

Abstract
Over the last decade there has been a resurgence in the use of plant protoplasts that range from model species to crop species, for analysis of signal transduction pathways, transcriptional regulatory networks, gene expression, genome-editing, and gene-silencing. Furthermore, significant progress has been made in the regeneration of plants from protoplasts, which has generated even more interest in the use of these systems for plant genomics. In this work, a protocol has been developed for automation of protoplast isolation and transformation from a 'Bright Yellow' 2 (BY-2) tobacco suspension culture using a robotic platform. The transformation procedures were validated using an orange fluorescent protein (OFP) reporter gene (pporRFP) under the control of the Cauliflower mosaic virus 35S promoter (35S). OFP expression in protoplasts was confirmed by epifluorescence microscopy. Analyses also included protoplast production efficiency methods using propidium iodide. Finally, low-cost food-grade enzymes were used for the protoplast isolation procedure, circumventing the need for lab-grade enzymes that are cost-prohibitive in high-throughput automated protoplast isolation and analysis. Based on the protocol developed in this work, the complete procedure from protoplast isolation to transformation can be conducted in under 4 hr, without any input from the operator. While the protocol developed in this work was validated with the BY-2 cell culture, the procedures and methods should be translatable to any plant suspension culture/protoplast system, which should enable acceleration of crop genomics research.
Keywords: Genetics, Issue 115, Tobacco, Protoplasts, Transformation, Enzymatic Digestion, High-throughput Screening, Automation, Robotics
Introduction
In recent years there has been significant impetus placed on the design of transgenic crops to overcome various diseases1, endow herbicide resistance2, confer drought3,4 and salt tolerance5, prevent herbivory6, increase biomass yield7, and decrease cell wall recalcitrance8. This trend has been aided by the development of new molecular tools for generating transgenic plants, including genome-editing using CRISPR and TALENs9, and gene silencing through dsRNA10, miRNA11, and siRNA12. While these technologies have simplified the generation of transgenic plants, they have also created a bottleneck, where the sheer number of transgenic plants generated cannot be screened using traditional systems that rely on plant regeneration. Related to this bottleneck, while silencing and genome-editing constructs can be rapidly inserted into plants, many of the targeted traits fail to produce the desired effect, which is often not discovered until plants are analyzed in the greenhouse. In this work, we have developed a method for rapid, automated, high-throughput screening of plant protoplasts, specifically to address the current bottleneck in early screening of large numbers of genome-editing and gene silencing targets.
The use of protoplasts, as opposed to intact plant cells, has several advantages for the development of an automated platform. First, protoplasts are isolated after digestion of the plant cell wall, and with this barrier no longer present, transformation efficiency is increased13. In intact plant cells there are only two well established methods for transformation, biolistics14 and Agrobacterium-mediated transformation15. Neither of these methods can be easily translated to liquid handling platforms, as biolistics requires specialized equipment for transformation, whereas Agrobacterium-mediated transformation requires co-culture and subsequent removal of the bacteria. Neither are amenable for high throughput methods. In the case of protoplasts, transformation is routinely conducted using polyethylene glycol (PEG)-mediated transfection16, which only requires several solution exchanges, and is ideally suited for liquid handling platforms. Second, protoplasts, by definition, are single-cell cultures, and thus the problems associated with clumping and chain formation in plant cell cultures, are not observed in protoplasts. In terms of rapid screening using a plate-based spectrophotometer, clumping of cells, or cells in multiple planes will lead to difficulty in acquiring consistent measurements. Since protoplasts are also denser than their culture media, they sediment to the bottom of wells, forming a monolayer, which is conducive for plate-based spectrophotometry. Finally, while plant cell suspension cultures are primarily derived from callus17, protoplasts can be harvested from a number of plant tissues, leading to the ability to identify tissue-specific expression. For example, the ability to analyze root- or leaf-specific expression of a gene can be very important to phenotype prediction. For these reasons, the protocols developed in this work were validated using protoplasts isolated from the widely-used tobacco (Nicotiana tabacum L.) 'Bright Yellow' 2 (BY-2) suspension culture.
The BY-2 suspension culture has been described as the "HeLa" cell of higher plants, owing to its ubiquitous use in molecular analysis of plant cells18. Recently, BY-2 cells have been used to study the effects of plant stressors19-22, intracellular protein localization23,24, and basic cell biology25-27 demonstrating the broad utility of these cultures in plant biology. An additional advantage of BY-2 cultures is the ability to synchronize the cultures with aphidicolin, which can lead to enhanced reproducibility for gene expression studies28. Furthermore, methods have been developed for the extraction of BY-2 protoplasts using low-cost enzymes29,30, as enzymes traditionally used for generating protoplasts are cost prohibitive for high-throughput systems. As such, the protocol described below has been validated using the BY-2 suspension culture, but it should be amendable to any plant cell suspension culture. Proof-of-concept experiments are performed using an orange fluorescence protein (OFP) reporter gene (pporRFP) from the hard coral Porites porites31 under the control of the CAMV 35S promoter.
Protocol
1. Establishment of Suspension Cell Cultures
Prepare liquid BY-2 media by adding 4.43 g Linsmaier & Skoog Basal media, 30 g of sucrose, 200 mg KH2PO4, and 200 µg of 2,4-dichlorophenoxyacetic acid (2,4-D) to 900 ml of distilled water and pH to 5.8 with 0.1 M KOH. After adjusting pH, adjust final volume to 1,000 ml with distilled water and autoclave. Media can be stored up to 2 weeks at 4 °C.
Inoculate a 250 ml Erlenmeyer flask with 100 ml of liquid BY-2 media and a single piece of BY-2 callus (>1 cm in diameter) grown on solid BY-2 media (liquid BY-2 media with the addition of 1% agar) and seal with aluminum foil. Incubate culture at 28-30 °C with shaking for 5 days. NOTE: BY-2 callus is maintained on solid media for long term storage, as liquid cultures will rapidly overgrow. Culture volume can be adjusted as necessary, typically a 100 ml culture will be capable of loading thirty-three 6-well plates.
Transfer 2 ml of log phase BY-2 culture to 98 ml of fresh BY-2 media and incubate for 5-7 days at 28-30 °C with shaking. NOTE: Sub-culturing of established liquid cultures can be carried out regularly using 1:100 dilutions of 5-7 day old cultures, rather than inoculation with callus.
Mix the culture thoroughly, as cells will rapidly settle, and transfer 6 ml of the cell culture to a 15 ml conical bottom centrifuge tube and let the cells settle for at least 10 min. Adjust the packed cell volume to 50% of the total volume by removing supernatant.
Shake tube by inverting and pipet 500 µl into each well of a 6-well plate for digestion. Use wide-bore pipets to transfer the cells at this stage, as they are dense and will clog standard pipet tips.
2. Protoplast Isolation
Turn on all components of the robotic system (Figure 1A) and open the plate mover task scheduling software. The task scheduling program integrates the microplate mover with the other equipment to enable transfer of plates between each piece of equipment.
- Prior to experimentation, define the technical specifications for labware (e.g., plates, lids) used in the protocol, as well as starting and ending positions for each piece of labware, in the plate mover software, as follows:
- Click Setup from the main tool bar in the plate mover software and select the Manage Container Types command.
- If the container type is listed in the existing container type library, then select the appropriate container.
- If the container type is not listed in the existing container type library then click Add to specify a new container type.
- After adding a new container type, insert the dimensions of the new container (height, width, stacking dimensions, and gripper offset) into the appropriate tabs and click Save.
- If the correct dimensions are not available from the manufacturer, then accurately determine all dimensions to the nearest millimeter using calipers. Errors in measuring the container dimensions will result in failure of the plate mover.
- Repeat steps 2.2.2 through 2.2.4.1 for all container types that the plate mover will encounter. After completion of this step for all labware, it is necessary to define the start and end location for each container (step 2.2.6). NOTE: For this protocol the labware includes the 6-well plate, the deep-well plate, and the 96-well fluorescent screening plate.
- To define the start and end position of each container, click the Start/End tab in a specific protocol. First, select the start position of each container. Next, check the box for Lidded/Unlidded at both the start and end position. Repeat for all containers. NOTE: If the container will be left on another piece of equipment (instead of the plate mover) the end location can be left blank.
- At this stage manually load all plates into the starting position for the entire workflow. Load the 96-well fluorescent screening plates into Hotel 2, 6-well plates with BY-2 cells into Hotel 3, a 96 deep-well plate that will be used for transformation onto plate heater/chiller nest 2, and a 50 ml conical tube containing the enzyme solution (see Supplemental File, Section 1.2.2) onto the multi-mode dispenser.
- If transformation will be carried out in the protocol, pre-load the 96 deep-well plate with 10 µl of plasmid DNA per well (1 µg/µl, A260/280 > 1.8) containing the OFP reporter construct and incubate on the plate heater/chiller at 4 °C. Each well will be used for a single transformation, thus the number of wells filled will depend on the experimental setup.
- Load all supplies and plates on the automated liquid handling platform in the designated locations and define the position of each item in the automation control software (Figure 1B).
- Load a 96-well plate preloaded with 200 µl of 40% PEG in MMg (0.4 M mannitol, 100 mM MgCl2, 4 mM MES, pH 5.7) in column 1, 200 µl of propidium iodide (PI, 1 µg/ml) in column 2, and 200 µl of ethanol in column 3 (nest 2), and a box of pipette tips in nest 8.
- To define the location of the labware in the automated liquid handling platform's software, select Tools from the menu and click Labware Editor.
- Select the type of labware from the pulldown menu or define a new labware type using the New Labware button.
- Define the position of each piece of labware selected by selecting the Main Protocol tab and clicking Configure Labware. Ensure that each piece of labware placed on the automated liquid handling platform is defined prior to running the protocol.
- To trigger the automated protocols, click the Profile Explorer window and select the name of the protocol to be run (Protoplast Isolation, Cell Count, and PEG-mediated Transformation).
- Click Run to initialize all devices that will be used in the protocol.
- Finally, in the Work Explorer window, click Add Work Unit to verify all containers/labware in the system and begin the automated protocol. NOTE: Full descriptions of the automated protocols are contained in the Supplemental File.
3. Establish Standard Curve for Cell Counts
Manually concentrate protoplasts at a concentration of 1 x 106 protoplasts/ml in a volume of 1 ml. Centrifuge protoplasts at 1,000 x g for 10 min, and remove supernatant.
Manually add 900 µl of 70% ethanol (maintained at 4 °C) to the protoplast pellet and re-suspend the pellet. Incubate for 10 min on ice to fix cells.
Add 100 µl of PI (1 µg/ml) to the protoplasts to label the nuclei and allow detection by the plate reader.
Load 80 µl of liquid BY-2 media into each column and row of the 96-well fluorescent screening plate starting at column 2.
In column 1 of a 96-well fluorescent screening plate, add 70 µl of protoplasts and 50 µl BY-2 media to each well in the column.
- Place the plate in nest 6 of the automated liquid handling platform, and run a 2-fold serial dilution.
- To conduct a serial dilution on the automated liquid handling platform, click Launch Serial Dilution Wizard and adjust the transfer volume (60 µl), number of mixes and volume (2, 70 µl), initial wells (Column 1, Rows A-F), dilution wells (Columns 2-11, Rows A-F), and aspirate and dispense properties (0.5 mm from well bottom).
- After setting the parameters, save and run the protocol. NOTE: Since all protoplasts are labeled with PI (due to fixation of cells), the fluorescence is proportionate to the protoplast concentration.
- After the serial dilution is completed, remove the plate from nest 9 and insert into the plate reader and run the standard curve protocol in the plate reader software. NOTE: A standard curve will then be generated by comparing the fluorescence from PI (excitation 536 nm/emission 620 nm) in each well with the known protoplast concentration.
Upon completion of the plate reader protocol, remove the plate and collect any transgenic cells for autoclaving or other de-vitalization procedures as defined in biosafety protocols'.
4. Microscopic Analysis of Transformation
Remove plate with transformed protoplasts (the product of Automated Protocol 3, Supplemental File) from Hotel 1 of the robotic system.
Turn on inverted microscope, camera, and fluorescent lamp.
Select the 10X objective for initial focusing on the protoplasts, turn on the halogen lamp, and close the shutter for the fluorescent lamp.
Load plate onto microscopic system and focus on the protoplasts using brightfield.
After focusing on protoplasts, turn off the halogen bulb and open the shutter for the fluorescent lamp.
Select the CY3/TRITC filter set for visualization of the pporRFP protein expressed by the transgenic protoplasts.
Scan each well to determine the number of protoplasts expressing the pporRFP fluorescent marker.
Calculate the transformation efficiency as the total number of protoplasts expressing the fluorescent marker the total number of protoplasts.
Collect any plates containing transgenic cells in approved biosafety bags for autoclaving or other de-vitalization procedures as defined in biosafety protocols'.
Representative Results
In the current study, the doubling rate of BY-2 varied from 14-18 hr dependent on the temperature at which the cultures were incubated, consistent with previous reports of a mean cell cycle length of 15 hr. With this doubling rate, a 1:100 starting inoculum was used to initiate cultures, leading to cultures with a packed cell volume (PCV) of 50% in 5-7 days. In the current protocol, in which cultures were grown in 200 ml of media, a PCV of 100 ml was generated in 7 days, which provided enough cells to fill 33 6-well plates. In terms of protoplast yield, digestion at the conditions specified in the protocol generated 4.70 x 105 ± 5.77 x 103 protoplasts per well, resulting in 2.82 x 106 protoplasts per 6-well plate (Figure 2A). Based on these data, 214 96-well plates (70 µl of protoplasts per well) could be loaded from a single 6-well plate digestion, with the potential for two 6-well plates to be digested simultaneously, utilizing the two plate heater/chiller stations. Thus, the maximum number of protoplasts that can be generated during a single three-hour digestion protocol is 5.64 x 106.
While the total number of protoplasts generated per digestion protocol provides one metric for the system, it is also necessary to evaluate the loss in protoplast concentration as the protoplasts are transferred through the different steps of the protocol. As such, the concentration of protoplasts was evaluated after each transfer step in the protocol. After the digestion protocol, 1.15 x 104 ± 1.35 x 102 protoplasts were transferred to each well of the 96 deep-well plates, if conducting the transformation protocol, or the 96-well fluorescent screening plate (Figure 2B). Once the transformation protocol was completed, 1.23 x 103 ± 4.66 x 101 protoplasts were transferred to the 96-well fluorescent screening plate (Figure 2C). In addition to the number of protoplasts transferred, it was important to determine the viability of the protoplasts to ensure that multiple pipetting stages did not damage the protoplasts.
Based on the results from the viability assay, 95.1 ±0.04% of protoplasts were viable after digestion, 92.1 ± 0.06% after transfer to the 96-well fluorescent screening plate, and 91.7 ± 16.67% after the transformation protocol. Despite multiple pipetting steps, there was no significant difference (p > 0.44) between any of the samples. In addition to this measure of viability, a protocol was developed for determining the concentration of protoplasts at any stage of the procedure by measuring the total number of protoplasts stained with PI. As shown in Figure 3, a standard curve was generated by measuring the fluorescence intensity for known concentrations of protoplasts from the system. The standard curve was a good fit (R2 = 0.967) with the total number of protoplasts spanning the range from 0 to 30,000 protoplasts per well. To validate the standard curve, the fluorescence intensity of samples with a known number of protoplasts (8,125 and 24,375 calculated using a hemocytometer) was obtained and the number of protoplasts calculated from the equation of the standard curve. Based on these results, the standard curve predicted 9,560 and 28,200 protoplasts in each well, a 15 and 14% error. Considering that there is also variability in measuring the cell number using a hemocytometer, this error was deemed acceptable.
In addition to the results of the cell number and viability, the transformation protocol was successful in generating transgenic cells that express OFP (Figure 2D-F). Unfortunately the protocol used resulted in low, ~2%, transformation efficiency, due to the large 16,028 bp binary plasmid used in this study and since the protocol was not optimized specifically for BY-2 protoplasts. Optimization of the transformation protocol, in terms of reagents and conditions specifically for BY-2 protoplasts, is expected to increase the transformation efficiency, as elucidated in the Discussion. In terms of this protocol, the transformation procedure was designed to demonstrate the proof-of-concept path from protoplast isolation to transformation, and was successful in achieving this goal.
After successfully validating all of the individual procedures, metrics were obtained for the timecourse of the protocol from protoplast isolation to transformation. As demonstrated in Figure 4, the major investment of time is spent during the digestion stage, in which 3 hr was required for complete digestion of the cell wall and release of the protoplasts. During this procedure, 18 loops are run between the plate shaker and plate heater/chiller 1 incubation station, with the plate mover moving the plate between the two stations. The transfer time between the plate shaker and plate heater/chiller 1 takes just 8.9 sec, and ensures that the majority of the digestion procedure is spent incubating and shaking. The total time from initiation of the procedure to complete loading of the enzymes by the multi-mode dispenser is 2.2 min. Overall, the complete protoplast isolation protocol is completed in 3 hr and 22.8 min. Based on these results, 88% of the protoplast isolation protocol is spent digesting. After completion of the protoplast isolation protocol, the transformation protocol is initiated, and completed within 27.5 min. Similar to the protoplast isolation protocol, 72% of the transformation procedure is spent incubating the reaction. The only other step in the protocol that has a significant time component (>10% of the total procedure time) is initialization of the protocol by the automated liquid handling platform, which accounts for 14% of the total time in the procedure. In this way, 86% of the protocol is spent in initialization and incubation, with only 14% of the time spent on pipetting and transfer steps. The complete duration of protoplast isolation and transformation was 3 hr, 50 min, and 53 sec, with no external input required from the operator.
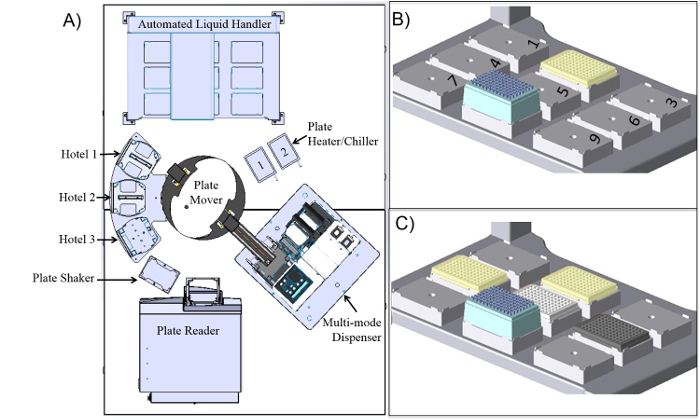 Figure 1:Schematic of the robotic platform and configuration of the automated liquid handling platform during the protocol. (A) The robotic system is composed of 3 plate stacks/hotels that can be accessed by the plate mover. Hotel 1 is the designated lidding/delidding station, Hotel 2 is the 96-well fluorescent plate stack, and Hotel 3 is the 6-well plate stack. The system has two liquid handlers, an automated liquid handling platform, and a nozzle-based multi-mode dispenser. For heating/cooling the robotic platform has two plate heating/chilling units, along with a plate shaker for mixing the plate. Finally, the system has a monochromator-based plate reader for measuring fluorescence. All components are housed in a custom built Biosafety Cabinet. (B) Starting configuration of the automated liquid handling platform with nest 2 occupied by the reagent plate and the tip box placed on nest 8. (C) Configuration of automated liquid handling platform prior to calling an experimental protocol. A deep-well plate is located on nest 4, a 96-well fluorescent screening plate is located on nest 6, and the 6-well plate containing protoplasts is located on nest 5. Please click here to view a larger version of this figure.
Figure 1:Schematic of the robotic platform and configuration of the automated liquid handling platform during the protocol. (A) The robotic system is composed of 3 plate stacks/hotels that can be accessed by the plate mover. Hotel 1 is the designated lidding/delidding station, Hotel 2 is the 96-well fluorescent plate stack, and Hotel 3 is the 6-well plate stack. The system has two liquid handlers, an automated liquid handling platform, and a nozzle-based multi-mode dispenser. For heating/cooling the robotic platform has two plate heating/chilling units, along with a plate shaker for mixing the plate. Finally, the system has a monochromator-based plate reader for measuring fluorescence. All components are housed in a custom built Biosafety Cabinet. (B) Starting configuration of the automated liquid handling platform with nest 2 occupied by the reagent plate and the tip box placed on nest 8. (C) Configuration of automated liquid handling platform prior to calling an experimental protocol. A deep-well plate is located on nest 4, a 96-well fluorescent screening plate is located on nest 6, and the 6-well plate containing protoplasts is located on nest 5. Please click here to view a larger version of this figure.
 Figure 2:Representative images of BY-2 protoplasts after each transfer and after transformation with the orange fluorescent protein reporter gene. (A-C) Micrographs of BY-2 protoplast density in 6-well plate, 96-well plate after transfer of 70 µl from the 6-well plate, and 96-well fluorescent screening plate post-transformation. (D) Brightfield micrograph of BY-2 protoplasts after transformation. (E) Fluorescent BY-2 protoplast expressing the OFP gene visualized using a Cy3/TRITC filter set. (F) Overlay of brightfield and fluorescent micrographs demonstrating the low efficiency of transformation. Scale bar = 50 µm. Please click here to view a larger version of this figure.
Figure 2:Representative images of BY-2 protoplasts after each transfer and after transformation with the orange fluorescent protein reporter gene. (A-C) Micrographs of BY-2 protoplast density in 6-well plate, 96-well plate after transfer of 70 µl from the 6-well plate, and 96-well fluorescent screening plate post-transformation. (D) Brightfield micrograph of BY-2 protoplasts after transformation. (E) Fluorescent BY-2 protoplast expressing the OFP gene visualized using a Cy3/TRITC filter set. (F) Overlay of brightfield and fluorescent micrographs demonstrating the low efficiency of transformation. Scale bar = 50 µm. Please click here to view a larger version of this figure.
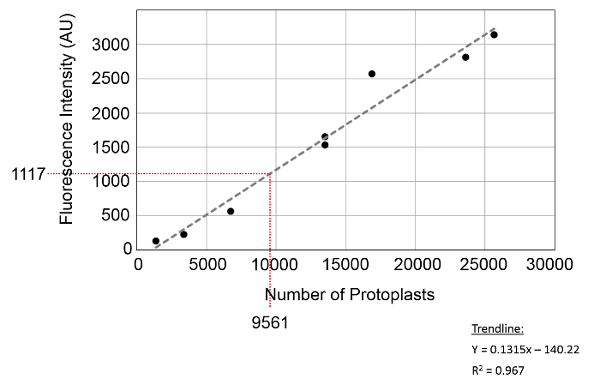 Figure 3:Standard curve for calculating the number of protoplasts. The number of protoplasts in a given well can be calculated by comparing the fluorescence intensity, in arbitrary units (AU), to the equation for the standard curve. Validation of the protoplast number was compared between the standard curve and hemocytometer at the point indicated on the graph, with 9,561 protoplasts identified from the plate reader, and 8,125 counted on the hemocytometer. Overall, an error of 15% was found between the two techniques. Please click here to view a larger version of this figure.
Figure 3:Standard curve for calculating the number of protoplasts. The number of protoplasts in a given well can be calculated by comparing the fluorescence intensity, in arbitrary units (AU), to the equation for the standard curve. Validation of the protoplast number was compared between the standard curve and hemocytometer at the point indicated on the graph, with 9,561 protoplasts identified from the plate reader, and 8,125 counted on the hemocytometer. Overall, an error of 15% was found between the two techniques. Please click here to view a larger version of this figure.
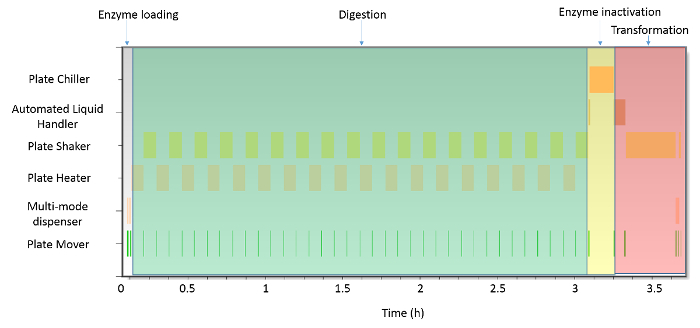 Figure 4:Gantt chart for the described procedure for the production of BY-2 protoplasts and genetic transformation. The majority of the protocol, ~3 hr, is spent on digestion of the BY-2 cells to generate the protoplasts. Loading of the enzyme solution and inactivation require only 12% of the overall procedure time. Similarly, incubation of the transformation reaction on the plate shaker accounts for 72% of the transformation protocol. Overall, the procedure from protoplast isolation until genetic transformation is achieved is under 4 hr (3 hr, 50 min, and 53 sec). Please click here to view a larger version of this figure.
Figure 4:Gantt chart for the described procedure for the production of BY-2 protoplasts and genetic transformation. The majority of the protocol, ~3 hr, is spent on digestion of the BY-2 cells to generate the protoplasts. Loading of the enzyme solution and inactivation require only 12% of the overall procedure time. Similarly, incubation of the transformation reaction on the plate shaker accounts for 72% of the transformation protocol. Overall, the procedure from protoplast isolation until genetic transformation is achieved is under 4 hr (3 hr, 50 min, and 53 sec). Please click here to view a larger version of this figure.
Supplemental File: Automated protocols for protoplast isolation, cell counting, and PEG-mediated transformation. Complete protocols developed for use in the robotic platform to accomplish each of the tasks listed above are included in this file. Please click here to download this file.
Discussion
The protocol described above has been successfully validated for protoplast isolation, enumeration, and transformation using the BY-2 tobacco suspension cell culture; however, the protocol could easily be extended to any plant suspension culture. At present, protoplast isolation and transformation has been achieved in numerous plants, including maize (Zea mays)10, carrot (Daucus carota)32, poplar (Populus euphratica)33, grape (Vitis vinifera)34, oil palm (Elaeis guineensis)35, lettuce (Lactuca sativa)36, mustard (Brassica juncea)37, rice (Oryza sativa)38, and switchgrass (Panicum virgatum)39,40. While variation in the culture and digestion conditions occur from species to species, in terms of the protocol developed in this work, replacing culture media or altering the conditions of digestion should not change the fundamental protocol. To adjust to different species, culture media and enzymes required for digestion are loaded by the operator prior to initialization of the protocol, thus swapping these components is trivial. As an added benefit, the protocol is amendable to screening multiple digestion conditions with the yield of protoplasts measured by the system. This is an essential component, as the use of low-cost enzymes are necessary for high-throughput applications, thus it is important to minimize the amount of enzymes used in digestion.
Similar to culture and digestion, modifications to the transformation protocol would be required when adopting the system for transformation of protoplasts isolated from different plant species. Considerable work has been conducted on optimization of PEG-mediated transformation in plant protoplasts16,35, with numerous factors affecting the efficiency of transformation. In the current work one limiting factor affecting the transformation efficiency was the large size of the binary test plasmid, 16,028 bp, which was used for the purposes of modeling a genome-editing plasmid size. In previous studies on PEG-mediated transformation of BY-2 protoplasts, 40-60% transformation has been achieved; however, these studies used small 5,000 bp plasmids41,42. The primary modifications to PEG-mediated transformation among species include the concentration of PEG and MgCl2, amount and concentration of DNA, and reaction duration. As the PEG and MgCl2 solutions are introduced into the system on the reagent plate, and the DNA is pre-loaded into the deep-well plate, these conditions can be easily modified in the protocol described above. Additionally, increasing or decreasing the incubation time, along with modification of mixing speeds, can be precisely controlled by the robotic platform. The current limitation of the transformation protocol is the lack of screening using the plate reader to detect the fluorescent reporter, due to the low efficiency of PEG-mediated transformation in BY-2 protoplasts. In other species, such as Arabidopsis thaliana and maize16, PEG-mediated transformation is 40-90% efficient. In such systems transformants could be rapidly characterized using the plate reader on the robot. Thus, it would be possible to identify wells with positive transformants, and also quantify the level of expression. For applications, such as promoter screening, this feature would increase the utility of the current protocol, and will be added in later iterations of the system.
In addition to optimization of the transformation efficiency for BY-2 protoplasts, several modifications may increase the efficiency and overall throughput of the system. Previous studies on automation of protoplast transformation allowed protoplasts to settle to the bottom of wells, prior to the addition of wash solutions43. In the current protocol, protoplasts were mixed on the plate shaker to prevent settling and keep the protoplasts in suspension using flat bottom well plates. Furthermore, during transformation, protoplasts were not allowed to settle in the deep-well plates, to reduce the exposure to PEG, which can be toxic over prolonged exposure. As such, the protoplasts were diluted ~10-fold with W5 solution when transferred from the deep-well plate to the 96-well fluorescent screening plate, resulting in 1.23 x 103 ± 4.66 x 101 protoplasts per well. In order to increase the number of protoplasts transferred after transformation, an incubation step could be added to allow the protoplasts to settle, and the supernatant removed followed by addition of a smaller volume of liquid BY-2 media. Additionally, if a plate-based centrifuge was added to the system, this step could be achieved more rapidly. Another modification that could add functionality to the system would be the use of a live/dead fluorescence assay to determine the viability of the isolated protoplasts prior to transformation. In the current protocol, fluorescein diacetate (FDA) was tested as a live dye; however, the BY-2 protoplast background fluorescence obscured the FDA signal. If a more stable viability dye was used, then protoplast viability could be determined as the ratio of red (PI) to green (Calcein AM, etc.) fluorescence, and standardized similar to the cell counting protocol. These modifications would add further functionality to the system, and will be included in future protocols.
With regards to the current protocol, there are several critical steps that must be followed to ensure the success of the protocol. First, the age/health of the BY-2 cultures is essential to guarantee that viable protoplasts can be isolated for subsequent steps. As explained in step 1.3, by transferring a log phase culture to a fresh flask at a 1:50 ratio of cells to fresh media, and allowing the culture to grow for 5-7 days, the maximum number of viable protoplasts can be isolated. Allowing the cultures to grow for longer or shorter durations using the specified inoculum will significantly reduce the protoplast yield, and cause failure of subsequent protocols. A second critical step is step 1.5, since the use of standard pipet tips, instead of the wide-bore pipet tips specified, will cause the tips to clog and lead to the incorrect volumes being transferred. With regard to the setup of the robotic system, the arrangement of the plates on the automated liquid handling platform, step 2.4, is critical, with a modified setup preventing the head of the liquid handler from reaching all wells on the 6-well plate. Due to the design of the automated liquid handling platform, the 6-well plate must be placed in nest 5, or protocols selected for this instrument will fail. Similarly, failure to correctly define the labware in the plate mover software, step 2.2, will cause the instrument to fail to grab, or drop the labware resulting in termination of the protocol. For the cell count protocol, the critical step involves the fixation of the cells with ethanol, step 3.2. If cells are not fixed prior to labeling with PI, only nonviable cells will be labeled, instead of the entire population of protoplasts, and it will not be possible to count the number of protoplasts isolated. The final critical step, step 4.6, involves selection of the fluorescent filters for microscopic analysis of the expression of the pporRFP fluorescent marker. The use of other "red" filter sets allows for background fluorescence from chlorophyll, significantly increasing the signal in all chloroplast containing protoplasts, preventing calculation of the transformation efficiency. Carefully adhering to the specifications in these critical steps guarantees the greatest opportunity for success and the generation of the most reproducible dataset.
Disclosures
The authors declare that they have no competing financial interest.
Acknowledgments
This research was supported by Advanced Research Projects Agency - Energy (ARPA-E) Award No. DE-AR0000313.
References
- Atkinson HJ, Lilley CJ, Urwin PE. Strategies for transgenic nematode control in developed and developing world crops. Curr. Opin. Biotech. 2012;23(2):251–256. doi: 10.1016/j.copbio.2011.09.004. [DOI] [PubMed] [Google Scholar]
- Duke SO. Perspectives on transgenic, herbicide-resistant crops in the United States almost 20 years after introduction. Pest Manag. Sci. 2015;71(5):652–657. doi: 10.1002/ps.3863. [DOI] [PubMed] [Google Scholar]
- Mir R, Zaman-Allah M, Sreenivasulu N, Trethowan R, Varshney R. Integrated genomics, physiology and breeding approaches for improving drought tolerance in crops. Theor. Appl. Genet. 2012;125(4):625–645. doi: 10.1007/s00122-012-1904-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Xiong L. Genetic engineering and breeding of drought-resistant crops. Annu. Rev. Plant Bio. 2014;65:715–741. doi: 10.1146/annurev-arplant-050213-040000. [DOI] [PubMed] [Google Scholar]
- Marco F, et al. Plant Biology and Biotechnology. Springer; 2015. pp. 579–609. [Google Scholar]
- Edgerton MD, et al. Transgenic insect resistance traits increase corn yield and yield stability. Nat. Biotechnol. 2012;30(6):493–496. doi: 10.1038/nbt.2259. [DOI] [PubMed] [Google Scholar]
- Vanhercke T, et al. Metabolic engineering of biomass for high energy density: oilseed-like triacylglycerol yields from plant leaves. Plant Biotech. J. 2014;12(2):231–239. doi: 10.1111/pbi.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter HL, et al. Two-year field analysis of reduced recalcitrance transgenic switchgrass. Plant Biotech. J. 2014;12(7):914–924. doi: 10.1111/pbi.12195. [DOI] [PubMed] [Google Scholar]
- Xing HL, et al. A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biol. 2014;14(1):327. doi: 10.1186/s12870-014-0327-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Yao D, Lin F, Jiang M. PEG-mediated transient gene expression and silencing system in maize mesophyll protoplasts: a valuable tool for signal transduction study in maize. Acta Physio. Plant. 2014;36(5):1271–1281. [Google Scholar]
- Martinho C, et al. Dissection of miRNA pathways using Arabidopsis mesophyll protoplasts. Mol. Plant. 2015;8(2):261–275. doi: 10.1016/j.molp.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Bart R, Chern M, Park CJ, Bartley L, Ronald PC. A novel system for gene silencing using siRNAs in rice leaf and stem-derived protoplasts. Plant Methods. 2006;2(1):13. doi: 10.1186/1746-4811-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F, Zhu J, Liu H-L. Protoplasts: a useful research system for plant cell biology, especially dedifferentiation. Protoplasma. 2013;250(6):1231–1238. doi: 10.1007/s00709-013-0513-z. [DOI] [PubMed] [Google Scholar]
- Martin-Ortigosa S, Valenstein JS, Lin VSY, Trewyn BG, Wang K. Nanotechnology meets plant sciences: Gold functionalized mesoporous silica nanoparticle mediated protein and DNA codelivery to plant cells via the biolistic method. Adv. Funct. Mater. 2012;22(17):3529–3529. [Google Scholar]
- Křenek P, et al. Transient plant transformation mediated by Agrobacterium tumefaciens: Principles, methods and applications. Biotechnol. Adv. 2015. [DOI] [PubMed]
- Yoo SD, Cho YH, Sheen J. Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat. Protoc. 2007;2(7):1565–1572. doi: 10.1038/nprot.2007.199. [DOI] [PubMed] [Google Scholar]
- Mustafa NR, de Winter W, van Iren F, Verpoorte R. Initiation, growth and cryopreservation of plant cell suspension cultures. Nat. Protoc. 2011;6(6):715–742. doi: 10.1038/nprot.2010.144. [DOI] [PubMed] [Google Scholar]
- Nagata T, Nemoto Y, Hasezawa S. Tobacco BY-2 cell line as the "HeLa" cell in the cell biology of higher plants. Int. Rev. Cytol. 1992;132(1):1–30. [Google Scholar]
- Centomani I, et al. Involvement of DNA methylation in the control of cell growth during heat stress in tobacco BY-2 cells. Protoplasma. 2015. pp. 1–9. [DOI] [PubMed]
- Sgobba A, et al. Cyclic AMP deficiency stimulates a stress condition in tobacco BY-2 cells. BioTechnologia. 2013;94(2) [Google Scholar]
- Väisänen EE, et al. Coniferyl alcohol hinders the growth of tobacco BY-2 cells and Nicotiana benthamiana seedlings. Planta. 2015;242(3):747–760. doi: 10.1007/s00425-015-2348-7. [DOI] [PubMed] [Google Scholar]
- Ortiz-Espìn A, et al. Over-expression of Trxo1 increases the viability of tobacco BY-2 cells under H2O2 treatment. Ann. Botany. 2015;116(4):571–582. doi: 10.1093/aob/mcv076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, et al. cis-Golgi proteins accumulate near the ER exit sites and act as the scaffold for Golgi regeneration after brefeldin A treatment in tobacco BY-2 cells. Mol. Bio. Cell. 2012;23(16):3203–3214. doi: 10.1091/mbc.E12-01-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison SL, Nebenführ A. Live-cell imaging of dual-labeled Golgi stacks in tobacco BY-2 cells reveals similar behaviors for different cisternae during movement and brefeldin A treatment. Mol. Plant. 2011;4(5):896–908. doi: 10.1093/mp/ssr067. [DOI] [PubMed] [Google Scholar]
- de Pinto MC, et al. S-nitrosylation of ascorbate peroxidase is part of programmed cell death signaling in tobacco Bright Yellow-2 cells. Plant Physiol. 2013;163(4):1766–1775. doi: 10.1104/pp.113.222703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanamata S, et al. In vivo imaging and quantitative monitoring of autophagic flux in tobacco BY-2 cells. Plant Signa. Behav. 2013;8(1):22510. doi: 10.4161/psb.22510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai A, Takusagawa M, Nio A, Sawai Y. Cytological Studies on proliferation, differentiation, and death of BY-2 cultured tobacco cells. Cytologia. 2015;80(2):133–141. [Google Scholar]
- Yasuhara H, Kitamoto K. Aphidicolin-induced nuclear elongation in tobacco BY-2 cells. Plant Cell Physiol. 2014;55(5):913–927. doi: 10.1093/pcp/pcu026. [DOI] [PubMed] [Google Scholar]
- Buntru M, Vogel S, Stoff K, Spiegel H, Schillberg S. A versatile coupled cell-free transcription-translation system based on tobacco BY-2 cell lysates. Biotechnol. Bioeng. 2015;112(5):867–878. doi: 10.1002/bit.25502. [DOI] [PubMed] [Google Scholar]
- Buntru M, Vogel S, Spiegel H, Schillberg S. Tobacco BY-2 cell-free lysate: an alternative and highly-productive plant-based in vitro translation system. BMC Biotechnol. 2014;14(1):37. doi: 10.1186/1472-6750-14-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alieva NO, et al. Diversity and evolution of coral fluorescent proteins. PLoS ONE. 2008;3(7):2680. doi: 10.1371/journal.pone.0002680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maćkowska K, Jarosz A, Grzebelus E. Plant regeneration from leaf-derived protoplasts within the Daucus genus: effect of different conditions in alginate embedding and phytosulfokine application. Plant Cell Tiss. Org. 2014;117(2):241–252. [Google Scholar]
- Guo Y, Song X, Zhao S, Lv J, Lu M. A transient gene expression system in Populus euphratica Oliv. protoplasts prepared from suspension cultured cells. Acta Physio. Plant. 2015;37(8):1–8. [Google Scholar]
- Wang H, Wang W, Zhan J, Huang W, Xu H. An efficient PEG-mediated transient gene expression system in grape protoplasts and its application in subcellular localization studies of flavonoids biosynthesis enzymes. Sci. Hort. 2015;191:82–89. [Google Scholar]
- Masani MYA, Noll GA, Parveez GKA, Sambanthamurthi R, Pruefer D. Efficient transformation of oil palm protoplasts by PEG-mediated transfection and DNA microinjection. PLoS One. 2014;9(5):96831. doi: 10.1371/journal.pone.0096831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasamoto H, Ashihara H. Effect of nicotinic acid, nicotinamide and trigonelline on the proliferation of lettuce cells derived from protoplasts. Phytochem. Lett. 2014;7:38–41. [Google Scholar]
- Uddin MJ, Robin AHK, Raffiand S, Afrin S. Somatic embryo formation from co-cultivated protoplasts of Brassica rapa & B. juncea. Am. J. Exp. Ag. 2015;8(6):342–349. [Google Scholar]
- Hayashimoto A, Li Z, Murai N. A polyethylene glycol-mediated protoplast transformation system for production of fertile transgenic rice plants. Plant Physiol. 1990;93(3):857–863. doi: 10.1104/pp.93.3.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazarei M, Al-Ahmad H, Rudis MR, Stewart CN. Protoplast isolation and transient gene expression in switchgrass, Panicum virgatum L. Biotechnol. J. 2008;3(3):354–359. doi: 10.1002/biot.200700189. [DOI] [PubMed] [Google Scholar]
- Mazarei M, Al-Ahmad H, Rudis MR, Joyce BL, Stewart CN. Switchgrass (Panicum virgatum L.) cell suspension cultures: Establishment, characterization, and application. Plant Sci. 2011;181(6):712–715. doi: 10.1016/j.plantsci.2010.12.010. [DOI] [PubMed] [Google Scholar]
- Locatelli F, Vannini C, Magnani E, Coraggio I, Bracale M. Efficiency of transient transformation in tobacco protoplasts is independent of plasmid amount. Plant Cell Rep. 2003;21(9):865–871. doi: 10.1007/s00299-003-0593-x. [DOI] [PubMed] [Google Scholar]
- Di Sansebastiano GP, Paris N, Marc-Martin S, Neuhaus JM. Specific accumulation of GFP in a non-acididc vacuolar compartment via a C-terminal propeptide-mediated sorting pathway. Plant J. 1998;15(4):449–457. doi: 10.1046/j.1365-313x.1998.00210.x. [DOI] [PubMed] [Google Scholar]
- De Sutter V, et al. Exploration of jasmonate signalling via automated and standardized transient expression assays in tobacco cells. Plant J. 2005;44(6):1065–1076. doi: 10.1111/j.1365-313X.2005.02586.x. [DOI] [PubMed] [Google Scholar]