

Abstract
There are increasing demands for simple but still effective methods that can be used to detect specific pathogens for point-of-care or field applications. Such methods need to be user-friendly and produce reliable results that can be easily interpreted by both specialists and non-professionals. The litmus test for pH is simple, quick, and effective as it reports the pH of a test sample via a simple color change. We have developed an approach to take advantage of the litmus test for bacterial detection. The method exploits a bacterium-specific RNA-cleaving DNAzyme to achieve two functions: recognizing a bacterium of interest and providing a mechanism to control the activity of urease. Through the use of magnetic beads immobilized with a DNAzyme-urease conjugate, the presence of bacteria in a test sample is relayed to the release of urease from beads to solution. The released urease is transferred to a test solution to hydrolyze urea into ammonia, resulting in an increase of pH that can be visualized using the classic litmus test.
Keywords: Cellular Biology, Issue 115, Bacterial detection, Colorimetric assay, Litmus test, Biosensor, DNAzyme and E. coli
Introduction
Bacterial pathogens are one of the major causes of global morbidity and mortality. Outbreaks from hospital-acquired infections, food-borne pathogens, and bacterial contaminants in the environment pose serious and on-going threats to public health and safety. To prevent these outbreaks, effective tools are needed that permit pathogen detection in a timely fashion under a variety of settings. Simple but still effective tests that are portable and cost-effective are greatly coveted, especially in regions that are susceptible to outbreaks but cannot afford expensive testing facilities.1-3 Although there exists a multitude of methods to detect bacteria, many of them are not suitable as screening or on-site testing tools because they require long test times, expensive instruments and complicated testing procedures.
Colorimetric tests are particularly attractive for point-of-care or field applications as color changes can be easily detected by the naked eye. The litmus test for pH is simple, quick, and effective. Although it is a very old technology, it is still widely used today because of its simplicity and effectiveness. Surprisingly, this simple test had never been modified to achieve the detection of other analytes before we recently developed an approach of modifying this test for E. coli testing.4
The expanded litmus test for E. coli employs three additional components: an E. coli activated RNA-cleaving DNAzyme (EC1), 5 urease, and magnetic beads. DNAzymes refer to synthetic single-stranded DNA molecules with catalytic activity.6 They can be isolated from random-sequence DNA pools using in vitro selection.7,8 They are highly stable and can be produced cost-effectively using high-efficiency automated DNA synthesis.9 For these reasons, DNAzymes, particularly RNA-cleaving DNAzymes, have been widely examined for biosensing applications.6,10,11 RNA-cleaving DNAzyme sensors have been developed to detect metal ions,12-16 small molecules,17,18 bacterial pathogens5,19-21 and cancer cells.22 Given the great availability of target-induced RNA-cleaving DNAzymes, any assay that utilizes a DNAzyme can be potentially expanded to detect a diverse range of analytes.
Urease is chosen for its ability to hydrolyze urea into ammonia,23,24 resulting in a pH increase. Urease is also highly efficient, stable and amenable for conjugation to other biomolecules. Therefore, we postulated that a conjugate of an RNA-cleavage DNAzyme with urease would allow the use of litmus test for the detection of other targets.5
The action of the RNA-cleaving DNAzyme is relayed to urease-mediated increase of pH through the use of magnetic beads that are immobilized with the DNAzyme-urease conjugate. Because the activity of the DNAzyme under investigation is strictly dependent on E. coli, the presence of this bacterium in the test solution will result in the release of urease from the magnetic beads to the solution, which is then taken and used to hydrolyze urea in a reporter solution that contains a pH-sensitive dye. The final outcome of this procedure is a color change that can be conveniently reported by the dye or pH paper.
Protocol
1. Preparation of Reagents and Buffers
- 0.5 M Ethylenediaminetetraacetic Acid (EDTA)
- In a 2 L beaker, add 186.1 g EDTA to 800 ml of deionized-distilled water (ddH2O). Adjust the pH of the solution to 8.0 using NaOH pellets. Add ddH2O to a final volume of 1.0 L and transfer the solution to an autoclavable glass bottle for autoclaving and store at 4 °C.
- 10× Tris-borate EDTA (10x TBE)
- In a 4 L plastic beaker, add 432 g Tris-base, 200 g boric acid, 80 ml of 0.5 M EDTA (pH 8.0) and ddH2O to a final volume of 4 L. Mix using a stir bar until all the components are dissolved. Transfer the solution to 1 L glass bottles for autoclaving and store at 4 °C.
- 10% Denaturing Polyacrylamide Stock
- In a 4 L plastic beaker, add 1681.7 g urea, 400 ml 10x TBE, 1 L of 40% acrylamide/bisacrylamide (29:1) solution and ddH2O until the final volume is 4 L. Mix using a stir bar until the urea is dissolved. Transfer the solution to 1 L amber glass bottles and store at 4 °C.
- 2x Gel Loading Buffer (2x GLB)
- In a 200 ml glass beaker, add 44 g urea, 8 g sucrose, 10 mg bromophenol blue, 10 mg xylene cyanol FF, 400 µl of 10% sodium dodecyl sulfate, and 4 ml of 10× TBE. Add ddH2O to a final volume of 40 ml and dissolve the components with mild heating at 50 °C while mixing with a magnetic stir bar. Transfer 1 ml aliquot to 1.5 ml microfuge tubes and store at 4 °C. NOTE: Prior use requires brief heating at 90 °C to redissolve any solids.
- 1 M Tris-hydrochloride (Tris-HCl) (pH 7.5)
- In a glass bottle, add 12.1 g of Tris-base and 70 ml of ddH2O and mix until the solid is dissolved. Adjust the pH to 7.5 using 1 M hydrochloric acid (HCl). Add ddH2O to a final volume of 100 ml, autoclave and store at 4 °C
- 5 M Sodium Chloride (NaCl)
- In a glass bottle, dissolve 58.4 g of NaCl in 150 ml of ddH2O. Adjust the volume to 200 ml with ddH2O. Store at 4 °C.
- DNA Elution Buffer
- In a glass bottle, mix 2.0 ml of 1 M Tris-HCl (pH 7.5), 8.0 ml of 5 M NaCl and 0.4 ml of 0.5 M EDTA (pH 8.0). Adjust the volume to 200 ml with ddH2O, autoclave and store at 4 °C.
- 1 M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 7.4)
- In a glass bottle, dissolve 2.38 g of HEPES in 80 ml of ddH2O. Adjust the pH to 7.4 using 5 N NaOH and add ddH2O to a final volume of 100 ml.
- 1 M Magnesium Chloride (MgCl2)
- In a glass bottle, add 2.03 g of MgCl2-6H2O and bring the volume to 100 ml with ddH2O.
- Reaction Buffer (RB)
- In a 50 ml conical tube, add 50 µl of 1 M HEPES (pH 7.4), 1.5 ml of 5 M NaCl, 0.75 ml of 1 M MgCl2, and 5 µl of Tween-20. Add ddH2O to a final volume of 50 ml. Mix the solution and filter into another conical tube using a syringe-driven filter (0.22 µm) and store at 4 °C until use.
- Binding Buffer (BB)
- In a 50 ml conical tube, add 500 µl of 1 M Tris-HCl (pH 7.5), 8.8 g of NaCl, 50 µl of 1 M of MgCl2, and 5 µl of Tween 20. Add ddH2O to a final volume of 50 ml. Mix the solution and filter into another conical tube using a syringe-driven filter (0.22 µm) and store at 4 °C until use.
- Substrate Solution (SS)
- In a 50 ml conical tube, add 5.8 g of NaCl, 3 ml of 1 M MgCl2, 1.5 g urea, and 40 ml of ddH2O. Adjust the pH of the solution to 5.0 using 10 mM HCl. Because the solution is not buffered, adjust pH using HCl carefully through the addition of small HCl aliquots. Add ddH2O to a final volume of 50 ml.
- Mix the solution and filter into another conical tube using a syringe-driven filter (0.22 µm) and store at 4 °C until use.
- Luria Bertani (LB) Broth
- In a beaker, dissolve 20.0 g LB powder in 1 L of ddH2O. Transfer to a glass bottle, autoclave, and store at 4 °C.
- 1.5% LB agar
- In a 250 ml flask, add 1.5 g agar and 100 ml of LB broth. Autoclave and store at 4 °C.
- Agar Plates
- Redissolve the LB agar in a microwave and cool the solution to ~50 °C. Pour the solution into petri dishes, making ~5 plates and allow them to solidify.
2. Synthesis and Purification of E. coli-responsive DNAzyme EC1
- Synthesis of EC1 by Template Mediated Enzymatic Ligation
- Purify commercially synthesized oligonucleotides BS1, DE1, and T1 (sequences provided in Table 1) by 10% denaturing polyacrylamide gel electrophoresis (dPAGE) according to standard protocols.
- Prepare a 100 µM stock of BS1, DE1, and T1. Store at -20 °C until use.
- In a 1.5 ml microfuge tube, add 38.5 µl of ddH2O, 5 µl of DE1, and 5 µl of 10x T4 polynucleotide kinase reaction buffer provided by enzyme supplier (500 mM Tris-HCl (pH 7.6), 100 mM MgCl2, 50 mM dithiothreitol (DTT), 1.0 mM spermidine).
- Add 1 µl of adenosine triphosphate (ATP) (100 mM). Add 5 units of T4 polynucleotide kinase (10 U/µl). Mix by carefully pipetting the reaction mixture.
- Incubate the reaction at 37 °C for 30 min.
- Quench the reaction by heating to 90 °C for 5 min.
- Add 118 µl of ddH2O, 5 µl of BS1, and 5 µl of T1. Heat the reaction to 90 °C for 2 min and then cool to room temperature over 10 min.
- Add 20 µl of 10× T4 DNA ligase reaction buffer provided by the enzyme supplier (400 mM Tris-HCl (pH 7.8), 100 mM MgCl2, 100 mM DTT, 5 mM ATP). Add 10 units of T4 DNA ligase (5 U/µl) and carefully mix by pipetting.
- Incubate at room temperature for 2 hr.
- Add 20 µl of 3 M sodium acetate (NaOAc) (pH 5.2), 500 µl of cold 100% ethanol. Mix the solution by vortexing and place the tube in a -20 °C freezer for 30 min.
- Centrifuge the microfuge at 20,000 x g for 20 min at 4 °C. Carefully remove the supernatant by pipetting.
- Wash the pellet with cold 70% ethanol and centrifuge again at 20,000 x g for 10 min at 4 °C. Once again remove the supernatant and dry the pellet under vacuum for 10 min.
- Resuspend the DNA with 15 µl of ddH2O and then add 15 µl of 2x GLB.
- Vortex vigorously and heat to 90 °C for 2 min. Purify the full-length DNA by 10% dPAGE as described below.
- Setting Up 10% dPAGE
- Clean two glass plates (one full plate and one notched), two 0.75 mm thick spacers, and a 12-well comb. Lay one glass plate on a flat surface with two spacers on each side and the notched plate on top. Clip the two plates together using the four clips provided by the supplier.
- In a 150 ml plastic beaker, pour 40 ml of 10% dPAGE, 40 µl of tetramethylethylenediamine (TEMED), and 400 µl of 10% ammonium persulfate (APS). Mix the components and pour the solution between the plates slowly.
- Insert the comb and then allow the gel to polymerize over 10 min. Once the gel is polymerized, slowly remove the comb and flush the wells with ddH2O.
- Mount the plates onto the gel electrophoresis apparatus. Use a metal plate to dissipate heat generated to prevent overheating.
- Pour 1x TBE onto the top and bottom chambers and ensure that the wells are well submerged in the buffer. Flush the wells with 1x TBE using a pipette or syringe.
- Set the apparatus to run at 35 mA and pre-run for 15 min before loading samples.
- Elution of Ligated EC1 from 10% dPAGE
- Following step 2.2.6, run the gel at 35 mA until the bottom dye (bromophenol blue) reaches the bottom of the glass plate. This should take approximately 1.5 hr. Turn off the power and remove the plates from the apparatus.
- Lay the plates on a few sheets of paper towels and carefully remove the spacers from the glass plates. Carefully remove the top glass plate and wrap the gel with plastic wrap.
- Flip the gel over to remove the second glass plate and cover with plastic wrap again. Take care to avoid wrinkles of the plastic wrap.
- Visualize the ligated product by using UV shadowing (260 nm), which will produce 4 distinct DNA bands (Fully ligated EC1, DE1, BS1, and T1).
- Excise the top band (EC1) with a sterile razor blade and transfer to a 1.5 ml microfuge tube. Crush the gel with a sterile pipette tip (200 µl tip size) until it turns into a fine paste. Add 550 µl of DNA elution buffer, cover the tube with aluminum foil to protect the embedded fluorophore, and shake for 15 min.
- Centrifuge the gel solution at 20,000 x g for 5 min and carefully transfer 400 µl of the supernatant to another 1.5 ml microfuge tube. Avoid withdrawing gel pieces during pipetting.
- To the microfuge tube with the transferred supernatant add 40 µl of 3 M NaOAc (pH 5.2), and 1.0 ml of cold 100% ethanol. Mix the solution by vortexing and place the tube in a -20 °C freezer for 30 min.
- Centrifuge the microfuge at 20,000 x g for 20 min at 4 °C. Carefully remove the supernatant by pipetting.
- Wash the pellet with cold 70% ethanol and centrifuge again at 20,000 x g for 10 min at 4 °C. Once again, remove the supernatant and dry the pellet under vacuum for 10 min.
- Determine the concentration of EC1 by measuring UV absorbance at 260 nm. Make a 10 µM stock and store the sample at -20 °C until use.
3. Conjugation of Urease to DNA
- Preparation of Succinimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC) Stock
- Dissolve 1 mg SMCC in 676 µl of DMSO. Vortex and place on ice until use.
- Preparation of Urease Stock
- Dissolve 1 mg urease in 1 ml of 1× PBS (no Mg2+ or Ca2+). Place on ice until use. NOTE: Crystallized urease is slow to dissolve and gentle mixing is needed to avoid denaturation.
- Synthesis of Urease-DNA (UrDNA)
- Add 10 µl of 100 µM LD1 to a 2.5 ml microfuge tube. Add 140 µl of ddH2O, 40 µl of 10x PBS, and vortex.
- Add 80.5 µl of SMCC stock, 159.5 µl of DMSO, vortex, and briefly centrifuge using a benchtop centrifuge.
- Incubate at 37 °C for 60 min. Avoid condensation under the microfuge cap.
- Add 200 µl of 1x PBS, 60 µl of 3 M NaOAc (pH 5.2), and 1.5 ml of cold 100% ethanol. Mix the solution by vortexing and incubate at -20 °C for 30 min.
- Centrifuge the solution at 20,000 x g for 20 min at 4 °C. Remove the supernatant and dry the pellet under vacuum. Avoid over-drying.
- To the dried pellet add 400 µl of urease stock and incubate at room temperature for 5 hr.
- Transfer 200 µl of crude conjugate to a pre-washed 100k MWCO centrifugal filter column. Centrifuge the column at 14,000 x g for 5 min.
- Transfer the remaining 200 µl of crude conjugate to the column and centrifuge at 14,000 x g for 5 min. Remove the column and place it upside down in a new 2.0 ml microfuge tube ("collection tube").
- Centrifuge the collection tube (with the inverted column) at 1,000 x g for 2 min. Remove the collection tube and add 30 µl of 1x PBS to the centrifugal column to wash the membrane for additional recovery of conjugates.
- Once again invert the column and place back into the collection tube. Centrifuge the collection tube (with the inverted column) at 1,000 x g for 2 min. Remove and dispose the column.
- Store the UrDNA at 4 °C until use.
4. Assembly of EC1 and UrDNA onto Magnetic Beads
Mix the magnetic bead (MB) stock well and transfer 100 µl of MB suspension to a 1.5 ml microfuge tube. Place the microfuge tube on a magnetic rack holder for isolating the MB.
Remove the supernatant by pipetting and add 150 µl of binding buffer (BB) to the tube. Remove the tube from the holder and carefully tap the tube to resuspend the MB to a homogeneous solution. Avoid splashing the suspension to the top of the tube or cap. If this happens, use a benchtop centrifuge to briefly spin the residue back to the suspension.
Repeat step 4.2 two more times.
To this suspension add 10 µl of 10 µM EC1. Carefully mix by tapping on the tube.
Incubate the solution with mild shaking for 30 min. Tap the tube every 2-3 min to avoid the precipitation of the MB.
Place the microfuge tube back on the magnetic rack to isolate the MB and remove the supernatant by pipetting. Wash the MB three times with 150 µl of BB (as described in step 4.2).
Once the washing is complete, suspend the MB in a total of 150 µl of BB. To this solution, add 15 µl of UrDNA and heat the solution to 45 °C for 2 min. Cool the solution to room temperature and incubate for 2 hr.
Place the microfuge tube back on the magnetic rack to isolate the MB and remove the supernatant by pipetting.
Add 100 µl of reaction buffer (RB). Remove the microfuge tube from the magnetic rack and carefully resuspend the MB.
Wash the MB three more times by repeating step 4.9. NOTE: The washed supernatant can be quickly tested to determine if unhybridized UrDNA is still present, which may result in a false positive signal. The test can be done by adding 10 µl of 50 mM urea and 10 µl of 0.04% phenol red to the wash solution. Continue to wash the MB until the wash solution does not cause a color change. The 100 µl suspension is stored at 4 °C until use.
5. Preparation of Bacterial Cells20
- Culturing E. coli from Stocks
- Plate E. coli K12 (MG1655) onto LB agar plates from a glycerol stock under a flame or in a biological safety cabinet.
- Using a sterile pipette tip, gently touch the glycerol stock and lightly streak the surface of an agar plate to avoid puncturing the LB agar.
- Invert the streaked plate and incubate at 37 °C for 14 hr.
- Seal the plate with Parafilm and store at 4 °C for a maximum of 3 weeks.
- Culturing E. coli for Cell Counting
- In a sterile 14 ml culture tube, dispense 2 ml of LB broth.
- Using a sterile pipette tip, pick a single colony from the streaked agar plate prepared in step 5.1 and transfer it to the culture tube.
- Incubate the culture at 37 °C and shake at 230 rpm for 14 hr.
- Serially dilute the bacterial culture in 10-fold intervals.
- For each diluted sample, evenly plate five 100 µl aliquots on separate LB agar plates. Invert the plates and incubate at 37 °C for 14 hr.
- Count the cells of each sample to obtain the average cell concentration of each dilution. NOTE: 107 cells are often used to set up a reference litmus test for E. coli as this level of E. coli can trigger a quick color change. However, a properly executed litmus test can detect as low as 500 cells, as discussed in the Results section.
- Preparing E. coli Cells for Testing
- For a desired cell suspension, transfer 1 ml of cultured stock to a 1.5 ml microfuge tube.
- Centrifuge the cells at 6,000 x g for 10 min at 4 °C. Carefully remove the supernatant without disturbing the cell pellet.
- Add 10 µl of reaction buffer to the cell pellet and resuspend the cells. Sonicate the cell suspension for 5 min. Transfer the cell suspension to an ice box for 5 min.
- Sonicate the cell suspension for another 5 min.
- Centrifuge the cell suspension at 13,000 x g for 10 min at 4 °C. Use the supernatant for testing (10 µl).
6. Litmus Test
In a 1.5 ml microfuge tube, prewash the tube by adding and vortexing 100 µl of reaction buffer (RB) in the microfuge tube and discarding the buffer.
Transfer 15 µl of assembled EC1 (protocol 4) to the washed microfuge tube.
Wash the magnetic beads by placing the microfuge tube on a magnetic rack. Remove the supernatant by pipetting. Remove the microfuge tube from the rack, add 100 µl of RB, and carefully resuspend the magnetic beads.
Wash the MB two more times by repeating step 6.3.
Place the microfuge tube back on the magnetic rack, remove the supernatant and add the 10 µl E. coli sample prepared from step 5.3.
Mix the sample and magnetic beads carefully by gently tapping on the microfuge tube.
Incubate the reaction at room temperature for 1 hr.
To the reaction, add 90 µl of ddH2O and place the microfuge tube onto a magnetic rack.
After approximately 3 min of magnetic separation, carefully transfer 85 µl of the supernatant to a 0.5 ml microfuge tube. Withdraw the supernatant slowly to avoid collecting any magnetic beads.
To the above microfuge tube add 15 µl of 0.04% phenol red, and 100 µl of substrate solution.
Take a photograph at specific time intervals to record color change. NOTE: The change in pH can also be monitored using a pH meter with a microelectrode. The starting pH should be approximately 5.2-5.5 (the solution is yellow). If not, the solution can be adjusted by addition of 1 mM Acetate Buffer pH 5.0).
Representative Results
The principle of the bacterial litmus test is explained in Figure 1. The test uses three key materials: an RNA-cleaving DNAzyme that is activated by a specific bacterium, urease and magnetic beads. The DNAzyme is used as the molecular recognition element to achieve highly specific detection of a bacterium of interest. Urease and magnetic beads are used to achieve signal transduction of the RNA-cleavage activity of the DNAzyme. This involves the creation of magnetic beads that contain urease-DNAzyme conjugates. In the presence of the target bacterium, the DNAzyme cleaves its RNA linkage. This action gives rise to the dissociation of urease from magnetic beads. The released urease can be easily separated from magnetic beads and used to generate a color change in a reporter solution, which contains urea and a pH-sensitive dye. Urease hydrolyzes urea into ammonia, accompanied by the increase of pH that triggers the color change of the dye.
Figure 2 presents a bacterial litmus test where EC1, an E. coli-responsive RNA-cleaving DNAzyme, was used as the DNAzyme, and phenol red was used as the pH-reporting dye. EC1 was previously isolated by our group from a random-sequence DNA pool using the technique of in vitro selection.5 Our previous studies have shown that EC1 is highly specific for E. coli and exhibits minimal activity towards other bacteria.5,19 It has been found that EC1 is activated by a protein molecule from E. coli. Although the identity of this protein biomarker has not been deciphered, the high recognition specificity suggests that this protein is unique to E. coli. The reporter solution is set up to have an initial pH of 5.5. At this pH, phenol red exhibits a yellow color. As urease hydrolyzes urea into ammonia, the basicity of the reporter solution increases. This is reflected by the gradual change of color from yellow to pink. The depth of color change is dependent on the following two parameters, as illustrated by Figure 2: the number of E. coli cells used in the DNAzyme activation step and the time allowed for the urea hydrolysis step. More E. coli cells resulted in stronger color changes, reflected by the observation of a progressive yellow-to-pink color transition when E. coli cells were serially increased from 5 to 5 x 107 (10-fold increase each time). Meanwhile, a longer time for urea hydrolysis allowed for the detection of smaller numbers of E. coli cells (5,000 cells in the 1 hr reaction and 500 cells in the 2-hour reaction).
The pH change of the bacterial litmus test can also be monitored using a handheld pH meter and representative results are illustrated in Figure 3. It was found that the presence of 107 E. coli cells resulted in gradual increase of pH by 3 units within 10 min. In contrast, the absence of E. coli cells did not cause detectable pH changes under the same setting.
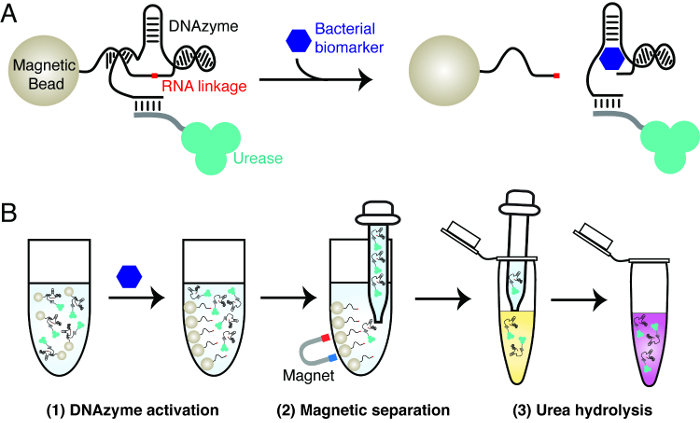 Figure 1:The design principle of bacterial litmus test. (A) Activation of an RNA-cleaving DNAzyme by a specific biomarker from a bacterium of interest. In the presence of the biomarker, the RNA-cleaving DNAzyme immobilized on magnetic beads cleaves the RNA linkage, resulting in the release of the tagged urease from magnetic beads to solution. (B) Three-step assay procedure. Step 1: DNAzyme activation, as described in panel A. Step 2: Magnetic separation — the released urease is separated from magnetic beads. Step 3: Urea hydrolysis — the released urease is added into a urea-containing reporter solution. Urease hydrolyzes urea into ammonia, resulting in a change in pH that can be reported by a pH-sensitive dye. Please click here to view a larger version of this figure.
Figure 1:The design principle of bacterial litmus test. (A) Activation of an RNA-cleaving DNAzyme by a specific biomarker from a bacterium of interest. In the presence of the biomarker, the RNA-cleaving DNAzyme immobilized on magnetic beads cleaves the RNA linkage, resulting in the release of the tagged urease from magnetic beads to solution. (B) Three-step assay procedure. Step 1: DNAzyme activation, as described in panel A. Step 2: Magnetic separation — the released urease is separated from magnetic beads. Step 3: Urea hydrolysis — the released urease is added into a urea-containing reporter solution. Urease hydrolyzes urea into ammonia, resulting in a change in pH that can be reported by a pH-sensitive dye. Please click here to view a larger version of this figure.
 Figure 2:Litmus test with E. coli using E. coli-responsive DNAzyme EC1. Representative color-changing results with varying numbers of E. coli cells provided above each test-tube. Phenol red was used as the pH-sensitive dye. A test without E. coli was used as a negative control. More E. coli cells are expected to cause the release of more urease molecules, accompanied by stronger color changes. Please click here to view a larger version of this figure.
Figure 2:Litmus test with E. coli using E. coli-responsive DNAzyme EC1. Representative color-changing results with varying numbers of E. coli cells provided above each test-tube. Phenol red was used as the pH-sensitive dye. A test without E. coli was used as a negative control. More E. coli cells are expected to cause the release of more urease molecules, accompanied by stronger color changes. Please click here to view a larger version of this figure.
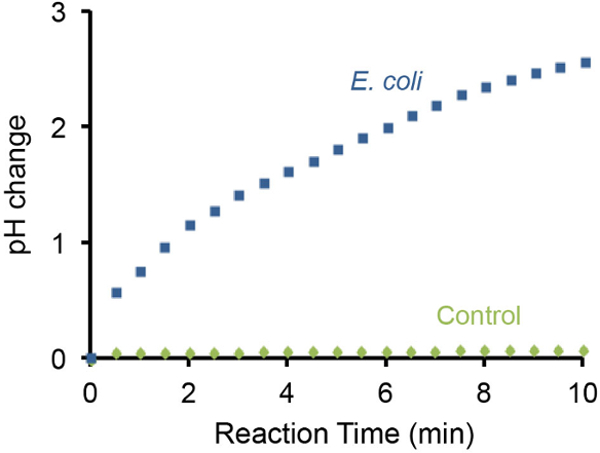 Figure 3:Monitoring pH increase using a pH meter. The change of pH caused by 107
E. coli cells was monitored using a portable pH meter. A test without E. coli was used as a negative control. The presence of 107
E. coli cells in the test solution can increase the basicity by ~3 pH units in 10 min. Please click here to view a larger version of this figure.
Figure 3:Monitoring pH increase using a pH meter. The change of pH caused by 107
E. coli cells was monitored using a portable pH meter. A test without E. coli was used as a negative control. The presence of 107
E. coli cells in the test solution can increase the basicity by ~3 pH units in 10 min. Please click here to view a larger version of this figure.
| Name | Sequence (5'-3') | Note |
| BS1 | BTTTT TTTTT TTTAC TCTTC CTAGC FRQGG TTCGA TCAAG A | B: 5'-Biotin; R: adenine ribonucleotide; F: fluorescein-dT; Q: dabcyl-dT |
| DE1 | GATGT GCGTT GTCGA GACCT GCGAC CGGAA CACTA CACTG TGTGG GGATG GATTT CTTTA CAGTT GTGTG TTGAA CGCTG TGTCA AAAAA AAAA | |
| T1 | GACAA CGCAC ATCTC TTGAT CGAAC C | |
| LD1 | XTTTT TTTTT TTTTT TTGAC ACAGC GTTCA A | X: 5'-NH2 |
Table 1: Sequences of synthetic oligonucleotides.
Discussion
The translation of the action of the RNA cleavage activity of a bacterium-responsive DNAzyme to a litmus test is made possible through the use of urease and magnetic separation, as illustrated by Figure 1. Although the demonstration of the modified litmus test for bacterial detection is done with an E. coli-dependent RNA-cleaving DNAzyme,5,19,20 the design can be generally extended for any RNA-cleaving DNAzyme. Given the great availability of RNA-cleaving DNAzymes for different analytes and various methodologies to isolate new RNA-cleaving DNAzymes from random-sequence pools for new targets, we expect that the modified litmus test platform can be extended to the detection of diverse targets of interest.
The litmus test for E. coli detection can detect 5,000 and 500 cells when the reporting reaction time is set to be 1 and 2 hr, respectively. The popular polymerase chain reaction (PCR) and sandwich enzyme-linked immunosorbent assay (ELISA) methods can achieve detection limits of approximately 104-105 E. coli cells in similar testing times.25,26 Thus, the bacterial litmus test offers comparable detection sensitivity.
Although the bacterial litmus test is easy to carry out and can produce vibrant color changes, several factors can significantly affect test results. Firstly, the quality of urease is very important. We have used urease from different sources and found the test results can vary significantly. We recommend the use of urease from the source specified in the Materials section.
The assembly of DNAzyme/urease/magnetic beads needs special attention. Thorough washing of magnetic beads to remove unhybridized UrDNA is necessary to prevent false-positive results. Care also needs to be taken to avoid the accumulation of residual magnetic beads on the inside surface of the lid of the microfuge tube, which may be difficult to see. Once there, the magnetic beads are no longer subjected to magnetic separation and thus, could carry some unhybridized UrDNA that can lead to false-positive signals in the reporter reaction. It is also important to avoid leaving the microfuge tube on the magnetic rack for longer than 10 min during the magnetic separation step. The beads may aggregate or stick to the microfuge tube, which may reduce the washing efficiency and introduce batch-to-batch inconsistency. Inclusion of 0.01% Tween-20 in the washing solution can improve batch-to-batch consistency and should be implemented.
The magnetic beads are coated with streptavidin, which was used as the anchor to assemble DNAzyme-urease conjugates onto the magnetic beads. Both streptavidin and urease are protein molecules that can be denatured during storage. We typically store the assembled DNAzyme-urease-magnetic beads at 4 °C for up to 4 weeks and make fresh batches regularly to achieve more consistent results.
Care also needs to be taken to avoid accidently taking magnetic beads in the magnetic separation step (step 6.9) following DNAzyme activation. From our experience, cellular debris and other particulates in the solution can reduce the magnetic separation efficiency, and therefore, some magnetic beads may be unintentionally taken out during pipetting. This will result in false-positive results. We recommend the following measures to alleviate the problem: a longer separation time (such as 5-10 min), a slower release of pressure on the pipette to allow gentle withdrawal of the supernatant, and subjecting the supernatant to an additional round of magnetic separation.
Finally, it is important to avoid accident contamination of the reporter stock solution by urease during the course of an experiment where multiple samples are tested. Given the high reactivity of urease, contamination of this nature can lead to false-positive results.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The funding for this research project was provided by the Natural Sciences and Engineering Research Council of Canada (NSERC) via a Discovery Grant to YL.
References
- Daar AS, et al. Top ten biotechnologies for improving health in developing countries. Nat. Genet. 2002;32:229–232. doi: 10.1038/ng1002-229. [DOI] [PubMed] [Google Scholar]
- Newman JD, Turner AP. Home blood glucose biosensors: a commercial perspective. Biosens. Bioelectron. 2005;20:2435–2453. doi: 10.1016/j.bios.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Turner AP. Biosensors: sense and sensibility. Chem. Soc. Rev. 2013;42:3184–3196. doi: 10.1039/c3cs35528d. [DOI] [PubMed] [Google Scholar]
- Tram K, Kanda P, Salena BJ, Huan SY, Li YF. Translating Bacterial Detection by DNAzymes into a Litmus Test. Angew. Chem. Int. Ed. 2014;53:12799–12802. doi: 10.1002/anie.201407021. [DOI] [PubMed] [Google Scholar]
- Ali MM, Aguirre SD, Lazim H, Li Y. Fluorogenic DNAzyme probes as bacterial indicators. Angew. Chem. Int. Ed. 2011;50:3751–3754. doi: 10.1002/anie.201100477. [DOI] [PubMed] [Google Scholar]
- Schlosser K, Li Y. Biologically inspired synthetic enzymes made from DNA. Chem. Biol. 2009;16:311–322. doi: 10.1016/j.chembiol.2009.01.008. [DOI] [PubMed] [Google Scholar]
- Tuerk C, Gold L. Systematic evolution of ligand by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- Breaker RR. Making catalytic DNAs. Science. 2000;290:2095–2096. doi: 10.1126/science.290.5499.2095. [DOI] [PubMed] [Google Scholar]
- Liu J, Cao Z, Lu Y. Functional nucleic acid sensors. Chem. Rev. 2009;109:1948–1998. doi: 10.1021/cr030183i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navani NK, Li Y. Nucleic acid aptamers and enzymes as sensors. Curr. Opin. Chem. Biol. 2006;10:272–281. doi: 10.1016/j.cbpa.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Li J, Lu Y. A highly sensitive and selective catalytic DNA biosensor for lead ions. J. Am. Chem. Soc. 2000;122:10466–10467. [Google Scholar]
- Liu J, Lu Y. A colorimetric lead biosensor using DNAzyme-directed assembly of gold nanoparticles. J. Am. Chem. Soc. 2003;125:6642–6643. doi: 10.1021/ja034775u. [DOI] [PubMed] [Google Scholar]
- Liu Z, Mei SHJ, Brennan JD, Li Y. Assemblage of signaling DNA enzymes with intriguing metal specificity and pH dependence. J. Am. Chem. Soc. 2003;125:7539–7545. doi: 10.1021/ja035208+. [DOI] [PubMed] [Google Scholar]
- Liu J, et al. A catalytic beacon sensor for uranium with parts-per-trillion sensitivity and millionfold selectivity. Proc. Natl. Acad. Sci. USA. 2007;104:2056–2061. doi: 10.1073/pnas.0607875104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang PJ, Vazin M, Liu J. In vitro selection of a new lanthanide-dependent DNAzyme for ratiometric sensing lanthanides. Anal. Chem. 2014;86:9993–9999. doi: 10.1021/ac5029962. [DOI] [PubMed] [Google Scholar]
- Chiuman W, Li Y. Simple fluorescent sensors engineered with catalytic DNA 'MgZ' based on a non-classic allosteric design. PLoS One. 2007;2:e1224. doi: 10.1371/journal.pone.0001224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Lu Y. Using personal glucose meters and functional DNA sensors to quantify a variety of analytical targets. Nat. Chem. 2011;3:697–703. doi: 10.1038/nchem.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre SD, Ali MM, Salena BJ, Li Y. A sensitive DNA enzyme-based fluorescent assay for bacterial detection. Biomolecules. 2013;3:563–577. doi: 10.3390/biom3030563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre SD, Ali MM, Kanda P, Li YF. Detection of Bacteria Using Fluorogenic DNAzymes. J. Vis. Exp. 2012. p. e3961. [DOI] [PMC free article] [PubMed]
- Shen Z, et al. A catalytic DNA activated by a specific strain of bacterial pathogen. Angew. Chem. Int. Ed. 2015;54 doi: 10.1002/anie.201510125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, et al. Highly specific recognition of breast tumors by an RNA-cleaving fluorogenic DNAzyme probe. Anal. Chem. 2015;87:569–577. doi: 10.1021/ac5031557. [DOI] [PubMed] [Google Scholar]
- Sumner JB, Hand DB. Isoelectric point of crystalline urease. J. Am. Chem. Soc. 1929;51:1255–1260. [Google Scholar]
- Karplus PA, Pearson M, Hausinger RP. 70 Years of crystalline urease: What have we learned. Acc. Chem. Res. 1997;30:330–337. [Google Scholar]
- Omiccioli E, Amagliani G, Brandi G, Magnani M. A new platform for Real-Time PCR detection of Salmonella spp., Listeria monocytogenes and Escherichia coli O157 in milk. Food Microbiol. 2009;26:615–622. doi: 10.1016/j.fm.2009.04.008. [DOI] [PubMed] [Google Scholar]
- Cui S, Schroeder CM, Zhang DY, Meng J. Rapid sample preparation method for PCR-based detection of Escherichia coli O157:H7 in ground beef. J. Appl. Microbiol. 2003;95:129–134. doi: 10.1046/j.1365-2672.2003.01951.x. [DOI] [PubMed] [Google Scholar]
- Ibekwe AM, Watt PM, Grieve CM, Sharma VK, Lyons SR. Multiplex fluorogenic real-time PCR for detection and quantification of Escherichia coli O157:H7 in dairy wastewater wetlands. Appl. Environ. Microbiol. 2002;68:4853–4862. doi: 10.1128/AEM.68.10.4853-4862.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strachan NJ, Ogden ID. A sensitive microsphere coagulation ELISA for Escherichia coli O157:H7 using Russell's viper venom. FEMS Microbiol Lett. 2000;186:79–84. doi: 10.1111/j.1574-6968.2000.tb09085.x. [DOI] [PubMed] [Google Scholar]
- de Boer E, Beumer RR. Methodology for detection and typing of foodborne microorganisms. Int. J. Food Microbiol. 1999;50:119–130. doi: 10.1016/s0168-1605(99)00081-1. [DOI] [PubMed] [Google Scholar]
- Gracias KS, McKillip JL. A review of conventional detection and enumeration methods for pathogenic bacteria in food. Can. J. Microbiol. 2004;50:883–890. doi: 10.1139/w04-080. [DOI] [PubMed] [Google Scholar]