peroxiredoxin family and are involved in oxidative stress defence in Helicobacter and Campylobacter species (Nguyen et al. 2010).
Helicobacter pylori Tpx detoxifies a wide range of peroxide substrates including H2O2 and cumene hydroperoxide (Comtois, Gidley and Kelly 2003; Olczak et al. 2003). The Δtpx mutant exhibits hypersensitivity to H2O2, cumene hydroperoxide, paraquat and atmospheric O2 (Comtois, Gidley and Kelly 2003; Olczak et al. 2003) and reduced colonisation ability of a mouse stomach (Olczak et al. 2003).
Helicobacter pylori Δbcp has a moderate increase in the sensitivity to paraquat, cumene hydroperoxide and tert-butyl hydroperoxide but not to H2O2 (Comtois, Gidley and Kelly 2003; Wang et al. 2005b). Enzymatic assays with purified BCP showed minor thiol peroxidase activity using H2O2 or tert-butyl hydroperoxide, which is in accordance with the in vitro growth sensitivity assays (Wang et al. 2005b). Interestingly, H. pylori BCP has a significant peroxidase activity in the presence of linoleic acid hydroperoxide and is seems to be able to protect against lipid peroxidation (Wang et al. 2005b). Despite the minor role of BCP for in vitro ROS protection, in vivo murine gastric colonisation studies revealed diminished long-term colonisation and lower survival of the Δbcp mutant relative to the wild-type (Wang et al. 2005b).
Campylobacter jejuni Tpx and BCP are cytoplasmic proteins with distinct peroxide substrate specificities (Atack et al. 2008). Purified Tpx was reported to specifically detoxify H2O2, whereas BCP catalyses the detoxification of H2O2 as well as the organic peroxides, cumene hydroperoxide and tert-butyl hydroperoxide, but not linoleic acid hydroperoxide (Atack et al. 2008). Both Δtpx and Δbcp single mutants are less resistant to organic peroxides when compared with the wild-type, and also had elevated levels of lipid peroxides (Atack et al. 2008). However, under H2O2 stress conditions, the Δtpx and Δbcp single mutants exhibited a growth behaviour similar to that of the parental strain and only the double mutant ΔtpxΔbcp was significantly impaired (Atack et al. 2008). Furthermore, stationary phase assays with, microaerobically grown strains that were transferred to aerobic conditions showed that the double mutant ΔtpxΔbcp loses viability more rapidly than either the wild-type or the respective single mutants Δtpx and Δbcp (Atack et al. 2008). Taken together, these findings suggest compensatory roles for Tpx and BCP in H2O2 defence and O2 survivability.
Overall, these results suggest that although C. jejuni and H. pylori AhpC, Tpx and BCP enzymes have overlapping functions, each individual protein still imparts protection against ROS and provides a fitness advantage during host colonisation.
NADPH-specific quinone reductase MdaB
Liposoluble quinones (for example, in the form of ubiquinone or menaquinone) are important components of the bacterial respiratory chains that conduct electron transfer between respiratory complexes (Soballe and Poole 1999). However, one electron reduction of quinones produces semiquinone radicals, which can subsequently reduce O2 to superoxide. Furthermore, soluble cytosolic quinones and derivatives can be quite toxic to cells. Two-electron reduction of quinones to quinols by cytosolic NADPH:quinone oxidoreductase enzymes (distinct from the respiratory type II NADH:quinone oxidoreductases) is one strategy utilised by cells to reduce the toxicity of quinones (Dinkova-Kostova and Talalay 2000).
Helicobacter pylori encodes a NADPH-specific quinone oxidoreductase, MdaB. Substrate specificity assays using purified protein showed that MdaB reduces a broad range of water-soluble quinones including coenzyme Q0, coenzyme Q1, menadione and 1,4-naphthoquinone (Wang and Maier 2004; Hong, Wang and Maier 2008). Deletion of mdaB resulted in augmented sensitivity to O2, H2O2, organic peroxides and paraquat (Wang and Maier 2004). Similar findings have also been reported for a H. hepaticus ΔmdaB mutant, which presented hypersensitivity towards the same compounds (Hong, Wang and Maier 2008). In vivo assays revealed that the H. pylori ΔmdaB mutant had diminished colonisation ability of the mouse stomach relative to the wild-type strain (Wang and Maier 2004).
For C. jejuni, transcriptome analysis showed that the pathogen expresses a mdaB homologue (Parkhill et al. 2000) that is induced in response to oxidant exposure generated by H2O2 and cumene hydroperoxide (Palyada et al. 2009). However, deletion of mdaB did not result in aggravated sensitivity to any of the oxidants assayed (Flint et al. 2014). Therefore, the role of MdaB in C. jejuni oxidative stress defence remains unclear.
Other oxidative stress protective systems
In addition to the major direct detoxifiers SodB, KatA, AhpC, BCP, Tpx and MdaB active in Helicobacter and Campylobacter, other less well-characterised proteins have been shown to be important for oxidative stress defence, such as Dps-like proteins, DRbr and Ccp.
The DNA-binding protein from starved cells (Dps) belongs to a subgroup within the ferritin family (Ishikawa et al. 2003). Dps consists of 12 identical subunits that form a dodecamer and each monomer can store up to 500 iron atoms (Ishikawa et al. 2003, Sanchuki et al. 2015). Dps binds Fe2+ ions that are oxidised to Fe3+ by the Dps ferroxidase centre (Ishikawa et al. 2003, Sanchuki et al. 2015). Sequestration of free Fe2+ ions by Dps helps prevent the formation of the hydroxyl radical through the Fenton reaction that occurs during periods of cellular H2O2 exposure (Ishikawa et al. 2003). In addition to scavenging free Fe2+ ions, Dps binds to DNA to protect it against oxidative damage (Cooksley et al. 2003; Ceci et al. 2004; Wang et al. 2006).
Helicobacter pylori expresses a Dps-like protein, the neutrophil-activating protein NapA, that also sequesters iron and binds DNA (Zanotti et al. 2002). NapA has a positive surface charge that is not present in other Dps proteins (Zanotti et al. 2002). Interestingly, H. pylori NapA appears to have a dual function as it binds to the outer membrane (Namavar et al. 1998) and is involved in the activation of host neutrophils. It is speculated that the positive surface charge plays a role in binding to and activating neutrophils (Evans et al. 1995; Montecucco and de Bernard 2003). Although NapA is involved in activating neutrophils, competitive mouse colonisation in vivo assays using wild-type and a ΔnapA strain revealed that NapA is required for successful H. pylori pathogenesis (Wang et al. 2006).
Similarly, H. hepaticus Dps was described to confer oxidative stress resistance and protect from DNA damage. Purified H. hepaticus Dps binds both iron and DNA, and the iron-loaded form of the protein presented increased DNA-binding ability (Hong, Wang and Maier 2006).
Campylobacter jejuni also encodes a Dps protein that was reported to bind DNA (Huergo et al. 2013, Sanchuki et al. 2015), protect cells from H2O2 stress (Ishikawa et al. 2003) and contribute for successful colonisation of chicks and neonate piglets (Theoret et al. 2011, 2012).
In C. jejuni, a multidomain protein from the family of rubrerythrins named desulforubrerythrin (Drbr) was isolated and characterised; it contains a desulforedoxin-like domain fused to a canonical rubrerythrin (Yamasaki et al. 2004). Drbr exhibits a significant NADH-dependent hydrogen peroxide reductase activity within the range of those observed for rubrerythrins from other species (Pinto et al. 2011). Deletion of the Drbr coding gene in C. jejuni resulted in a modest increase in sensitivity towards H2O2 and menadione (Flint et al. 2014). Interestingly, the mutant strain was not significantly attenuated in chick colonisation studies (Flint et al. 2014). Thus, although Drbr exhibits H2O2 detoxification activity in vitro, it seems to play only a minor role (in contrast to KatA and AhpC) in cellular protection against host innate immunity.
Campylobacter jejuni encodes two periplasmic cytochrome c peroxidases (Ccp), namely cj0020c and cj0358 that detoxify H2O2 (Parkhill et al. 2000; Bingham-Ramos and Hendrixson 2008). Characterisation of Cj0020c and Cj0358 in vitro has been carried out in C. jejuni 81–176 (designated as DocA and Cjj0382, respectively) which are periplasmic proteins with apparent haem-dependent peroxidase activity (Bingham-Ramos and Hendrixson 2008). However, deletion of docA or cjj0382 did not significantly impaired the resistance of the strain to H2O2 (Bingham-Ramos and Hendrixson 2008), similar to what was observed for the C. jejuni Δcj0358 mutant (Flint et al. 2014). In vivo, the ΔdocA and Δcjj0382 mutants were attenuated in chick colonisation assays relative to the wild-type, but the Δcjj0382 mutant revealed lower infection ability only at low inoculum doses (102 organisms) (Bingham-Ramos and Hendrixson 2008). Thus, the actual role of Ccps in resistance to ROS during C. jejuni colonisation remains unclear (Bingham-Ramos and Hendrixson 2008).
Macromolecule repair
Bacteria use several molecular mechanisms to repair the oxidative damage that is inflicted to DNA, proteins and lipids. In Campylobacter and Helicobacter, proteins such as MsrAB and MutSHL have been identified and characterised for their roles in macromolecule repair and shown to be important for colonisation of the host.
Methionine sulfoxide reductase MsrAB
Methionine residues are common targets of oxidative damage due to the presence of an oxidisable sulphur atom (Moskovitz 2005). Two isomers of methionine are produced from oxidation, R- and S-methionine sulfoxide (R- and S-MetSO), which imply conformational changes that may result in loss of function (Ciorba et al. 1997; Gao et al. 1998; Sigalov and Stern 1998). Bacteria contain methionine sulfoxide reductase MsrA and MsrB that reduce the oxidised S- and R-MetSO residues to methionine, respectively (Boschi-Muller, Gand and Branlant 2008).
In H. pylori, the msrA and msrB genes are fused to encode a single Msr enzyme (Alamuri and Maier 2004). Helicobacter pylori Msr is reported to be specific for the R-isomer of sulfoxide and unable to reduce the S- isomer form (Alamuri and Maier 2006). Msr plays an important role in repairing protein oxidation by protecting key cellular proteins that are rich in methionine residues including KatA, AhpC, GroEL, Trx1 and urease maturation protein (UreG) (Alamuri and Maier 2006; Benoit et al. 2013; Kuhns et al. 2013). Accordingly, the Δmsr mutant had increased levels of protein oxidation relative to the wild-type (Alamuri and Maier 2004). Moreover, deletion of msr or inactivation of only the msrB domain conferred hypersensitivity to H2O2 and paraquat (Alamuri and Maier 2004). The H. pylori Δmsr strain was described to be more susceptible to killing by neutrophils than the wild-type (Mahawar et al. 2011). Furthermore, Δmsr was shown to be attenuated for colonisation of mice stomachs at 7 days post inoculation, and no mice could be colonised by Δmsr at later time points (14 and 21 days post inoculation) in contrast to the parental strain (Alamuri and Maier 2004).
Campylobacter jejuni has both MsrA and MsrB which exhibit methionine reductase activity (Parkhill et al. 2000). Deletion of these genes resulted in aggravated sensitivity towards H2O2, cumene hydroperoxide and paraquat (Atack and Kelly 2008).
DNA repair
Under oxidative or chemical stress, bacteria use the SOS response to halt bacterial cell division and induce DNA enzymes that repair harmful modifications (Eisen and Hanawalt 1999).
Some bacteria contain a set of gene products MutSHL for methyl-directed mismatch repair. MutL along with MutS, which recognises and binds to the misincorporated nucleotide, activates MutH that cleaves specific unmethylated daughter strand DNA and allows access for single-strand exonucleases that remove the defective DNA region (Harfe and Jinks-Robertson 2000). Helicobacter pylori and C. jejuni lack the classical SOS system, and mutL and mutH genes, but do encode a mutS homologue (Wang et al. 2005a; Gaasbeek, van der Wal et al. 2009). However, the MutS protein is not thought to be functionally involved in methyl-directed mismatch repair. In H. pylori, the ΔmutS strain had increased sensitivity to oxygen, H2O2 and paraquat. Furthermore, upon exposure to oxidative stress, the ΔmutS mutant exhibited a higher 8-hydroxyguanine mutation rate (a base mutation that usually occurs during periods of oxidant exposure (Wang et al. 2005a; Imlay 2008). Recent structural and mechanistic studies of H. pylori MutS have demonstrated that the protein is a bifunctional single-strand specific nuclease with both DNA nicking and RNA nuclease activity; however, how MutS recognises 8-hydroxyguanine mutations is still unknown. In vivo, MutS contributes to survival and colonisation as inactivation of mutS made H. pylori less efficient for colonisation of the mouse intestine (Wang et al. 2005a). On the contrary, the same deletion did not significantly change mutation rates in C. jejuni (Gaasbeek et al. 2009).
The base excision repair system of DNA repair involves glycosidases that recognise mismatched DNA and specifically cleave the N-glycosylic bond to remove the mismatched base. Helicobacter pylori and C. jejuni encode DNA repair-related glycosidases including the G/U and A/U glycosidase Ung (Gaasbeek et al. 2009) and A/G or A/8-hydroxyguanine glycosidase MutY (Parkhill et al. 2000). The later was shown to participate in oxidative DNA damage repair (Huang, Kang and Blaser 2006; Eutsey, Wang and Maier 2007). Interestingly, H. pylori MutY presented activity specifically against A/8-hydroxyguanine mismatches, but not for A/G mismatch repair. Although a ΔmutY deletion strain did not have any observable sensitivity towards H2O2, cumene hydroperoxide, t-butyl hydroperoxide or paraquat, it did exhibit an increased mutation rate in the presence of atmospheric oxygen (Eutsey, Wang and Maier 2007). Furthermore, the ΔmutY strain was attenuated for colonisation of the stomach of mice, indicative of the protective role that MutY plays against inflammatory induced DNA damage (Eutsey, Wang and Maier 2007).
Helicobacter pylori expresses endonuclease III, a DNA-repairing enzyme that removes oxidised pyrimidine and contributes to ROS resistance. In the absence of the gene, H. pylori presents elevated spontaneous and induced mutation rates and higher sensitivity to killing by exposure to oxidative agents or activated macrophages. Furthermore, endonuclease III potentiates the colonisation capacity of H. pylori (O'Rourke et al. 2003).
NITRIC OXIDE STRESS DEFENCES OF HELICOBACTER PYLORI AND CAMPYLOBACTER JEJUNI
Bacteria submitted to nitrosative stress activate responsive genes that encode enzymes involved in detoxification, export, repair as well as other homeostatic functions. The prokaryotic responses include at least five distinct types of enzymes/proteins: (i) enzymes that directly remove NO and S-nitrosothiols; (ii) enzymes that detoxify ROS, which also avoid indirectly the formation of RNS due to blockage of the reaction of ROS with NO; (iii) enzymes that regenerate reduced pyridine nucleotides; (iv) DNA-repairing enzymes; and (v) regulators of iron homeostasis that by controlling the formation of iron-nitrosyl species hinder their catalytic role in nitrosylation reactions. In the following sections, the systems currently known to contribute to nitrosative stress resistance in Helicobacter and Campylobacter spp. will be described (Fig. 2, Tables 1 and 2), with major emphasis on two of the most prominent members of this family, namely H. pylori and C. jejuni.
Figure 2.

Biological targets damaged by ROS and RNS, regulators, direct detoxification defences and indirect protective systems against RNS and ROS in Campylobacter and Helicobacter species. RNS and ROS species damage important biological molecules such as DNA, lipids, haem and Fe-S clusters. Transcriptional and post-transcriptional regulators important for ROS/RNS defence are shown. Fur, AcnB and CrsA are present in H. pylori and C. jejuni; ArsRS, HsrA, CrsRS and Hp110 are expressed in H. pylori and regulators indicated in the right column are active in C. jejuni. Detoxification and repair enzymes of H. pylori (white), C. jejuni (blue) and common to both pathogens (white-blue gradient) that protect against oxidative and nitrosative stress are indicated in black and blue, respectively. ETC, respiratory electron transfer chain.
Detoxification
In a large number of bacteria, two major families of proteins promote the enzymatic removal of NO, namely the flavohaemoglobins and flavodiiron NO reductases (Saraiva, Vicente and Teixeira 2004; Forrester and Foster 2012; Stern and Zhu 2014; Romao et al. 2016). Although H. pylori is able to thrive in NO-enriched environments (Park et al. 2003), such enzymes are apparently absent in the Helicobacter and Campylobacter genomes. These pathogens also lack homologues of other bacterial NO detoxification/reduction systems like the respiratory membrane-bound NO reductases and c-type cytochromes. Recently, it was shown that H. pylori contains a new type of NO-reducing enzyme, NorH reductase (Justino et al. 2012), which is encoded in a large number of Helicobacter and Campylobacter species. RNS detoxification in Campylobacter is also dependent on haemoglobin-like and nitrite reductase enzymes, as described below.
Globins: flavohaemoglobin (FHb), single domain haemoglobins (SDHb) and truncated haemoglobins (TrHb)
In several bacteria, resistance to NO and RNS has been attributed to haemoglobins (Hbs), a diverse group of proteins that belong to the myoglobin superfamily and that include flavohaemoglobins (FHb), single domain haemoglobins (SDHb) and truncated haemoglobins (TrHb) (Vinogradov et al. 2013). One of the best-studied enzymes is flavohaemoglobin (also known as Hmp), which is composed of two domains: a N-terminal globin domain that is fused with a C-terminal NAD(P)H and FAD-binding reductase domain (Bonamore and Boffi 2008). The role of flavohaemoglobin in mediating NO protection is well established for a wide range of bacteria, and its NO-detoxifying activity may occur under aerobic or anaerobic conditions, albeit through different enzymatic mechanisms (Justino et al. 2005; Goncalves et al. 2006; Forrester and Foster 2012). Although homologues of flavohaemoglobins seem to be missing in the Campylobacter and Helicobacter genomes, ∼50% of the Campylobacterales species express single domain and truncated haemoglobins (Vinogradov et al. 2013).
In C. jejuni and C. coli, a single domain haemoglobin designated cgb was described to be induced by nitrosative stress, nitrate and nitrite, via the NssR regulator (see Section Nitrosative-stress response regulator NssR). In a periplasmic nitrate reductase (nap) C. jejuni-deficient mutant, cgb expression was upregulated in response to nitrite (Pittman et al. 2007). While a C. jejuni Δcgb mutant displayed no major differences relative to the wild-type when exposed to oxidative stress agents, it was hypersensitive to the NO donor spermine NONOate and GSNO (Elvers et al. 2004). Furthermore, GSNO-treated C. jejuni expressing higher levels of Cgb had increased NO consumption and aerobic respiration activities, and consequently was relatively insensitive to NO (Elvers et al. 2004; Kern, Winkler and Simon 2011; Smith et al. 2011; Vinogradov et al. 2013). Expression of C. jejuni cgb in an Escherichia coli Δhmp mutant strain abolished the RNS-sensitive phenotype of the E. coli hmp mutant under aerobic conditions (Smith et al. 2011; Tinajero-Trejo et al. 2013). However, data suggest that Cgb has no role in NO resistance when C. jejuni is under oxygen-limited conditions (Avila-Ramirez et al. 2013).
Campylobacter jejuni also expresses a truncated haemoglobin protein (Ctb), which is induced by treatment with NO donors and GSNO (Wainwright et al. 2005, 2006). However, the Δctb mutant did not have augmented sensitivity to nitrosative or oxidative stress-generating compounds neither restore the RNS-sensitive phenotype of the E. coli hmp mutant strain. Ctb was shown to bind O2 under normoxic growth conditions and NO under hypoxic conditions (Smith et al. 2011), which is thought to influence NssR regulation and downstream expression of cgb. Specifically, Ctb-NO binding under low oxygen conditions would limit NssR sensing of NO and decrease the NssR-dependent expression of cgb (Smith et al. 2011). Given that Cgb utilises O2 for activity, Ctb would play a role in reducing expression of cgb under conditions where it would be less enzymatically active. Thus, Ctb is considered to have an indirect role in nitrosative stress defence.
Although apparently absent from H. pylori, other Helicobacter species such as H. hepaticus and H. pullorum express haemoglobin-like proteins. Helicobacter hepaticus encodes a truncated haemoglobin (Nothnagel et al. 2011) while H. pullorum relies on haemoglobin-like proteins such as HPMG_00979 and HPMG_00954 to survive nitrosative stress exposure (Saraiva, Vicente and Teixeira 2004, unpublished results). More work is still required to establish the function of these proteins in Helicobacter spp.
Nitric oxide reductase NorH
Recent studies have shown that the H. pylori hp0013 gene encodes a novel type of nitric oxide reductase that was named NorH. Inactivation of norH generated H. pylori cells that had reduced ability to resist nitrosative stress conditions and exhibited a significantly reduced NADPH-dependent NO reductase activity (Justino et al. 2012). Consistent with these results, the recombinant NorH displayed significant NO reductase activity. Under NO stress conditions, heterologous expression of H. pylori NorH rescued the growth of the E. coli NO reductase-deficient strain (ΔnorV) to resistance levels similar those of the wild-type. In vivo studies showed that deletion of norH renders H. pylori more vulnerable to nitric oxide synthase-dependent macrophage killing, and decreases the ability of the pathogen to colonise mice stomachs (Justino et al. 2012). Extensive amino acid sequence comparisons indicated that NorH, which shares no significant amino acid sequence similarity to other known microbial NO detoxifiers, belongs to a novel family of proteins that are widespread in the microbial world (Justino et al. 2012).
Nitroreductase FrxA
Treatment of H. pylori infection is achieved by double or triple antibiotic-based therapies that include the prodrug metronidazole. This prodrug belongs to the nitroimidazole family of antibiotics, which are activated by microbial nitroreductases (Mendz and Megraud 2002). Helicobacter pylori contains at least two nitroreductases: the oxygen-independent NADPH nitroreductase, RdxA, and the NADH-flavin oxidoreductase, FrxA (Han et al. 2007). While it is well established that RdxA activates metronidazole, the in vivo role of FrxA has remained obscure due the rare occurrence of frxA mutations in metronidazole-resistant clinical strains (Bereswill et al. 2003; Llanes et al. 2010; Tanih, Ndip and Ndip 2011). Recent work has shown that frxA transcription is induced by nitrosative stress. Helicobacter pylori frxA mutant was hypersensitive to GSNO and had decreased survival during macrophage infection and mouse colonisation (Justino et al. 2014). Although retaining the ability to activate nitrofurans, recombinant FMN-binding FrxA was also able to reduce GSNO. This first GSNO reductase identified in H. pylori is one of the few examples of nitroreductases with a role in nitrosative stress (Justino et al. 2014). Analysis of the available Campylobacter genomes predicts that homologous gene products are expressed in these microorganisms (Ribardo, Bingham-Ramos and Hendrixson 2010); however, studies remain to be performed to confirm a stress-related function.
Other nitrosative stress protective systems
Some of the oxidative stress protective proteins previously mentioned also contribute to nitrosative stress defence in Helicobacter and Campylobacter, namely AhpC and Msr.
Helicobacter pylori AhpC reduces peroxynitrite to nitrite and is proposed to contribute to H. pylori resistance to RNS killing (Bryk, Griffin and Nathan 2000). The C. jejuni methionine sulphoxide reductases are also involved in NO protection as mutation of msrA and msrB generated a strain with reduced resistance to nitrosative stress (Atack and Kelly 2008).
Campylobacter jejuni constitutively expresses the periplasmic pentahaem nitrite reductase NrfA (cj1357c) that detoxifies nitrite as well as NO (Pittman et al. 2007), and protects the bacterium from nitrosative stress (Sellars, Hall and Kelly 2002; Pittman and Kelly 2005). Although the ΔnrfA mutant was not found to be attenuated for chick colonisation, it has been suggested that the constitutive expression of nrfA might confer significant protection against initial exposure to NO during host colonisation until the NssR-dependent induction of cgb (Pittman et al. 2007). Given its periplasmic location, NrfA could shield from exogenous NO diffusing across the outer membrane into the cell.
Arginase modulation of the host NO production
Helicobacter pylori arginase (RocF) catalyses the hydrolysis of arginine to urea (and to ornithine), which is then hydrolysed by urease to carbon dioxide and ammonium; the latter guards the pathogen by neutralising the stomach acidic environment. Moreover, H. pylori arginase competes for the same L-arginine substrate of mammalian iNOS (Fig. 1), therefore lowering the amount of toxic NO produced by the immune system (Wu and Morris 1998). Macrophages infected with rocF-inactivated H. pylori had augmented NO levels and increased ability to kill the pathogen (McGee et al. 1999; Gobert et al. 2001; Wang, Alamuri and Maier 2006; Das et al. 2010). Mutation of the rocF in H. pylori was reported to hamper survival and attenuate colonisation in a mouse model (Gobert et al. 2001; Chaturvedi et al. 2007). Interestingly, H. pylori arginase activity has been proposed to be post-translationally stimulated by the chaperone activity of thioredoxin Trx1 that promotes renaturation of actively damaged arginase (Baker et al. 2001; Comtois, Gidley and Kelly 2003; McGee et al. 2006).
Mammals express two isoforms of arginase, namely arginase I produced in the liver and arginase II (Arg2) formed in the kidney (Li et al. 2001; Nissim et al. 2005). Infection by H. pylori has also been reported to induce the Arg2 expression in murine and peritoneal macrophage cell lines, and in the gastric mucosa of mice and humans, which leads to decreased intracellular levels of L-arginine (Gobert et al. 2001, 2002; Lewis et al. 2010, 2011). A chronic H. pylori infection mice model showed an augmented expression of Arg2 and impaired host response, while Arg2 defective mice had higher levels of iNOS and NO, and lower bacterial counts (Lewis et al. 2010).
Helicobacter pylori modifies the polyamine levels, such as putrescine, spermidine and spermine (which are polycationic amino acids synthesised along the arginase–ornithine decarboxylase (ODC) pathway) that attenuate host immune responses by inhibiting proinflammatory cytokine expression and modulating apoptosis (Gobert et al. 2002; Lewis et al. 2011; Chaturvedi et al. 2012, 2014; Hardbower, Peek and Wilson 2014). One of the products of the H. pylori-induced Arg2 activity within macrophages is L-ornithine that is used by the ODC pathway to form putrescine, which can be converted to spermidine and spermine (Pegg and McCann 1992). Macrophages infected by H. pylori had higher expression levels of spermine oxidase (SMO) that back-converts spermine to spermidine. Also, in murine macrophages the knockdown of SMO elevated the spermine level, and lowered the iNOS expression and NO content. Moreover, the results indicated that during H. pylori infection the polyamine catabolism by SMO generates oxidative damage in gastric epithelial cells and causes cell apoptosis (Gobert et al. 2002; Lewis et al. 2011; Chaturvedi et al. 2012, 2014; Hardbower, Peek and Wilson 2014).
STRESS DEFENCES OF OTHER HELICOBACTER AND CAMPYLOBACTER SPP.
Helicobacter hepaticus was first isolated in the 1990s from the liver, colon and caecum of mice. It is associated with chronic intestinal infection that in susceptible mice can lead to hepatitis and colonic and hepatic carcinomas (Falsafi and Mahboubi 2013; Segura-Lopez, Guitron-Cantu and Torres 2015). Helicobacter hepaticus is resistant to cephalothin and nalidixic acid but sensitive to metronidazole (Tanaka et al. 2007). Helicobacter cinaedi, formerly named Campylobacter cinaedi, infects the intestine of humans and is transferred from animals as a zoonosis. The first case was identified in 1984 in homosexual men and several cases have been reported to date in patients suffering from several diseases, such as AIDS, haematological malignancy, diabetes mellitus, chronic liver and renal insufficiency (Kamimura et al. 2015).
In H. hepaticus, two proteins have been identified as protective against ROS, namely catalase KatA and the alkyl hydroperoxide reductase TsaA. Helicobacter hepaticus katA mutant strain proved to be impaired by H2O2 and exhibited a higher degree of DNA fragmentation (Hong, Wang and Maier 2007). The H. hepaticus tsaA mutant was also less resistant to paraquat, cumene hydroperoxide and t-butylhydroperoxide, but more resistant to hydrogen peroxide. It was proposed that a compensatory response of the bacterium upon loss of TsaA may led to an increase of the catalase expression and activity. Yet, the wild-type and tsaA mutant strains exhibited comparable colonising abilities and caused similar lesions in the mice hepatic tissue (Mehta et al. 2007).
The H. hepaticus NADPH quinone reductase (MdaB) was also reported to contribute to oxygen and oxidative stress tolerance. The mdaB mutant had increased sensitivity to oxidative stress-generating molecules, and expressed higher levels of other oxidative stress-combating enzymes, such as superoxide dismutase (Hong, Wang and Maier 2008). Also, the H. hepaticus dps mutant strain shows higher sensitivity to peroxides than the wild-type strain, and the mutant cells present a higher percentage of coccoid or lysed cells when exposed to oxidative stress (Hong, Wang and Maier 2006).
Helicobacter cinaedi expresses an ahpC- like gene, and the knockout strain was less resistant to hydrogen peroxide and more rapidly killed by macrophages. Furthermore, in vivo mice experiments indicated that deletion of ahpC generated a H. cinaedi strain with reduced caecal colonising ability and diminished ability to induce bacterial-specific immune responses (Charoenlap et al. 2012).
While C. jejuni is the primary cause of human gastrointestinal diseases, C. coli is responsible for the second highest number of intestinal infections (Skarp, Hanninen and Rautelin 2016). Like C. jejuni, C. coli encodes two of the major detoxification enzymes, namely SodB and KatA (Grant and Park 1995; Purdy et al. 1999). Deletion of sodB impaired the ability of C. coli to colonise the ceca of chicks, and had a significantly attenuated ability to survive O2 exposure.
Despite the importance of KatA for oxidative stress resistance in C. jejuni and C. coli, catalase is absent in several other species of Campylobacter, including C. concisus, C. mucosalis, C. sputorum, C. helveticus, C. curvus, C. rectus, C. upsaliensis and C. hominis (Bourke, Chan and Sherman 1998; Lawson et al. 2001). Furthermore, these catalase-negative strains lack homologues of the haem-trafficking protein, cj1386 (Flint and Stintzi 2015). Recently, Koolman et al. have investigated the presence of sodB and katA by PCR in 24 Campylobacter isolates obtained from poultry or human origin belonging to the C. jejuni, C. coli and C. lari species. Of the isolates, sodB was present in all samples whereas katA was identified in 19 samples (Koolman et al. 2015). The isolates lacking katA were identified in both C. jejuni and C. coli species (Koolman et al. 2015). Thus, sodB appears to be indispensable to Campylobacter strains, while other H2O2 detoxification enzymes are expected to compensate for the absence katA, such as those present in C. jejuni NCTC11168 or 81–176 (i.e. ahpC and tpx).
GLOBAL RESPONSES TO STRESS IN HELICOBACTER PYLORI AND CAMPYLOBACTER JEJUNI
Several works have investigated the global effects of oxidative and nitrosative stresses by whole transcriptomic and proteomic studies.
Global transcriptional profiling of H. pylori treated with spermine NONOate (20 μM, 4 h) revealed upregulation of 145 genes that belong to a wide variety of categories (Hung et al. 2015). The classes with the highest number of differentially expressed genes were those of the cell envelope (n = 17), followed by transport and binding (n = 13), cellular processes (n = 11) and DNA metabolism (n = 5). Under nitrosative stress, the genes of the iron(III) dicitrate transport protein (fecA), cytosine-specific DNA methyltransferase (hp0051) and ABC transporter-permease (glnP) had the highest levels of expression. Among the several knockout strains examined, namely hp0351 (fliF), hp0751 (flaG), fecA, hp0916 (frpB), glnP and hp1326 (crdA), the strains lacking flagellar proteins, ΔfliF and ΔflaG, were the strains most susceptible to nitrosative stress (Hung et al. 2015). Nevertheless, it was unexpected that the gene products that are known to be related with nitrosative stress defences were not detected in this study.
To identify genes that contribute to stomach colonisation, Salama and co-workers (Baldwin et al. 2007) used microarray-based tracking of transposon mutants of two mouse-adapted strains, H. pylori NSH79 and NSH57, to monitor 758 different gene loci in a C57BL/6 mouse infection model. Approximately 29% of the mutant strains showed colonisation defects, including those lacking the ferric uptake regulator fur, sodB, catalase and peroxiredoxin genes confirming the importance of oxidative stress resistance genes in colonisation (Baldwin et al. 2007).
In 2005, Chiou and co-workers (Chuang et al. 2005) analysed proteome alterations due to oxygen stress in H. pylori clinical isolates from patients with duodenal ulcers and gastric cancer. When exposed to 20% O2, the cells acquired a coccoid morphology and exhibited lower urease activity. Interestingly, the formation of coccoid-shaped cells was also observed in H. pylori cells exposed to NO donors (Tecder-Unal et al. 2008). The whole protein expression profiling revealed 11 differentially expressed proteins in response to high oxygen levels, which included AhpC, NapA and urease-related proteins UreE and urease β-subunit (Chuang et al. 2005). In a subsequent study, a clinical strain of H. pylori HC28 treated with 10 mM H2O2 exhibited increased expression of the following proteins: cytotoxin-associated protein A (CagA), vacuolating cytotoxin (VacA), adherence-associated protein (AlpA), two antioxidant enzymes (AhpC and KatA), serine protease (HtrA), aconitate hydratase (AcnB) and fumarate reductase (FrdA) (Huang and Chiou 2011).
Proteomic analysis was also used to explore the impact of SNP on H. pylori 26695 (0.5 mM SNP for 6 h) (Qu et al. 2009). Approximately 38 proteins were differentially expressed, including proteins involved in cell division and central metabolism. In particular, TrxR, SodB, TsaA and NapA were highly induced. Notably, the protein with highest elevated expression was flavodoxin (FldA), a protein essential to H. pylori viability that acts as an electron acceptor for pyruvate: flavodoxin oxidoreductase during the oxidative decarboxylation of pyruvate (Hughes et al. 1995; Freigang et al. 2002; Cremades et al. 2005). The downregulation of the major virulence cytotoxin-associated protein A (CagA) and the flagella protein PflA was also observed (Qu et al. 2009).
Another proteomic study of H. pylori treated with GSNO (100 μM for 20 min) detected five differentially expressed proteins including the chaperone and heat shock protein (GroEL), urease alpha subunit (UreA), alkyl hydroperoxide reductase (designated as TsaA), sialic acid-specific adhesion protein HP0721 and a protein of unknown function HP0129 (Qu et al. 2011). Consistent with these results, deletion of tsaA in H. pylori increased the sensitivity of the bacterium to RNS. Additionally, the urease activity was found to be inhibited by GSNO (Bryk, Griffin and Nathan 2000).
Exposure of C. jejuni to cumene hydroperoxide induced expression of genes coding for detoxification enzymes and genes involved in general cellular stress response. In particular, significant upregulation of one of the major regulators of the heat shock response, hrcA, and the nucleotide exchange factor grpE was observed (Palyada et al. 2009). Several genes involved in DNA replication and repair were found downregulated in response to cumene hydroperoxide including dnaA (chromosomal replication initiator protein), urvC (exonuclease ABC subunit C) and cj1669c (ATP-dependent DNA ligase) (Palyada et al. 2009). Therefore, it seems that in contrast to other bacteria which display induction of DNA repair mechanisms as a protective measure against stress (Chang et al. 2006; Wolf et al. 2008), C. jejuni may slow growth and replication as a strategy to avoid accumulation of DNA mutations and to provide more time for DNA repair to occur (given the absence of an SOS response and smaller number of DNA repair genes in C. jejuni). Additional global responses to oxidant exposure included up-regulation of the multidrug efflux pump, cmeABC. The induction of these genes, which are involved in pumping out toxic substances such as bile salts and antimicrobial compounds, is also thought to be a cross-protective response of C. jejuni when encountering unfavourable conditions during virulence.
Based on the microarray data of C. jejuni under oxidative stress as well as from a ΔperR mutant strain, several genes (putative guanosine-3′,5′-bis[diphosphate]3’pyrophosphohydrolase spoT, dihydropteroate synthase folP and tRNA pseudouridine synthase B (truB)) displaying meaningful changes in their gene expression patterns were targeted for further characterisation (Flint et al. 2014). Surprisingly, the ΔspoT, ΔfolP and ΔtruB mutants did not reveal different sensitivity towards the oxidants relative to the parental strain (Flint et al. 2014), suggesting that they likely play a role in cross-protection against nutrient stress (spoT), sulphonamide resistance (folP) and potential osmotic/temperature resistance (truB).
STRESS-RELATED REGULATORS OF HELICOBACTER PYLORI AND CAMPYLOBACTER JEJUNI
A great wealth of knowledge on transcriptional regulation of bacterial oxidative and nitrosative stress systems has been generated over many decades of research. However, C. jejuni and H. pylori lack many typical key regulatory proteins found in other Gram-negative bacteria, including homologues of stress-related regulators such as SoxRS, OxyR, Crp and FNR (Hillion and Antelmann 2015; Miller and Auerbuch 2015). Nevertheless, it has been shown that several global transcriptional regulators such as Fur, PerR and NssR sense and respond to changes in environmental conditions and are important for host colonisation and pathogenesis of C. jejuni (Butcher et al. 2012; Carpenter et al. 2013; Kim et al. 2015), as described below. Interestingly, H. pylori seems to contain only the Fur regulator and to rely in less common regulators to sense ROS and RNS stresses (see below).
Ferric uptake regulator Fur
The ferric uptake regulator (Fur) is a widely spread intracellular iron-sensor regulator that controls expression of iron acquisition and storage systems, and that has been shown to contribute to oxidative and nitrosative stress resistance of bacteria.
Campylobacter jejuni and H. pylori Fur regulons include over 50 genes, some of which are related to stress protection (Ernst et al. 2005a,b; Danielli et al. 2006; Miles et al. 2010; Butcher et al. 2012, 2015; Butcher and Stintzi 2013). Therefore, H. pylori Fur is described to have both features of iron-sensing (Fur function) and oxidation-sensing (PerR-like function) (Pelliciari et al. 2015). Specifically, Fur induces katA expression under iron-replete conditions (Harris et al. 2002), and apo-Fur regulates sodB (Ernst et al. 2005a,b). Additionally, iron-inducible, apo-Fur represses genes, such as pfr (iron storage gene) and hydA (hydrogenase (NiFe) small subunit), which are expressed shortly after oxidative stress, while holo-Fur represses genes, such as frpB1 (iron-regulated outer membrane protein) and fecA1 (iron(III) dicitrate transport protein), that vary modestly in response to oxidative stress. Therefore, Pelliciari et al. (2015) considered that Fur exerts an allosteric regulation to enable transduction of oxidative stress signals in H. pylori, and propose that apo-Fur-repressed genes can be considered oxidation-inducible Fur regulatory targets.
Since H. pylori strains mutated in the fur gene are less sensitive to NO, derepression of iron-regulated genes is considered an important physiological response of H. pylori to nitrosative damage. Furthermore, Fur contributes to successful colonisation of H. pylori in both murine and Mongolian gerbil models of infection (Danielli et al. 2006; Miles et al. 2010).
Peroxide-sensing regulator PerR
PerR, a member of the Fur family, is a peroxide-sensing regulator that controls oxidative stress-related genes (Dubbs and Mongkolsuk 2012).
Helicobacter pylori does not encode perR and the understanding of H. pylori oxidative stress regulation is still limited. On the contrary, H. hepaticus expresses a perR gene, which when inactivated results in a high expression level of katA and ahpC (Belzer et al. 2011).
Campylobacter jejuni PerR represses several peroxide resistance genes (e.g. katA, ahpC, trxB, dps and cj1386) and the perR mutation generates a strain hyperresistant to hydrogen peroxide (due to depression of H2O2 detoxification enzymes) (Palyada et al. 2009; Butcher et al. 2015). Iron was shown to downregulate perR transcription, which is suggested to occur by a PerR autoregulatory mechanism (Kim et al. 2011). Furthermore, deletion of perR lowered the C. jejuni chick colonisation ability relative to the parental strain indicating that PerR regulation reinforces colonisation (Palyada et al. 2009).
Nitrosative-stress response regulator NssR
Although Campylobacter and Helicobacter spp. are equipped with regulators that induce expression of oxidative stress-related proteins, nitrosative-sensing regulators seem to be scarce or have not yet been identified.
NssR is a global transcription factor that belongs to the Crp−Fnr superfamily of bacterial regulators. The protein contains an N-terminal sensory domain that uses a Fe-S cluster as sensor of nitrosative and oxidative stresses, and a C-terminal DNA-binding domain that recognises target sequences by means of a helix–turn–helix motif. In C. jejuni, NssR (Cj0466) controls a small regulon that includes the two NO-inducible single domain and truncated haemoglobins, Cgb and Ctb. For cgb, NssR regulation occurs via a cis–acting motif located in the promoter region of the gene (Elvers et al. 2005; Monk et al. 2008).
In Helicobacter species, NssR homologues are apparently absent from the genomes. However, proteomic studies indicate that H. pylori Fur plays a role in RNS stress resistance. The transcription of fur is induced by nitrosative stress and the fur mutant is more sensitive to nitrosative stress than the wild-type strain (Qu et al. 2009).
Copper resistance determinant CrdRS
Helicobacter pylori CrdRS is a two-component sensor for copper and acid. Yet, the role of CrdRS in acid sensing remains less clear as it has been demonstrated for H. pylori J99 but not for H. pylori 26695 (Waidner et al. 2005; Loh and Cover 2006). The crdS gene is upregulated by RNS, and single and double crdR/crdS deletion mutants display reduced viability under NO (Hung et al. 2015). In vivo studies reported that CrdRS facilitates H. pylori stomach colonisation in a murine infection model (Panthel et al. 2003). Moreover, a global transcriptional profiling analysis, which defined the CrdR regulon as including circa 100 genes, indicated that CrdS may regulate the ABC transporter permease glnP protein. The glnP gene is induced under stress conditions and its deletion resulted in a strain that grows deficiently when exposed to NO (Waidner et al. 2005; Loh and Cover 2006).
Aconitase
One of the few and notable examples of post-transcriptional regulation of genes involved in oxidative stress defence has been described in Escherichia coli. That is the case of the E. coli aconitase proteins AcnA and AcnB that have a dual function, acting both as enzymes and as post-transcriptional regulators in response to oxidative stress (Tang et al. 2002). During oxidant exposure or low intracellular iron levels, demetallation of the active site [4Fe-4S]2+/1+ clusters of AcnB renders the enzyme catalytically inactive (Gardner and Fridovich 1992). Upon inactivation, the apo-AcnB and apo-AcnA proteins act as post-transcriptional regulators by binding to the 3′ UTR of their cognate mRNAs (Tang and Guest 1999). Binding of apo-AcnA or apo-AcnB to the acnA and acnB mRNA increases transcript stability and synthesis of the AcnA and AcnB proteins during periods of Fe-S cluster inactivation due to exposure to oxidative stress (Tang et al. 2002).
Helicobacter pylori AcnB has also been shown to be a bifunctional protein with both catalytic (Pitson et al. 1999) and post-transcriptional regulatory roles (Austin and Maier 2013). Included in H. pylori's repertoire of oxidative stress responsive systems is a peptidoglycan deacetylase (pgdA), which is involved in peptidoglycan modification and bacterial cell-wall integrity (Wang et al. 2009). PgdA protects H. pylori during host colonisation by modifying peptidoglycan constituents to resist cleavage by host lysozyme and prevent subsequent cell lysis (Wang et al. 2009). Specifically, PgdA deacetylates N-acetylglucosamine residues (Wang et al. 2009), which along with N-acetylmuramic acid form β-1,4 bonds between these residues in peptidoglycan that are targeted for lysozyme-induced hydrolysis (Dziarski 2003). PgdA is induced by ROS (Wang et al. 2009) and during macrophage contact, and is important for survival within the murine stomach (Wang et al. 2010). Deletion of acnB in H. pylori significantly decrease the pgdA transcript levels in the presence or absence of oxidative stress. Oxidative conditions trigger binding of apo-AcnB to the mRNA 3′UTR of pgdA increasing the transcript stability and consequently elevating the PgdA expression levels (Austin and Maier 2013).
Additional regulatory targets of AcnB have been recently identified by mass spectrometry through examining changes to the H. pylori proteome in a ΔacnB mutant strain. In particular, the abundance of AhpC was significantly lower in the ΔacnB strain compared to the wild-type (Austin, Wang and Maier 2015). The ΔacnB mutant also displayed decreased survival during exposure to atmospheric O2 as well as hypersensitivity towards cumene hydroperoxide (Austin, Wang and Maier 2015). Binding of apo-AcnB to the 3′UTR of the ahpC transcript was confirmed by electrophoretic mobility shift assay (EMSA) (Austin, Wang and Maier 2015).
Like in H. pylori, the C. jejuni ΔacnB mutant revealed lower resistance to H2O2 and O2•− (Flint et al. 2014). However, whether the C. jejuni acnB gene has a post-transcriptional regulatory role for its own transcript or other oxidative stress protective genes, such as sodB, remains to be proved. Given that apo-AcnA increases SodA levels in E. coli in the presence of oxidative stress by post-transcriptional regulation (Tang et al. 2002), it is tempting to speculate that the absence of AcnB in the C. jejuni could lead to lower SodB production due to the loss of post-transcriptional regulation of the sodB transcript. Consequently, the decreased expression levels of SodB would explain the hypersensitivity towards superoxide observed in the C. jejuni ΔacnB mutant. Furthermore, the increased sensitivity towards H2O2 of the ΔacnB strain suggests post-transcriptional regulation of ahpC and/or katA. The established post-transcriptional regulatory function of apo-AcnB for the ahpC transcript in the closely related pathogen H. pylori (Austin and Maier 2013) supports this hypothesis. Altogether, these results demonstrate the important regulatory role of AcnB in oxidative stress resistance in H. pylori and C. jejuni.
Other regulators
Other C. jejuni regulators have been associated with oxidative stress control, namely CosR, Cj1000, RrpA and RrpB.
The C. jejuni oxidative stress sensor CosR is an OmpR-type regulator shown to be important for the viability of the bacterium. Microarray studies of a cosR knockdown strain defined the cosR regulon to consist of approximately 90 genes involved in various cellular functions. In particular, CosR was shown to bind the katA promoter and activate katA transcription, and knockdown of cosR led to reduced catalase activity of cell-free extracts (Garenaux et al. 2008; Hwang et al. 2012). The CosR regulon was also analysed by two-dimensional gel electrophoresis and shown to repress the expression of sodB and dps and to induce ahpC (Hwang et al. 2011).
Campylobacter jejuni cj1000 encodes a LysR-type regulator that controls O2 consumption (Dufour et al. 2013). Microarray analysis of the Δcj1000 mutant revealed several targets, including katA, ahpC and trxB. Although the Δcj1000 mutant did not show lower viability under oxidative stress, it had attenuated ability to colonise and was outcompeted by the wild-type strain during colonisation of the piglet intestine (Dufour et al. 2013).
RrpA and RrpB are two MarR family transcriptional regulators with roles in regulating C. jejuni's oxidative stress response (Gundogdu et al. 2011, 2015). RrpA has recently been shown to regulate oxidative stress defence genes (Gundogdu et al. 2015). The ΔrrpA mutant displayed increased sensitivity to H2O2, reduced katA expression and catalase activity in cell extracts, and decreased virulence during Galleria mellonella infection relative to the wild-type (Gundogdu et al. 2015). Microarray analysis indicated that rrpB is negatively autoregulated and induces expression of katA, perR and hspR (Gundogdu et al. 2011). The ΔrrpB mutant strain had lower resistance to H2O2 and oxygen stress, and diminished ability to adhere, invade and survive within Caco-2 human intestinal epithelial cells, reduced survivability in murine macrophages and lower virulence in the Galleria mellonella infection model (Gundogdu et al. 2011). Helicobacter pylori CrsA, ArsRS and HsrA proteins have been reported to regulate expression of oxidative stress-related genes.
Helicobacter pylori CrsA is an homologue of the E. coli global carbon starvation regulator CrsA; the crsA deletion strain exhibited significantly attenuated survival under H2O2 stress (Fields and Thompson 2008). However, the genes under the control of this transcription factor remain to be identified.
Helicobacter pylori also contains the two-component signal transduction system ArsRS that responds to acid stress. Protein expression studies revealed that the ArsRS system regulates the thioredoxin reductase gene trx1 (Loh et al. 2010).
Helicobacter pylori HsrA is a homeostatic global regulator that represses sodB and controls the cellular SOD activity (Olekhnovich et al. 2014). Autoregulation was suggested on the basis of the specific binding of HsrA-enriched protein fractions to the hsrA promoter (Roncarati et al. 2007).
Bacterial histone-like proteins regulate gene expression by binding to target DNA promotor sequences (Grainger et al. 2008). In H. pylori, the hp119 gene coding for a putative histone was shown, by EMSA, to bind DNA (Chen, Ghosh and Grove 2004; Wang, Lo and Maier 2012; Wang and Maier 2015). Two-dimensional gel analysis of an hp119 inactivated strain exposed to oxic conditions showed that the abundance of SodB, Tpx, TrxR, NapA, and PgdA in the mutant cells is lower than in the wild-type. Conversely, the AhpC levels were increased in the hp119 mutant (Wang and Maier 2015). Furthermore, the Δhp119 mutant exhibited decreased viability when grown under atmospheric O2, attenuated survival in murine macrophages and reduced colonisation of murine stomachs. Hence, Hp19 is proposed to be involved in stress tolerance (Wang and Maier 2015).
CONCLUSION
The success of bacterial pathogens in host colonisation and virulence is highly dependent on circumvention of innate immunological responses. Helicobacter and Campylobacter are both equipped with a repertoire of systems that enable survival under the toxic effects of oxidative and nitrosative compounds from innate immune cells, such as macrophages and neutrophils, and also present in the gastrointestinal tract (Fig. 2).
Characterisation of the oxidative stress protectors of Helicobacter and Campylobacter has revealed that, in contrast to other bacterial pathogens such as E. coli and Salmonella, these bacteria contain fewer direct detoxification enzymes. Nevertheless, the data show that reliance on only a key number of oxidant protectors confers Helicobacter and Campylobacter the ability to survive exposure to harsh conditions and be highly effective at inducing illness and chronic disease. The major oxidant detoxification enzymes include AhpC, KatA, SodB, Tpx, BCP and MdaB (Helicobacter specific), which respond to a wide range of oxidative compounds and have been demonstrated to be critical for in vivo colonisation. Indirect oxidant defences are also equally important for surviving the oxidative burst imposed by mammalian host immune cells. Macromolecule repair systems (MsrAB), DNA repair systems (MutS, MutY), iron sequestration and DNA-binding proteins (Dps, NapA) and chaperone/biogenesis proteins (KapA/Cj1386) all play a significant role in oxidative stress resistance in vitro and in vivo.
In Campylobacter jejuni, the fine-tuning of the gene expression by transcriptional regulators may provide an explanation for the success of a pathogen that encodes for a relatively small number of oxidative stress responsive systems and lacks an SOS response. In particular, katA has been shown to be controlled by more than one regulator (e.g. Fur, PerR and CosR). Clearly, C. jejuni is able to sense and respond to numerous environmental stimuli leading to differential expression of katA. This may enable C. jejuni to upregulate katA during periods of H2O2 stress while maintaining iron homeostasis during other phases of growth and/or infection.
When compared with the oxidative stress, the systems directly protecting Campylobacter and Helicobacter against NO are less well characterised. Genomic analyses indicate that these bacteria lack homologues of the major detoxifiers such as flavohaemoglobin and flavodiiron enzymes. However, these pathogens are exposed to bursts of RNS when establishing colonisation as well as throughout the infectious process within the host, and are clearly well adapted at surviving harsh gastrointestinal conditions. Our current understanding of NO detoxification in C. jejuni, Helicobacter pullorum and H. hepaticus consists of haemoglobins, which are absent in H. pylori. Indeed, H. pylori employs unique strategies for protection against an NO-rich environment by depleting one of the substrates specifically required by the host to generate RNS, namely arginine. Helicobacter pylori also utilises a novel nitroreductase system (NorH) and uses the prodrug-activating nitroreductase FrxA protein to detoxify GSNO. The identification of nitrosative-related sensors and regulators also remains in its infancy. To date, only two regulators, NssR in C. jejuni and CrdRS in H. pylori, have been linked to NO sensing.
It is expected that future next-generation sequencing experiments in Campylobacter and Helicobacter species would help drive novel discoveries aimed at identifying and characterising the ROS/RNS regulatory and defence systems in these pathogens, and provide new potential targets to use for therapeutics against these pathogens.
Acknowledgments
The authors thank Luís Morgado of ITQB-UNL for helping in the Figures’ preparation.
FUNDING
Research has been supported by the CIHR grant MOP#84224 (Alain Stintzi) and Project LISBOA-01-0145-FEDER-007660 (Microbiologia Molecular, Estrutural e Celular) funded by FEDER funds through COMPETE2020 - Programa Operacional Competitividade e Internacionalização (POCI) and by national funds through Fundação para a Ciência e a Tecnologia (Lígia Saraiva).
Conflict of interest. None declared.
REFERENCES
- Alamuri P, Maier RJ. Methionine sulphoxide reductase is an important antioxidant enzyme in the gastric pathogen Helicobacter pylori. Mol Microbiol. 2004;53:1397–406. doi: 10.1111/j.1365-2958.2004.04190.x. [DOI] [PubMed] [Google Scholar]
- Alamuri P, Maier RJ. Methionine sulfoxide reductase in Helicobacter pylori: interaction with methionine-rich proteins and stress-induced expression. J Bacteriol. 2006;188:5839–50. doi: 10.1128/JB.00430-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonso-Prieto M, Biarnes X, Vidossich P, et al. The molecular mechanism of the catalase reaction. J Am Chem Soc. 2009;131:11751–61. doi: 10.1021/ja9018572. [DOI] [PubMed] [Google Scholar]
- Algood HM, Cover TL. Helicobacter pylori persistence: an overview of interactions between H. pylori and host immune defenses. Clin Microbiol Rev. 2006;19:597–613. doi: 10.1128/CMR.00006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali A, Naz A, Soares SC, et al. Pan-genome analysis of human gastric pathogen H. pylori: comparative genomics and pathogenomics approaches to identify regions associated with pathogenicity and prediction of potential core therapeutic targets. Biomed Res Int. 2015;2015:139580. doi: 10.1155/2015/139580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altekruse SF, Stern NJ, Fields PI, et al. Campylobacter jejuni–an emerging foodborne pathogen. Emerg Infect Dis. 1999;5:28–35. doi: 10.3201/eid0501.990104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anjem A, Imlay JA. Mononuclear iron enzymes are primary targets of hydrogen peroxide stress. J Biol Chem. 2012;287:15544–56. doi: 10.1074/jbc.M111.330365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atack JM, Harvey P, Jones MA, et al. The Campylobacter jejuni thiol peroxidases Tpx and Bcp both contribute to aerotolerance and peroxide-mediated stress resistance but have distinct substrate specificities. J Bacteriol. 2008;190:5279–90. doi: 10.1128/JB.00100-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atack JM, Kelly DJ. Contribution of the stereospecific methionine sulphoxide reductases MsrA and MsrB to oxidative and nitrosative stress resistance in the food-borne pathogen Campylobacter jejuni. Microbiology. 2008;154:2219–30. doi: 10.1099/mic.0.2008/019711-0. [DOI] [PubMed] [Google Scholar]
- Augusto AC, Miguel F, Mendonca S, et al. Oxidative stress expression status associated to Helicobacter pylori virulence in gastric diseases. Clin Biochem. 2007;40:615–22. doi: 10.1016/j.clinbiochem.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Austin CM, Maier RJ. Aconitase-mediated posttranscriptional regulation of Helicobacter pylori peptidoglycan deacetylase. J Bacteriol. 2013;195:5316–22. doi: 10.1128/JB.00720-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin CM, Wang G, Maier RJ. Aconitase functions as a pleiotropic posttranscriptional regulator in Helicobacter pylori. J Bacteriol. 2015;197:3076–86. doi: 10.1128/JB.00529-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila-Ramirez C, Tinajero-Trejo M, Davidge KS, et al. Do globins in microaerophilic Campylobacter jejuni confer nitrosative stress tolerance under oxygen limitation? Antioxid Redox Signal 2013. 18 424–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillon ML, van Vliet AH, Ketley JM, et al. An iron-regulated alkyl hydroperoxide reductase (AhpC) confers aerotolerance and oxidative stress resistance to the microaerophilic pathogen Campylobacter jejuni. J Bacteriol. 1999;181:4798–804. doi: 10.1128/jb.181.16.4798-4804.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker LM, Raudonikiene A, Hoffman PS, et al. Essential thioredoxin-dependent peroxiredoxin system from Helicobacter pylori: genetic and kinetic characterization. J Bacteriol. 2001;183:1961–73. doi: 10.1128/JB.183.6.1961-1973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin DN, Shepherd B, Kraemer P, et al. Identification of Helicobacter pylori genes that contribute to stomach colonization. Infect Immun. 2007;75:1005–16. doi: 10.1128/IAI.01176-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu M, Czinn SJ, Blanchard TG. Absence of catalase reduces long-term survival of Helicobacter pylori in macrophage phagosomes. Helicobacter. 2004;9:211–6. doi: 10.1111/j.1083-4389.2004.00226.x. [DOI] [PubMed] [Google Scholar]
- Bauerfeind P, Garner R, Dunn BE, et al. Synthesis and activity of Helicobacter pylori urease and catalase at low pH. Gut. 1997;40:25–30. doi: 10.1136/gut.40.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belzer C, van Schendel BA, Hoogenboezem T, et al. PerR controls peroxide- and iron-responsive expression of oxidative stress defense genes in Helicobacter hepaticus. Eur J Microbiol Immunol (Bp) 2011;1:215–22. doi: 10.1556/EuJMI.1.2011.3.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit SL, Bayyareddy K, Mahawar M, et al. Alkyl hydroperoxide reductase repair by Helicobacter pylori methionine sulfoxide reductase. J Bacteriol. 2013;195:5396–401. doi: 10.1128/JB.01001-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereswill S, Krainick C, Stahler F, et al. Analysis of the rdxA gene in high-level metronidazole-resistant clinical isolates confirms a limited use of rdxA mutations as a marker for prediction of metronidazole resistance in Helicobacter pylori. FEMS Immunol Med Mic. 2003;36:193–8. doi: 10.1016/S0928-8244(03)00031-2. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya A, Chattopadhyay R, Mitra S, et al. Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol Rev. 2014;94:329–54. doi: 10.1152/physrev.00040.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingham-Ramos LK, Hendrixson DR. Characterization of two putative cytochrome c peroxidases of Campylobacter jejuni involved in promoting commensal colonization of poultry. Infect Immun. 2008;76:1105–14. doi: 10.1128/IAI.01430-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black RE, Levine MM, Clements ML, et al. Experimental Campylobacter jejuni infection in humans. J Infect Dis. 1988;157:472–9. doi: 10.1093/infdis/157.3.472. [DOI] [PubMed] [Google Scholar]
- Bogdan C. Nitric oxide synthase in innate and adaptive immunity: an update. Trends Immunol. 2015;36:161–78. doi: 10.1016/j.it.2015.01.003. [DOI] [PubMed] [Google Scholar]
- Bohr UR, Annibale B, Franceschi F, et al. Extragastric manifestations of Helicobacter pylori infection – other Helicobacters. Helicobacter. 2007;12(Suppl 1):45–53. doi: 10.1111/j.1523-5378.2007.00533.x. [DOI] [PubMed] [Google Scholar]
- Bonamore A, Boffi A. Flavohemoglobin: structure and reactivity. IUBMB Life. 2008;60:19–28. doi: 10.1002/iub.9. [DOI] [PubMed] [Google Scholar]
- Borlace GN, Keep SJ, Prodoehl MJ, et al. A role for altered phagosome maturation in the long-term persistence of Helicobacter pylori infection. Am J Physiol-Gastr L. 2012;303:G169–79. doi: 10.1152/ajpgi.00320.2011. [DOI] [PubMed] [Google Scholar]
- Boschi-Muller S, Gand A, Branlant G. The methionine sulfoxide reductases: Catalysis and substrate specificities. Arch Biochem Biophys. 2008;474:266–73. doi: 10.1016/j.abb.2008.02.007. [DOI] [PubMed] [Google Scholar]
- Bourke B, Chan VL, Sherman P. Campylobacter upsaliensis: waiting in the wings. Clin Microbiol Rev. 1998;11:440–9. doi: 10.1128/cmr.11.3.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman LA, McLean S, Poole RK, et al. The diversity of microbial responses to nitric oxide and agents of nitrosative stress close cousins but not identical twins. Adv Microb Physiol. 2011;59:135–219. doi: 10.1016/B978-0-12-387661-4.00006-9. [DOI] [PubMed] [Google Scholar]
- Broniowska KA, Diers AR, Hogg N. S-nitrosoglutathione. Biochim Biophys Acta. 2013;1830:3173–81. doi: 10.1016/j.bbagen.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broniowska KA, Hogg N. The chemical biology of S-nitrosothiols. Antioxid Redox Signal. 2012;17:969–80. doi: 10.1089/ars.2012.4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunder W, Schmidt H, Karch H. KatP, a novel catalase-peroxidase encoded by the large plasmid of enterohaemorrhagic Escherichia coli O157:H7. Microbiology. 1996;142(Pt 11):3305–15. doi: 10.1099/13500872-142-11-3305. [DOI] [PubMed] [Google Scholar]
- Bryk R, Griffin P, Nathan C. Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature. 2000;407:211–5. doi: 10.1038/35025109. [DOI] [PubMed] [Google Scholar]
- Butcher J, Handley RA, van Vliet AH, et al. Refined analysis of the Campylobacter jejuni iron-dependent/independent Fur- and PerR-transcriptomes. BMC Genomics. 2015;16:498. doi: 10.1186/s12864-015-1661-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher J, Sarvan S, Brunzelle JS, et al. Structure and regulon of Campylobacter jejuni ferric uptake regulator Fur define apo-Fur regulation. P Natl Acad Sci USA. 2012;109:10047–52. doi: 10.1073/pnas.1118321109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher J, Stintzi A. The transcriptional landscape of Campylobacter jejuni under iron replete and iron limited growth conditions. PLoS One. 2013;8:e79475. doi: 10.1371/journal.pone.0079475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter BM, Gilbreath JJ, Pich OQ, et al. Identification and characterization of novel Helicobacter pylori apo-fur-regulated target genes. J Bacteriol. 2013;195:5526–39. doi: 10.1128/JB.01026-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC. CDC Estimates of Foodborne Illness in the United States. Atlanta, USA: Centers for Disease Control and Prevention; 2014. [Google Scholar]
- Ceci P, Cellai S, Falvo E, et al. DNA condensation and self-aggregation of Escherichia coli Dps are coupled phenomena related to the properties of the N-terminus. Nucleic Acids Res. 2004;32:5935–44. doi: 10.1093/nar/gkh915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang W, Small DA, Toghrol F, et al. Global transcriptome analysis of Staphylococcus aureus response to hydrogen peroxide. J Bacteriol. 2006;188:1648–59. doi: 10.1128/JB.188.4.1648-1659.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charoenlap N, Shen Z, McBee ME, et al. Alkyl hydroperoxide reductase is required for Helicobacter cinaedi intestinal colonization and survival under oxidative stress in BALB/c and BALB/c interleukin-10-/- mice. Infect Immun. 2012;80:921–8. doi: 10.1128/IAI.05477-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi R, Asim M, Barry DP, et al. Spermine oxidase is a regulator of macrophage host response to Helicobacter pylori: enhancement of antimicrobial nitric oxide generation by depletion of spermine. Amino Acids. 2014;46:531–42. doi: 10.1007/s00726-013-1531-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi R, Asim M, Lewis ND, et al. L-arginine availability regulates inducible nitric oxide synthase-dependent host defense against Helicobacter pylori. Infect Immun. 2007;75:4305–15. doi: 10.1128/IAI.00578-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi R, de Sablet T, Coburn LA, et al. Arginine and polyamines in Helicobacter pylori-induced immune dysregulation and gastric carcinogenesis. Amino Acids. 2012;42:627–40. doi: 10.1007/s00726-011-1038-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelikani P, Fita I, Loewen PC. Diversity of structures and properties among catalases. Cell Mol Life Sci. 2004;61:192–208. doi: 10.1007/s00018-003-3206-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Ghosh S, Grove A. Substrate specificity of Helicobacter pylori histone-like HU protein is determined by insufficient stabilization of DNA flexure points. Biochem J. 2004;383:343–51. doi: 10.1042/BJ20040938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherdantseva LA, Potapova OV, Sharkova TV, et al. Association of Helicobacter pylori and iNOS production by macrophages and lymphocytes in the gastric mucosa in chronic gastritis. J Immunol Res. 2014;2014:762514. doi: 10.1155/2014/762514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang MH, Wu MS, Lin JT, et al. Proteomic analysis of proteins expressed by Helicobacter pylori under oxidative stress. Proteomics. 2005;5:3895–901. doi: 10.1002/pmic.200401232. [DOI] [PubMed] [Google Scholar]
- Ciorba MA, Heinemann SH, Weissbach H, et al. Modulation of potassium channel function by methionine oxidation and reduction. P Natl Acad Sci USA. 1997;94:9932–7. doi: 10.1073/pnas.94.18.9932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coker AO, Isokpehi RD, Thomas BN, et al. Human Campylobacteriosis in developing countries. Emerg Infect Dis. 2002;8:237–44. doi: 10.3201/eid0803.010233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comtois SL, Gidley MD, Kelly DJ. Role of the thioredoxin system and the thiol-peroxidases Tpx and Bcp in mediating resistance to oxidative and nitrosative stress in Helicobacter pylori. Microbiology. 2003;149:121–9. doi: 10.1099/mic.0.25896-0. [DOI] [PubMed] [Google Scholar]
- Cooke M. Causes and management of diarrhoea in children in a clinical setting. S Afr J Clin Nutr. 2010;23:S42–6. [Google Scholar]
- Cooksley C, Jenks PJ, Green A, et al. NapA protects Helicobacter pylori from oxidative stress damage, and its production is influenced by the ferric uptake regulator. J Med Microbiol. 2003;52:461–9. doi: 10.1099/jmm.0.05070-0. [DOI] [PubMed] [Google Scholar]
- Corcionivoschi N, Alvarez LA, Sharp TH, et al. Mucosal reactive oxygen species decrease virulence by disrupting Campylobacter jejuni phosphotyrosine signaling. Cell Host Microbe. 2012;12:47–59. doi: 10.1016/j.chom.2012.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa P, Piazuelo MB. Helicobacter pylori infection and gastric adenocarcinoma. US Gastroenterol Hepatol Rev. 2011;7:59–64. [PMC free article] [PubMed] [Google Scholar]
- Cremades N, Bueno M, Toja M, et al. Towards a new therapeutic target:Helicobacter pylori flavodoxin. Biophys Chem. 2005;115:267–76. doi: 10.1016/j.bpc.2004.12.045. [DOI] [PubMed] [Google Scholar]
- Crim SM, Griffin PM, Tauxe R, et al. Preliminary incidence and trends of infection with pathogens transmitted commonly through food - foodborne diseases active surveillance network, 10 u.s. Sites, 2006–2014. MMWR Morb Mortal Wkly Rep. 2015;64:495–9. [PMC free article] [PubMed] [Google Scholar]
- Danielli A, Roncarati D, Delany I, et al. In vivo dissection of the Helicobacter pylori Fur regulatory circuit by genome-wide location analysis. J Bacteriol. 2006;188:4654–62. doi: 10.1128/JB.00120-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das P, Lahiri A, Lahiri A, et al. Modulation of the arginase pathway in the context of microbial pathogenesis: a metabolic enzyme moonlighting as an immune modulator. PLoS Pathog. 2010;6:e1000899. doi: 10.1371/journal.ppat.1000899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day WA, Sajecki JL, Pitts TM, et al. Role of catalase in Campylobacter jejuni intracellular survival. Infect Immun. 2000;68:6337–45. doi: 10.1128/iai.68.11.6337-6345.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debruyne L, Gevers D, Vandamme P. Taxonomy of the family Campylobacteraceae. In: Nachemkin I, Blaser MJ, Szymanski CN, editors. Campylobacter. 3rd edn. Washington DC, USA: American Society of Microbiology; 2008. [Google Scholar]
- DeLeo FR, Allen LA, Apicella M, et al. NADPH oxidase activation and assembly during phagocytosis. J Immunol. 1999;163:6732–40. [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Talalay P. Persuasive evidence that quinone reductase type 1 (DT diaphorase) protects cells against the toxicity of electrophiles and reactive forms of oxygen. Free Radical Bio Med. 2000;29:231–40. doi: 10.1016/s0891-5849(00)00300-2. [DOI] [PubMed] [Google Scholar]
- Dubbs JM, Mongkolsuk S. Peroxide-sensing transcriptional regulators in bacteria. J Bacteriol. 2012;194:5495–503. doi: 10.1128/JB.00304-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour V, Li J, Flint A, et al. Inactivation of the LysR regulator Cj1000 of Campylobacter jejuni affects host colonization and respiration. Microbiology. 2013;159:1165–78. doi: 10.1099/mic.0.062992-0. [DOI] [PubMed] [Google Scholar]
- Dziarski R. Recognition of bacterial peptidoglycan by the innate immune system. Cell Mol Life Sci. 2003;60:1793–804. doi: 10.1007/s00018-003-3019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA. Scientific opinion on quantification of the risk posed by broiler meat to human Campylobacteriosis in the EU. EFSA J. 2010;8:1437–526. [Google Scholar]
- EFSA. The European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2013. EFSA J. 2015;13:162. doi: 10.2903/j.efsa.2018.5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen JA, Hanawalt PC. A phylogenomic study of DNA repair genes, proteins, and processes. Mutat Res. 1999;435:171–213. doi: 10.1016/s0921-8777(99)00050-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfvin A, Edebo A, Hallersund P, et al. Oxidative and nitrosative stress enzymes in relation to nitrotyrosine in Helicobacter pylori-infected humans. World J Gastrointest Pathophysiol. 2014;5:373–9. doi: 10.4291/wjgp.v5.i3.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elvers KT, Turner SM, Wainwright LM, et al. NssR, a member of the Crp-Fnr superfamily from Campylobacter jejuni, regulates a nitrosative stress-responsive regulon that includes both a single-domain and a truncated haemoglobin. Mol Microbiol. 2005;57:735–50. doi: 10.1111/j.1365-2958.2005.04723.x. [DOI] [PubMed] [Google Scholar]
- Elvers KT, Wu G, Gilberthorpe NJ, et al. Role of an inducible single-domain hemoglobin in mediating resistance to nitric oxide and nitrosative stress in Campylobacter jejuni and Campylobacter coli. J Bacteriol. 2004;186:5332–41. doi: 10.1128/JB.186.16.5332-5341.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst FD, Bereswill S, Waidner B, et al. Transcriptional profiling of Helicobacter pylori Fur- and iron-regulated gene expression. Microbiology. 2005a;151:533–46. doi: 10.1099/mic.0.27404-0. [DOI] [PubMed] [Google Scholar]
- Ernst FD, Homuth G, Stoof J, et al. Iron-responsive regulation of the Helicobacter pylori iron-cofactored superoxide dismutase SodB is mediated by Fur. J Bacteriol. 2005b;187:3687–92. doi: 10.1128/JB.187.11.3687-3692.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito L, Seydel A, Aiello R, et al. The crystal structure of the superoxide dismutase from Helicobacter pylori reveals a structured C-terminal extension. Biochim Biophys Acta. 2008;1784:1601–6. doi: 10.1016/j.bbapap.2008.04.024. [DOI] [PubMed] [Google Scholar]
- Eutsey R, Wang G, Maier RJ. Role of a MutY DNA glycosylase in combating oxidative DNA damage in Helicobacter pylori. DNA Repair. 2007;6:19–26. doi: 10.1016/j.dnarep.2006.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DJ, Evans DG, Takemura T, et al. Characterization of a Helicobacter pylori neutrophil-activating protein. Infect Immun. 1995;63:2213–20. doi: 10.1128/iai.63.6.2213-2220.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falsafi T, Mahboubi M. Helicobacter hepaticus, a new pathogenic species of the Helicobacter genus: Similarities and differences with H. pylori. Iran J Microbiol. 2013;5:185–94. [PMC free article] [PubMed] [Google Scholar]
- Fang FC. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat Rev Microbiol. 2004;2:820–32. doi: 10.1038/nrmicro1004. [DOI] [PubMed] [Google Scholar]
- Fehlings M, Drobbe L, Moos V, et al. Comparative analysis of the interaction of Helicobacter pylori with human dendritic cells, macrophages, and monocytes. Infect Immun. 2012;80:2724–34. doi: 10.1128/IAI.00381-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrero RL, Lee A. Motility of Campylobacter jejuni in a viscous environment: comparison with conventional rod-shaped bacteria. J Gen Microbiol. 1988;134:53–9. doi: 10.1099/00221287-134-1-53. [DOI] [PubMed] [Google Scholar]
- Fields JA, Thompson SA. Campylobacter jejuni CsrA mediates oxidative stress responses, biofilm formation, and host cell invasion. J Bacteriol. 2008;190:3411–6. doi: 10.1128/JB.01928-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flahou B, Rimbara E, Mori S, et al. The other Helicobacters. Helicobacter. 2015;20(Suppl 1):62–7. doi: 10.1111/hel.12259. [DOI] [PubMed] [Google Scholar]
- Flint A, Stintzi A. Cj1386, an atypical hemin-binding protein, mediates hemin trafficking to KatA in Campylobacter jejuni. J Bacteriol. 2015;197:1002–11. doi: 10.1128/JB.02346-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint A, Sun YQ, Butcher J, et al. Phenotypic screening of a targeted mutant library reveals Campylobacter jejuni defenses against oxidative stress. Infect Immun. 2014;82:2266–75. doi: 10.1128/IAI.01528-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint A, Sun YQ, Stintzi A. Cj1386 is an ankyrin-containing protein involved in heme trafficking to catalase in Campylobacter jejuni. J Bacteriol. 2012;194:334–45. doi: 10.1128/JB.05740-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flohe L, Toppo S, Cozza G, et al. A comparison of thiol peroxidase mechanisms. Antioxid Redox Signal. 2011;15:763–80. doi: 10.1089/ars.2010.3397. [DOI] [PubMed] [Google Scholar]
- Forrester MT, Foster MW. Protection from nitrosative stress: a central role for microbial flavohemoglobin. Free Radical Bio Med. 2012;52:1620–33. doi: 10.1016/j.freeradbiomed.2012.01.028. [DOI] [PubMed] [Google Scholar]
- Freigang J, Diederichs K, Schafer KP, et al. Crystal structure of oxidized flavodoxin, an essential protein in Helicobacter pylori. Protein Sci. 2002;11:253–61. doi: 10.1110/ps.28602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaasbeek EJ, van der Wal FJ, van Putten JP, et al. Functional characterization of excision repair and RecA-dependent recombinational DNA repair in Campylobacter jejuni. J Bacteriol. 2009;191:3785–93. doi: 10.1128/JB.01817-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Yin DH, Yao Y, et al. Loss of conformational stability in calmodulin upon methionine oxidation. Biophys J. 1998;74:1115–34. doi: 10.1016/S0006-3495(98)77830-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner PR, Fridovich I. Inactivation-reactivation of aconitase in Escherichia coli. A sensitive measure of superoxide radical. J Biol Chem. 1992;267:8757–63. [PubMed] [Google Scholar]
- Garenaux A, Guillou S, Ermel G, et al. Role of the Cj1371 periplasmic protein and the Cj0355c two-component regulator in the Campylobacter jejuni NCTC 11168 response to oxidative stress caused by paraquat. Res Microbiol. 2008;159:718–26. doi: 10.1016/j.resmic.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Gobert AP, McGee DJ, Akhtar M, et al. Helicobacter pylori arginase inhibits nitric oxide production by eukaryotic cells: a strategy for bacterial survival. P Natl Acad Sci USA. 2001;98:13844–9. doi: 10.1073/pnas.241443798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobert AP, Mersey BD, Cheng Y, et al. Cutting edge: urease release by Helicobacter pylori stimulates macrophage inducible nitric oxide synthase. J Immunol. 2002;168:6002–6. doi: 10.4049/jimmunol.168.12.6002. [DOI] [PubMed] [Google Scholar]
- Goldman CG, Mitchell HM. Helicobacter spp. other than Helicobacter pylori. Helicobacter. 2010;15(Suppl 1):69–75. doi: 10.1111/j.1523-5378.2010.00780.x. [DOI] [PubMed] [Google Scholar]
- Goncalves VL, Nobre LS, Vicente JB, et al. Flavohemoglobin requires microaerophilic conditions for nitrosative protection of Staphylococcus aureus. FEBS Lett. 2006;580:1817–21. doi: 10.1016/j.febslet.2006.02.039. [DOI] [PubMed] [Google Scholar]
- Grainger DC, Goldberg MD, Lee DJ, et al. Selective repression by Fis and H-NS at the Escherichia coli dps promoter. Mol Microbiol. 2008;68:1366–77. doi: 10.1111/j.1365-2958.2008.06253.x. [DOI] [PubMed] [Google Scholar]
- Grant KA, Park SF. Molecular characterization of katA from Campylobacter jejuni and generation of a catalase-deficient mutant of Campylobacter coli by interspecific allelic exchange. Microbiology. 1995;141(Pt 6):1369–76. doi: 10.1099/13500872-141-6-1369. [DOI] [PubMed] [Google Scholar]
- Gu M, Imlay JA. Superoxide poisons mononuclear iron enzymes by causing mismetallation. Mol Microbiol. 2013;89:123–34. doi: 10.1111/mmi.12263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundogdu O, da Silva DT, Mohammad B, et al. The Campylobacter jejuni MarR-like transcriptional regulators RrpA and RrpB both influence bacterial responses to oxidative and aerobic stresses. Front Microbiol. 2015;6:724. doi: 10.3389/fmicb.2015.00724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundogdu O, Mills DC, Elmi A, et al. The Campylobacter jejuni transcriptional regulator Cj1556 plays a role in the oxidative and aerobic stress response and is important for bacterial survival in vivo. J Bacteriol. 2011;193:4238–49. doi: 10.1128/JB.05189-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han F, Liu S, Ho B, et al. Alterations in rdxA and frxA genes and their upstream regions in metronidazole-resistant Helicobacter pylori isolates. Res Microbiol. 2007;158:38–44. doi: 10.1016/j.resmic.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Hardbower DM, Peek RM, Wilson KT. At the Bench: Helicobacter pylori, dysregulated host responses, DNA damage, and gastric cancer. J Leukocyte Biol. 2014;96:201–12. doi: 10.1189/jlb.4BT0214-099R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfe BD, Jinks-Robertson S. DNA mismatch repair and genetic instability. Annu Rev Genet. 2000;34:359–99. doi: 10.1146/annurev.genet.34.1.359. [DOI] [PubMed] [Google Scholar]
- Harris AG, Hazell SL. Localisation of Helicobacter pylori catalase in both the periplasm and cytoplasm, and its dependence on the twin-arginine target protein, KapA, for activity. FEMS Microbiol Lett. 2003;229:283–9. doi: 10.1016/S0378-1097(03)00850-4. [DOI] [PubMed] [Google Scholar]
- Harris AG, Hinds FE, Beckhouse AG, et al. Resistance to hydrogen peroxide in Helicobacter pylori: role of catalase (KatA) and Fur, and functional analysis of a novel gene product designated 'KatA-associated protein', KapA (HP0874) Microbiology. 2002;148:3813–25. doi: 10.1099/00221287-148-12-3813. [DOI] [PubMed] [Google Scholar]
- Harris AG, Wilson JE, Danon SJ, et al. Catalase (KatA) and KatA-associated protein (KapA) are essential to persistent colonization in the Helicobacter pylori SS1 mouse model. Microbiology. 2003;149:665–72. doi: 10.1099/mic.0.26012-0. [DOI] [PubMed] [Google Scholar]
- Hillion M, Antelmann H. Thiol-based redox switches in prokaryotes. Biol Chem. 2015;396:415–44. doi: 10.1515/hsz-2015-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofreuter D. Defining the metabolic requirements for the growth and colonization capacity of Campylobacter jejuni. Front Cell Infect Microbiol. 2014;4:137. doi: 10.3389/fcimb.2014.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Y, Wang G, Maier RJ. Helicobacter hepaticus Dps protein plays an important role in protecting DNA from oxidative damage. Free Radical Res. 2006;40:597–605. doi: 10.1080/10715760600618882. [DOI] [PubMed] [Google Scholar]
- Hong Y, Wang G, Maier RJ. A Helicobacter hepaticus catalase mutant is hypersensitive to oxidative stress and suffers increased DNA damage. J Med Microbiol. 2007;56:557–62. doi: 10.1099/jmm.0.46891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Y, Wang G, Maier RJ. The NADPH quinone reductase MdaB confers oxidative stress resistance to Helicobacter hepaticus. Microb Pathog. 2008;44:169–74. doi: 10.1016/j.micpath.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Bray MD, Osorio M, et al. Campylobacter jejuni induces maturation and cytokine production in human dendritic cells. Infect Immun. 2006;74:2697–705. doi: 10.1128/IAI.74.5.2697-2705.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CH, Chiou SH. Proteomic analysis of upregulated proteins in Helicobacter pylori under oxidative stress induced by hydrogen peroxide. Kaohsiung J Med Sci. 2011;27:544–53. doi: 10.1016/j.kjms.2011.06.019. [DOI] [PubMed] [Google Scholar]
- Huang S, Kang J, Blaser MJ. Antimutator role of the DNA glycosylase mutY gene in Helicobacter pylori. J Bacteriol. 2006;188:6224–34. doi: 10.1128/JB.00477-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huergo LF, Rahman H, Ibrahimovic A, et al. Campylobacter jejuni Dps protein binds DNA in the presence of iron or hydrogen peroxide. J Bacteriol. 2013;195:1970–8. doi: 10.1128/JB.00059-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes NJ, Chalk PA, Clayton CL, et al. Identification of carboxylation enzymes and characterization of a novel four-subunit pyruvate:flavodoxin oxidoreductase from Helicobacter pylori. J Bacteriol. 1995;177:3953–9. doi: 10.1128/jb.177.14.3953-3959.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey T, O'Brien S, Madsen M. Campylobacters as zoonotic pathogens: a food production perspective. Int J Food Microbiol. 2007;117:237–57. doi: 10.1016/j.ijfoodmicro.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Hung CL, Cheng HH, Hsieh WC, et al. The CrdRS two-component system in Helicobacter pylori responds to nitrosative stress. Mol Microbiol. 2015;97:1128–41. doi: 10.1111/mmi.13089. [DOI] [PubMed] [Google Scholar]
- Hurst JK. What really happens in the neutrophil phagosome? Free Radialc Bio Med. 2012;53:508–20. doi: 10.1016/j.freeradbiomed.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S, Kim M, Ryu S, et al. Regulation of oxidative stress response by CosR, an essential response regulator in Campylobacter jejuni. PLoS One. 2011;6:e22300. doi: 10.1371/journal.pone.0022300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S, Zhang Q, Ryu S, et al. Transcriptional regulation of the CmeABC multidrug efflux pump and the KatA catalase by CosR in Campylobacter jejuni. J Bacteriol. 2012;194:6883–91. doi: 10.1128/JB.01636-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima K, Grant J, McElroy K, et al. Novel mechanism of nitrosative stress from dietary nitrate with relevance to gastro-oesophageal junction cancers. Carcinogenesis. 2003;24:1951–60. doi: 10.1093/carcin/bgg168. [DOI] [PubMed] [Google Scholar]
- Imlay JA. Pathways of oxidative damage. Annu Rev Microbiol. 2003;57:395–418. doi: 10.1146/annurev.micro.57.030502.090938. [DOI] [PubMed] [Google Scholar]
- Imlay JA. Cellular defenses against superoxide and hydrogen peroxide. Annu Rev Biochem. 2008;77:755–76. doi: 10.1146/annurev.biochem.77.061606.161055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iovine NM, Pursnani S, Voldman A, et al. Reactive nitrogen species contribute to innate host defense against Campylobacter jejuni. Infect Immun. 2008;76:986–93. doi: 10.1128/IAI.01063-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Mizunoe Y, Kawabata S, et al. The iron-binding protein Dps confers hydrogen peroxide stress resistance to Campylobacter jejuni. J Bacteriol. 2003;185:1010–7. doi: 10.1128/JB.185.3.1010-1017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang S, Imlay JA. Micromolar intracellular hydrogen peroxide disrupts metabolism by damaging iron-sulfur enzymes. J Biol Chem. 2007;282:929–37. doi: 10.1074/jbc.M607646200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justino MC, Ecobichon C, Fernandes AF, et al. Helicobacter pylori has an unprecedented nitric oxide detoxifying system. Antioxid Redox Signal. 2012;17:1190–200. doi: 10.1089/ars.2011.4304. [DOI] [PubMed] [Google Scholar]
- Justino MC, Parente MR, Boneca IG, et al. FrxA is an S-nitrosoglutathione reductase enzyme that contributes to Helicobacter pylori pathogenicity. FEBS J. 2014;281:4495–505. doi: 10.1111/febs.12958. [DOI] [PubMed] [Google Scholar]
- Justino MC, Vicente JB, Teixeira M, et al. New genes implicated in the protection of anaerobically grown Escherichia coli against nitric oxide. J Biol Chem. 2005;280:2636–43. doi: 10.1074/jbc.M411070200. [DOI] [PubMed] [Google Scholar]
- Kaakoush NO, Castaño-Rodríguez N, Man SM, et al. Is Campylobacter to esophageal adenocarcinoma as Helicobacter is to gastric adenocarcinoma? Trends Microbiol 2015. 23 455–62. [DOI] [PubMed] [Google Scholar]
- Kamimura K, Kumaki D, Arita M, et al. First case of bacteremia caused by Helicobacter cinaedi in a patient with liver cirrhosis: a case report and literature review. Clin J Gastroenterol. 2015;8:306–17. doi: 10.1007/s12328-015-0600-0. [DOI] [PubMed] [Google Scholar]
- Kanizaj TF, Kunac N. Helicobacter pylori: future perspectives in therapy reflecting three decades of experience. World J Gastroenterol. 2014;20:699–705. doi: 10.3748/wjg.v20.i3.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara T, Kohjima M, Kuwano Y, et al. Helicobacter pylori lipopolysaccharide activates Rac1 and transcription of NADPH oxidase Nox1 and its organizer NOXO1 in guinea pig gastric mucosal cells. Am J Physiol-Cell Ph. 2005;288:C450–7. doi: 10.1152/ajpcell.00319.2004. [DOI] [PubMed] [Google Scholar]
- Kendall JJ, Barrero-Tobon AM, Hendrixson DR, et al. Hemerythrins in the microaerophilic bacterium Campylobacter jejuni help protect key iron-sulphur cluster enzymes from oxidative damage. Environ Microbiol. 2014;16:1105–21. doi: 10.1111/1462-2920.12341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern M, Winkler C, Simon J. Respiratory nitrogen metabolism and nitrosative stress defence in -proteobacteria: the role of NssR-type transcription regulators. Biochem Soc Trans. 2011;39:299–302. doi: 10.1042/BST0390299. [DOI] [PubMed] [Google Scholar]
- Kim JC, Oh E, Kim J, et al. Regulation of oxidative stress resistance in Campylobacter jejuni. a microaerophilic foodborne pathogen. Front Microbiol. 2015;6:751. doi: 10.3389/fmicb.2015.00751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Hwang S, Ryu S, et al. Regulation of perR expression by iron and PerR in Campylobacter jejuni. J Bacteriol. 2011;193:6171–8. doi: 10.1128/JB.05493-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebanoff SJ, Kettle AJ, Rosen H, et al. Myeloperoxidase: a front-line defender against phagocytosed microorganisms. J Leukocyte Biol. 2013;93:185–98. doi: 10.1189/jlb.0712349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koolman L, Whyte P, Burgess C, et al. Distribution of virulence-associated genes in a selection of Campylobacter isolates. Foodborne Pathog Dis. 2015;12:424–32. doi: 10.1089/fpd.2014.1883. [DOI] [PubMed] [Google Scholar]
- Kuhns LG, Mahawar M, Sharp JS, et al. Role of Helicobacter pylori methionine sulfoxide reductase in urease maturation. Biochem J. 2013;450:141–8. doi: 10.1042/BJ20121434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhns LG, Wang G, Maier RJ. Comparative roles of the two Helicobacter pylori thioredoxins in preventing macromolecule damage. Infect Immun. 2015;83:2935–43. doi: 10.1128/IAI.00232-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara H, Miyamoto Y, Akaike T, et al. Helicobacter pylori urease suppresses bactericidal activity of peroxynitrite via carbon dioxide production. Infect Immun. 2000;68:4378–83. doi: 10.1128/iai.68.8.4378-4383.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lah MS, Dixon MM, Pattridge KA, et al. Structure-function in Escherichia coli iron superoxide dismutase: comparisons with the manganese enzyme from Thermus thermophilus. Biochemistry. 1995;34:1646–60. doi: 10.1021/bi00005a021. [DOI] [PubMed] [Google Scholar]
- Lawson AJ, On SL, Logan JM, et al. Campylobacter hominis sp. nov., from the human gastrointestinal tract. Int J Syst Evol Micr. 2001;51:651–60. doi: 10.1099/00207713-51-2-651. [DOI] [PubMed] [Google Scholar]
- Lewis ND, Asim M, Barry DP, et al. Arginase II restricts host defense to Helicobacter pylori by attenuating inducible nitric oxide synthase translation in macrophages. J Immunol. 2010;184:2572–82. doi: 10.4049/jimmunol.0902436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis ND, Asim M, Barry DP, et al. Immune evasion by Helicobacter pylori is mediated by induction of macrophage arginase II. J Immunol. 2011;186:3632–41. doi: 10.4049/jimmunol.1003431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Meininger CJ, Hawker JR, et al. Regulatory role of arginase I and II in nitric oxide, polyamine, and proline syntheses in endothelial cells. Am J Physiol-Endoc M. 2001;280:E75–82. doi: 10.1152/ajpendo.2001.280.1.E75. [DOI] [PubMed] [Google Scholar]
- Llanes R, Soria C, Nagashima S, et al. Phenotypic and genetic characterization of antimicrobial profiles of Helicobacter pylori strains in Cuba. J Health Popul Nutr. 2010;28:124–9. doi: 10.3329/jhpn.v28i2.4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh JT, Cover TL. Requirement of histidine kinases HP0165 and HP1364 for acid resistance in Helicobacter pylori. Infect Immun. 2006;74:3052–9. doi: 10.1128/IAI.74.5.3052-3059.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh JT, Gupta SS, Friedman DB, et al. Analysis of protein expression regulated by the Helicobacter pylori ArsRS two-component signal transduction system. J Bacteriol. 2010;192:2034–43. doi: 10.1128/JB.01703-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee DJ, Kumar S, Viator RJ, et al. Helicobacter pylori thioredoxin is an arginase chaperone and guardian against oxidative and nitrosative stresses. J Biol Chem. 2006;281:3290–6. doi: 10.1074/jbc.M506139200. [DOI] [PubMed] [Google Scholar]
- McGee DJ, Radcliff FJ, Mendz GL, et al. Helicobacter pylori rocF is required for arginase activity and acid protection in vitro but is not essential for colonization of mice or for urease activity. J Bacteriol. 1999;181:7314–22. doi: 10.1128/jb.181.23.7314-7322.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahawar M, Tran V, Sharp JS, et al. Synergistic roles of Helicobacter pylori methionine sulfoxide reductase and GroEL in repairing oxidant-damaged catalase. J Biol Chem. 2011;286:19159–69. doi: 10.1074/jbc.M111.223677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masanta WO, Heimesaat MM, Bereswill S, et al. Modification of intestinal microbiota and its consequences for innate immune response in the pathogenesis of Campylobacteriosis. Clin Dev Immunol. 2013;2013:526860. doi: 10.1155/2013/526860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta NS, Benoit SL, Mysore J, et al. In vitro and in vivo characterization of alkyl hydroperoxide reductase mutant strains of Helicobacter hepaticus. Biochim Biophys Acta. 2007;1770:257–65. doi: 10.1016/j.bbagen.2006.09.022. [DOI] [PubMed] [Google Scholar]
- Menard A, Pere-Vedrenne C, Haesebrouck F, et al. Gastric and enterohepatic Helicobacters other than Helicobacter pylori. Helicobacter. 2014;19(Suppl 1):59–67. doi: 10.1111/hel.12162. [DOI] [PubMed] [Google Scholar]
- Mendz GL, Megraud F. Is the molecular basis of metronidazole resistance in microaerophilic organisms understood? Trends Microbiol. 2002;10:370–5. doi: 10.1016/s0966-842x(02)02405-8. [DOI] [PubMed] [Google Scholar]
- Miles S, Piazuelo MB, Semino-Mora C, et al. Detailed in vivo analysis of the role of Helicobacter pylori Fur in colonization and disease. Infect Immun. 2010;78:3073–82. doi: 10.1128/IAI.00190-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller HK, Auerbuch V. Bacterial iron-sulfur cluster sensors in mammalian pathogens. Metallomics. 2015;7:943–56. doi: 10.1039/c5mt00012b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monack DM. Helicobacter and Salmonella persistent infection strategies. Cold Spring Harb Perspect Med. 2013;3:a010348. doi: 10.1101/cshperspect.a010348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk CE, Pearson BM, Mulholland F, et al. Oxygen- and NssR-dependent globin expression and enhanced iron acquisition in the response of Campylobacter to nitrosative stress. J Biol Chem. 2008;283:28413–25. doi: 10.1074/jbc.M801016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecucco C, de Bernard M. Molecular and cellular mechanisms of action of the vacuolating cytotoxin (VacA) and neutrophil-activating protein (HP-NAP) virulence factors of Helicobacter pylori. Microbes Infect. 2003;5:715–21. doi: 10.1016/s1286-4579(03)00124-2. [DOI] [PubMed] [Google Scholar]
- Moskovitz J. Roles of methionine suldfoxide reductases in antioxidant defense, protein regulation and survival. Curr Pharm Des. 2005;11:1451–7. doi: 10.2174/1381612053507846. [DOI] [PubMed] [Google Scholar]
- Muller A, Oertli M, Arnold IC. H. pylori exploits and manipulates innate and adaptive immune cell signaling pathways to establish persistent infection. Cell Commun Signal. 2011;9:25. doi: 10.1186/1478-811X-9-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachamkin I. Chronic effects of Campylobacter infection. Microbes Infect. 2002;4:399–403. doi: 10.1016/s1286-4579(02)01553-8. [DOI] [PubMed] [Google Scholar]
- Nachamkin I, Szymanski CM, Blaser MJ. Campylobacter. Washington DC, USA: ASM Press; 2008. [Google Scholar]
- Nam KT, Oh SY, Ahn B, et al. Decreased Helicobacter pylori associated gastric carcinogenesis in mice lacking inducible nitric oxide synthase. Gut. 2004;53:1250–5. doi: 10.1136/gut.2003.030684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namavar F, Sparrius M, Veerman EC, et al. Neutrophil-activating protein mediates adhesion of Helicobacter pylori to sulfated carbohydrates on high-molecular-weight salivary mucin. Infect Immun. 1998;66:444–7. doi: 10.1128/iai.66.2.444-447.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson KJ, Knutson ST, Soito L, et al. Analysis of the peroxiredoxin family: using active-site structure and sequence information for global classification and residue analysis. Proteins. 2011;79:947–64. doi: 10.1002/prot.22936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HT, Nam KH, Saleem Y, et al. Characterization of Helicobacter pylori adhesin thiol peroxidase (HP0390) purified from Escherichia coli. J Biosci. 2010;35:241–8. doi: 10.1007/s12038-010-0028-0. [DOI] [PubMed] [Google Scholar]
- Nissim I, Luhovyy B, Horyn O, et al. The role of mitochondrially bound arginase in the regulation of urea synthesis: studies with [U-15N4]arginine, isolated mitochondria, and perfused rat liver. J Biol Chem. 2005;280:17715–24. doi: 10.1074/jbc.M500607200. [DOI] [PubMed] [Google Scholar]
- Nothnagel HJ, Winer BY, Vuletich DA, et al. Structural properties of 2/2 hemoglobins: the group III protein from Helicobacter hepaticus. IUBMB Life. 2011;63:197–205. doi: 10.1002/iub.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novik V, Hofreuter D, Galan JE. Identification of Campylobacter jejuni genes involved in its interaction with epithelial cells. Infect Immun. 2010;78:3540–53. doi: 10.1128/IAI.00109-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odenbreit S, Wieland B, Haas R. Cloning and genetic characterization of Helicobacter pylori catalase and construction of a catalase-deficient mutant strain. J Bacteriol. 1996;178:6960–7. doi: 10.1128/jb.178.23.6960-6967.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olczak AA, Olson JW, Maier RJ. Oxidative-stress resistance mutants of Helicobacter pylori. J Bacteriol. 2002;184:3186–93. doi: 10.1128/JB.184.12.3186-3193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olczak AA, Seyler RW, Olson JW, et al. Association of Helicobacter pylori antioxidant activities with host colonization proficiency. Infect Immun. 2003;71:580–3. doi: 10.1128/IAI.71.1.580-583.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olekhnovich IN, Vitko S, Valliere M, et al. Response to metronidazole and oxidative stress is mediated through homeostatic regulator HsrA (HP1043) in Helicobacter pylori. J Bacteriol. 2014;196:729–39. doi: 10.1128/JB.01047-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke EJ, Chevalier C, Pinto AV, et al. Pathogen DNA as target for host-generated oxidative stress: role for repair of bacterial DNA damage in Helicobacter pylori colonization. P Natl Acad Sci USA. 2003;100:2789–94. doi: 10.1073/pnas.0337641100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palyada K, Sun YQ, Flint A, et al. Characterization of the oxidative stress stimulon and PerR regulon of Campylobacter jejuni. BMC Genomics. 2009;10:481. doi: 10.1186/1471-2164-10-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan L, Zhang X, Song K, et al. Real-time imaging of autofluorescence NAD(P)H in single human neutrophils. Appl Opt. 2009;48:1042–6. doi: 10.1364/ao.48.001042. [DOI] [PubMed] [Google Scholar]
- Panthel K, Dietz P, Haas R, et al. Two-component systems of Helicobacter pylori contribute to virulence in a mouse infection model. Infect Immun. 2003;71:5381–5. doi: 10.1128/IAI.71.9.5381-5385.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parente MR, Monteiro JT, Martins GG, et al. Helicobacter pullorum induces nitric oxide release in murine macrophages that promotes phagocytosis and killing. Microbiology. 2016;162:503–12. doi: 10.1099/mic.0.000240. [DOI] [PubMed] [Google Scholar]
- Park AM, Nagata K, Sato EF, et al. Mechanism of strong resistance of Helicobacter pylori respiration to nitric oxide. Arch Biochem Biophys. 2003;411:129–35. doi: 10.1016/s0003-9861(02)00691-4. [DOI] [PubMed] [Google Scholar]
- Parkhill J, Wren BW, Mungall K, et al. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature. 2000;403:665–8. doi: 10.1038/35001088. [DOI] [PubMed] [Google Scholar]
- Pegg AE, McCann PP. S-adenosylmethionine decarboxylase as an enzyme target for therapy. Pharmacol Therapeut. 1992;56:359–77. doi: 10.1016/0163-7258(92)90025-u. [DOI] [PubMed] [Google Scholar]
- Pelliciari S, Vannini A, Roncarati D, et al. The allosteric behavior of Fur mediates oxidative stress signal transduction in Helicobacter pylori. Front Microbiol. 2015;6:840. doi: 10.3389/fmicb.2015.00840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesci EC, Cottle DL, Pickett CL. Genetic, enzymatic, and pathogenic studies of the iron superoxide dismutase of Campylobacter jejuni. Infect Immun. 1994;62:2687–94. doi: 10.1128/iai.62.7.2687-2694.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesci EC, Pickett CL. Genetic organization and enzymatic activity of a superoxide dismutase from the microaerophilic human pathogen, Helicobacter pylori. Gene. 1994;143:111–6. doi: 10.1016/0378-1119(94)90614-9. [DOI] [PubMed] [Google Scholar]
- PHAC. Enteric. Food and Waterborne Diseases, Reported Cases of Disease in Canada, Both Sexes (Including Unknown), All Ages. Canada: Public Health Agency of Canada; 2012. [Google Scholar]
- Phadnis SH, Parlow MH, Levy M, et al. Surface localization of Helicobacter pylori urease and a heat shock protein homolog requires bacterial autolysis. Infect Immun. 1996;64:905–12. doi: 10.1128/iai.64.3.905-912.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto AF, Todorovic S, Hildebrandt P, et al. Desulforubrerythrin from Campylobacter jejuni, a novel multidomain protein. J Biol Inorg Chem. 2011;16:501–10. doi: 10.1007/s00775-010-0749-4. [DOI] [PubMed] [Google Scholar]
- Pitson SM, Mendz GL, Srinivasan S, et al. The tricarboxylic acid cycle of Helicobacter pylori. Eur J Biochem. 1999;260:258–67. doi: 10.1046/j.1432-1327.1999.00153.x. [DOI] [PubMed] [Google Scholar]
- Pittman MS, Elvers KT, Lee L, et al. Growth of Campylobacter jejuni on nitrate and nitrite: electron transport to NapA and NrfA via NrfH and distinct roles for NrfA and the globin Cgb in protection against nitrosative stress. Mol Microbiol. 2007;63:575–90. doi: 10.1111/j.1365-2958.2006.05532.x. [DOI] [PubMed] [Google Scholar]
- Pittman MS, Kelly DJ. Electron transport through nitrate and nitrite reductases in Campylobacter jejuni. Biochem Soc Trans. 2005;33:190–2. doi: 10.1042/BST0330190. [DOI] [PubMed] [Google Scholar]
- Polk DB, Peek RM. Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer. 2010;10:403–14. doi: 10.1038/nrc2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole K. Bacterial stress responses as determinants of antimicrobial resistance. J Antimicrob Chemoth. 2012;67:2069–89. doi: 10.1093/jac/dks196. [DOI] [PubMed] [Google Scholar]
- Purdy D, Cawthraw S, Dickinson JH, et al. Generation of a superoxide dismutase (SOD)-deficient mutant of Campylobacter coli: evidence for the significance of SOD in Campylobacter survival and colonization. Appl Environ Microb. 1999;65:2540–6. doi: 10.1128/aem.65.6.2540-2546.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purdy D, Park SF. Cloning, nucleotide sequence and characterization of a gene encoding superoxide dismutase from Campylobacter jejuni and Campylobacter coli. Microbiology. 1994;140(Pt 5):1203–8. doi: 10.1099/13500872-140-5-1203. [DOI] [PubMed] [Google Scholar]
- Qu W, Zhou Y, Shao C, et al. Helicobacter pylori proteins response to nitric oxide stress. J Microbiol. 2009;47:486–93. doi: 10.1007/s12275-008-0266-0. [DOI] [PubMed] [Google Scholar]
- Qu W, Zhou Y, Sun Y, et al. Identification of S-nitrosylation of proteins of Helicobacter pylori in response to nitric oxide stress. J Microbiol. 2011;49:251–6. doi: 10.1007/s12275-011-0262-7. [DOI] [PubMed] [Google Scholar]
- Rain JC, Selig L, De Reuse H, et al. The protein-protein interaction map of Helicobacter pylori. Nature. 2001;409:211–5. doi: 10.1038/35051615. [DOI] [PubMed] [Google Scholar]
- Ribardo DA, Bingham-Ramos LK, Hendrixson DR. Functional analysis of the RdxA and RdxB nitroreductases of Campylobacter jejuni reveals that mutations in rdxA confer metronidazole resistance. J Bacteriol. 2010;192:1890–901. doi: 10.1128/JB.01638-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringe D, Petsko GA, Yamakura F, et al. Structure of iron superoxide dismutase from Pseudomonas ovalis at 2.9-A resolution. P Natl Acad Sci USA. 1983;80:3879–83. doi: 10.1073/pnas.80.13.3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe-Saule V, Coynault C, Ibanez-Ruiz M, et al. Identification of a non-haem catalase in Salmonella and its regulation by RpoS (sigmaS) Mol Microbiol. 2001;39:1533–45. doi: 10.1046/j.1365-2958.2001.02340.x. [DOI] [PubMed] [Google Scholar]
- Robinson DA. Campylobacter infection. Roy Soc Health J. 1981;101:138–40. doi: 10.1177/146642408110100404. [DOI] [PubMed] [Google Scholar]
- Rokutan K, Kawahara T, Kuwano Y, et al. Nox enzymes and oxidative stress in the immunopathology of the gastrointestinal tract. Semin Immunopathol. 2008;30:315–27. doi: 10.1007/s00281-008-0124-5. [DOI] [PubMed] [Google Scholar]
- Romao CV, Vicente JB, Borges PT, et al. The dual function of flavodiiron proteins: oxygen and/or nitric oxide reductases. J Biol Inorg Chem. 2016;21:39–52. doi: 10.1007/s00775-015-1329-4. [DOI] [PubMed] [Google Scholar]
- Roncarati D, Danielli A, Spohn G, et al. Transcriptional regulation of stress response and motility functions in Helicobacter pylori is mediated by HspR and HrcA. J Bacteriol. 2007;189:7234–43. doi: 10.1128/JB.00626-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi M, Hanninen ML. Helicobacter spp. other than H. pylori. Helicobacter. 2012;17(Suppl 1):56–61. doi: 10.1111/j.1523-5378.2012.00984.x. [DOI] [PubMed] [Google Scholar]
- Ruiz-Palacios GM. The health burden of Campylobacter infection and the impact of antimicrobial resistance: playing chicken. Clin Infect Dis. 2007;44:701–3. doi: 10.1086/509936. [DOI] [PubMed] [Google Scholar]
- Salama N, Guillemin K, McDaniel TK, et al. A whole-genome microarray reveals genetic diversity among Helicobacter pylori strains. P Natl Acad Sci USA. 2000;97:14668–73. doi: 10.1073/pnas.97.26.14668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama NR, Hartung ML, Muller A. Life in the human stomach: persistence strategies of the bacterial pathogen Helicobacter pylori. Nat Rev Microbiol. 2013;11:385–99. doi: 10.1038/nrmicro3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchuki HB, Valdameri G, Moure VR, et al. Purification of the Campylobacter jejuni Dps protein assisted by its high melting temperature. Protein Expres Purif. 2015;111:105–10. doi: 10.1016/j.pep.2014.12.011. [DOI] [PubMed] [Google Scholar]
- Saraiva LM, Vicente JB, Teixeira M. The role of the flavodiiron proteins in microbial nitric oxide detoxification. Adv Microb Physiol. 2004;49:77–129. doi: 10.1016/S0065-2911(04)49002-X. [DOI] [PubMed] [Google Scholar]
- Sarti P, Giuffre A, Barone MC, et al. Nitric oxide and cytochrome oxidase: reaction mechanisms from the enzyme to the cell. Free Radical Bio Med. 2003;34:509–20. doi: 10.1016/s0891-5849(02)01326-6. [DOI] [PubMed] [Google Scholar]
- Segura-Lopez FK, Guitron-Cantu A, Torres J. Association between Helicobacter spp. infections and hepatobiliary malignancies: a review. World J Gastroenterol. 2015;21:1414–23. doi: 10.3748/wjg.v21.i5.1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellars MJ, Hall SJ, Kelly DJ. Growth of Campylobacter jejuni supported by respiration of fumarate, nitrate, nitrite, trimethylamine-N-oxide, or dimethyl sulfoxide requires oxygen. J Bacteriol. 2002;184:4187–96. doi: 10.1128/JB.184.15.4187-4196.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyler RW, Olson JW, Maier RJ. Superoxide dismutase-deficient mutants of Helicobacter pylori are hypersensitive to oxidative stress and defective in host colonization. Infect Immun. 2001;69:4034–40. doi: 10.1128/IAI.69.6.4034-4040.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro KB, Hotchkiss JH. Induction of nitric oxide synthesis in murine macrophages by Helicobacter pylori. Cancer Lett. 1996;102:49–56. doi: 10.1016/0304-3835(96)04154-7. [DOI] [PubMed] [Google Scholar]
- Sheng Y, Abreu IA, Cabelli DE, et al. Superoxide dismutases and superoxide reductases. Chem Rev. 2014;114:3854–918. doi: 10.1021/cr4005296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigalov AB, Stern LJ. Enzymatic repair of oxidative damage to human apolipoprotein A-I. FEBS Lett. 1998;433:196–200. doi: 10.1016/s0014-5793(98)00908-9. [DOI] [PubMed] [Google Scholar]
- Singh A, Hodgson N, Yan M, et al. Screening Helicobacter pylori genes induced during infection of mouse stomachs. World J Gastroenterol. 2012;18:4323–34. doi: 10.3748/wjg.v18.i32.4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarp CP, Hanninen ML, Rautelin HI. Campylobacteriosis: the role of poultry-meat. Clin Microbiol Infec. 2016;22:103–9. doi: 10.1016/j.cmi.2015.11.019. [DOI] [PubMed] [Google Scholar]
- Slauch JM. How does the oxidative burst of macrophages kill bacteria? Still an open question. Mol Microbiol. 2011;80:580–3. doi: 10.1111/j.1365-2958.2011.07612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HK, Shepherd M, Monk C, et al. The NO-responsive hemoglobins of Campylobacter jejuni: concerted responses of two globins to NO and evidence in vitro for globin regulation by the transcription factor NssR. Nitric Oxide. 2011;25:234–41. doi: 10.1016/j.niox.2010.12.009. [DOI] [PubMed] [Google Scholar]
- Soballe B, Poole RK. Microbial ubiquinones: multiple roles in respiration, gene regulation and oxidative stress management. Microbiology. 1999;145(Pt 8):1817–30. doi: 10.1099/13500872-145-8-1817. [DOI] [PubMed] [Google Scholar]
- Sobota JM, Imlay JA. Iron enzyme ribulose-5-phosphate 3-epimerase in Escherichia coli is rapidly damaged by hydrogen peroxide but can be protected by manganese. P Natl Acad Sci USA. 2011;108:5402–7. doi: 10.1073/pnas.1100410108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegelhalder C, Gerstenecker B, Kersten A, et al. Purification of Helicobacter pylori superoxide dismutase and cloning and sequencing of the gene. Infect Immun. 1993;61:5315–25. doi: 10.1128/iai.61.12.5315-5325.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stent A, Every AL, Sutton P. Helicobacter pylori defense against oxidative attack. Am J Physiol-Gastr L. 2012;302:G579–87. doi: 10.1152/ajpgi.00495.2011. [DOI] [PubMed] [Google Scholar]
- Stern AM, Zhu J. An introduction to nitric oxide sensing and response in bacteria. Adv Appl Microbiol. 2014;87:187–220. doi: 10.1016/B978-0-12-800261-2.00005-0. [DOI] [PubMed] [Google Scholar]
- Sutton P, Mitchell H. Helicobacter pylori in the 21st Century. Sydney, Australia: CAB International; 2010. [Google Scholar]
- Tanaka T, Goto M, Okuzumi K, et al. Isolation and identification of Helicobacter cinaedi and H. cinaedi-like organisms isolated from blood culture in practical laboratory procedures. Kansenshogaku Zasshi. 2007;81:700–6. doi: 10.11150/kansenshogakuzasshi1970.81.700. [DOI] [PubMed] [Google Scholar]
- Tang Y, Guest JR. Direct evidence for mRNA binding and post-transcriptional regulation by Escherichia coli aconitases. Microbiology. 1999;145:3069–79. doi: 10.1099/00221287-145-11-3069. [DOI] [PubMed] [Google Scholar]
- Tang Y, Quail MA, Artymiuk PJ, et al. Escherichia coli aconitases and oxidative stress: post-transcriptional regulation of sodA expression. Microbiology. 2002;148:1027–37. doi: 10.1099/00221287-148-4-1027. [DOI] [PubMed] [Google Scholar]
- Tanih NF, Ndip LM, Ndip RN. Characterisation of the genes encoding resistance to metronidazole (rdxA and frxA) and clarithromycin (the 23S-rRNA genes) in South African isolates of Helicobacter pylori. Ann Trop Med Parasitol. 2011;105:251–9. doi: 10.1179/136485911X12899838683485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarantino M, Dionisi AM, Pistoia C, et al. Involvement of nitric oxide in the control of a mouse model of Campylobacter jejuni infection. FEMS Immunol Med Mic. 2009;56:98–101. doi: 10.1111/j.1574-695X.2009.00547.x. [DOI] [PubMed] [Google Scholar]
- Tecder-Unal M, Can F, Demirbilek M, et al. The bactericidal and morphological effects of peroxynitrite on Helicobacter pylori. Helicobacter. 2008;13:42–8. doi: 10.1111/j.1523-5378.2008.00583.x. [DOI] [PubMed] [Google Scholar]
- Theoret JR, Cooper KK, Glock RD, et al. A Campylobacter jejuni Dps homolog has a role in intracellular survival and in the development of Campylobacterosis in neonate piglets. Food Pathog Dis. 2011;8:1263–8. doi: 10.1089/fpd.2011.0892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theoret JR, Cooper KK, Zekarias B, et al. The Campylobacter jejuni Dps homologue is important for in vitro biofilm formation and cecal colonization of poultry and may serve as a protective antigen for vaccination. Clin Vaccine Immunol. 2012;19:1426–31. doi: 10.1128/CVI.00151-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MK, Murray R, Flockhart L, et al. Estimates of the burden of foodborne illness in Canada for 30 specified pathogens and unspecified agents, circa 2006. Food Pathog Dis. 2013;10:639–48. doi: 10.1089/fpd.2012.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinajero-Trejo M, Vreugdenhil A, Sedelnikova SE, et al. Nitric oxide reactivities of the two globins of the foodborne pathogen Campylobacter jejuni: roles in protection from nitrosative stress and analysis of potential reductants. Nitric Oxide. 2013;34:65–75. doi: 10.1016/j.niox.2013.06.002. [DOI] [PubMed] [Google Scholar]
- Vanet A, Labigne A. Evidence for specific secretion rather than autolysis in the release of some Helicobacter pylori proteins. Infect Immun. 1998;66:1023–7. doi: 10.1128/iai.66.3.1023-1027.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velayudhan J, Jones MA, Barrow PA, et al. L-serine catabolism via an oxygen-labile L-serine dehydratase is essential for colonization of the avian gut by Campylobacter jejuni. Infect Immun. 2004;72:260–8. doi: 10.1128/IAI.72.1.260-268.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradov SN, Bailly X, Smith DR, et al. Microbial eukaryote globins. Adv Microb Physiol. 2013;63:391–446. doi: 10.1016/B978-0-12-407693-8.00009-1. [DOI] [PubMed] [Google Scholar]
- Voisin S, Watson DC, Tessier L, et al. The cytoplasmic phosphoproteome of the Gram-negative bacterium Campylobacter jejuni: evidence for modification by unidentified protein kinases. Proteomics. 2007;7:4338–48. doi: 10.1002/pmic.200700483. [DOI] [PubMed] [Google Scholar]
- Waidner B, Melchers K, Stahler FN, et al. The Helicobacter pylori CrdRS two-component regulation system (HP1364/HP1365) is required for copper-mediated induction of the copper resistance determinant CrdA. J Bacteriol. 2005;187:4683–8. doi: 10.1128/JB.187.13.4683-4688.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainwright LM, Elvers KT, Park SF, et al. A truncated haemoglobin implicated in oxygen metabolism by the microaerophilic food-borne pathogen Campylobacter jejuni. Microbiology. 2005;151:4079–91. doi: 10.1099/mic.0.28266-0. [DOI] [PubMed] [Google Scholar]
- Wainwright LM, Wang Y, Park SF, et al. Purification and spectroscopic characterization of Ctb, a group III truncated hemoglobin implicated in oxygen metabolism in the food-borne pathogen Campylobacter jejuni. Biochemistry. 2006;45:6003–11. doi: 10.1021/bi052247k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Alamuri P, Humayun MZ, et al. The Helicobacter pylori MutS protein confers protection from oxidative DNA damage. Mol Microbiol. 2005a;58:166–76. doi: 10.1111/j.1365-2958.2005.04833.x. [DOI] [PubMed] [Google Scholar]
- Wang G, Alamuri P, Maier RJ. The diverse antioxidant systems of Helicobacter pylori. Mol Microbiol. 2006;61:847–60. doi: 10.1111/j.1365-2958.2006.05302.x. [DOI] [PubMed] [Google Scholar]
- Wang G, Hong Y, Olczak A, et al. Dual Roles of Helicobacter pylori NapA in inducing and combating oxidative stress. Infect Immun. 2006;74:6839–46. doi: 10.1128/IAI.00991-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Lo LF, Maier RJ. A histone-like protein of Helicobacter pylori protects DNA from stress damage and aids host colonization. DNA Repair. 2012;11:733–40. doi: 10.1016/j.dnarep.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Maier RJ. An NADPH quinone reductase of Helicobacter pylori plays an important role in oxidative stress resistance and host colonization. Infect Immun. 2004;72:1391–6. doi: 10.1128/IAI.72.3.1391-1396.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Maier RJ. A novel DNA-binding protein plays an important role in Helicobacter pylori stress tolerance and survival in the host. J Bacteriol. 2015;197:973–82. doi: 10.1128/JB.02489-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Maier SE, Lo LF, et al. Peptidoglycan deacetylation in Helicobacter pylori contributes to bacterial survival by mitigating host immune responses. Infect Immun. 2010;78:4660–6. doi: 10.1128/IAI.00307-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Olczak A, Forsberg LS, et al. Oxidative stress-induced peptidoglycan deacetylase in Helicobacter pylori. J Biol Chem. 2009;284:6790–800. doi: 10.1074/jbc.M808071200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Olczak AA, Walton JP, et al. Contribution of the Helicobacter pylori thiol peroxidase bacterioferritin comigratory protein to oxidative stress resistance and host colonization. Infect Immun. 2005b;73:378–84. doi: 10.1128/IAI.73.1.378-384.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse CA, Balbo PB, Pesci EC, et al. Campylobacter jejuni cytolethal distending toxin causes a G2-phase cell cycle block. Infect Immun. 1998;66:1934–40. doi: 10.1128/iai.66.5.1934-1940.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson KT, Ramanujam KS, Mobley HL, et al. Helicobacter pylori stimulates inducible nitric oxide synthase expression and activity in a murine macrophage cell line. Gastroenterology. 1996;111:1524–33. doi: 10.1016/s0016-5085(96)70014-8. [DOI] [PubMed] [Google Scholar]
- Windle HJ, Fox A, Ni Eidhin D, et al. The thioredoxin system of Helicobacter pylori. J Biol Chem. 2000;275:5081–9. doi: 10.1074/jbc.275.7.5081. [DOI] [PubMed] [Google Scholar]
- Wolf C, Hochgrafe F, Kusch H, et al. Proteomic analysis of antioxidant strategies of Staphylococcus aureus: diverse responses to different oxidants. Proteomics. 2008;8:3139–53. doi: 10.1002/pmic.200701062. [DOI] [PubMed] [Google Scholar]
- Wood ZA, Schroder E, Robin Harris J, et al. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- Wu G, Morris SM., Jr Arginine metabolism: nitric oxide and beyond. Biochem J. 1998;336(Pt 1):1–17. doi: 10.1042/bj3360001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaoka Y. Mechanisms of disease: Helicobacter pylori virulence factors. Nat Rev Gastroentero. 2010;7:629–41. doi: 10.1038/nrgastro.2010.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki M, Igimi S, Katayama Y, et al. Identification of an oxidative stress-sensitive protein from Campylobacter jejuni, homologous to rubredoxin oxidoreductase/rubrerythrin. FEMS Microbiol Lett. 2004;235:57–63. doi: 10.1016/j.femsle.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Young KT, Davis LM, Dirita VJ. Campylobacter jejuni: molecular biology and pathogenesis. Nat Rev Microbiol. 2007;5:665–79. doi: 10.1038/nrmicro1718. [DOI] [PubMed] [Google Scholar]
- Zamocky M, Gasselhuber B, Furtmuller PG, et al. Molecular evolution of hydrogen peroxide degrading enzymes. Arch Biochem Biophys. 2012;525:131–44. doi: 10.1016/j.abb.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotti G, Papinutto E, Dundon W, et al. Structure of the neutrophil-activating protein from Helicobacter pylori. J Mol Biol. 2002;323:125–30. doi: 10.1016/s0022-2836(02)00879-3. [DOI] [PubMed] [Google Scholar]