

Abstract
Development of potent small molecule inhibitors of protein–protein interactions with optimized druglike properties represents a challenging task in lead optimization process. Here, we report synthesis and structure-based optimization of new thienopyrimidine class of compounds, which block the protein–protein interaction between menin and MLL fusion proteins that plays an important role in acute leukemias with MLL translocations. We performed simultaneous optimization of both activity and druglike properties through systematic exploration of substituents introduced to the indole ring of lead compound 1 (MI-136) to identify compounds suitable for in vivo studies in mice. This work resulted in the identification of compound 27 (MI-538), which showed significantly increased activity, selectivity, polarity, and pharmacokinetic profile over 1 and demonstrated a pronounced effect in a mouse model of MLL leukemia. This study, which reports detailed structure–activity and structure–property relationships for the menin–MLL inhibitors, demonstrates challenges in optimizing inhibitors of protein–protein interactions for potential therapeutic applications.
Graphical abstract
INTRODUCTION
The protein–protein interaction between menin and MLL (mixed lineage leukemia) fusion proteins plays a critical role in acute leukemias with translocations of the MLL gene.1 The MLL leukemias constitute about 5–10% of acute leukemia cases in adults, both AML (acute myeloid leukemia) and ALL (acute lymphoblastic leukemia),2 and ~70% of acute leukemias in infants.3 Patients with MLL leukemias do not respond well to currently available treatments,4–6 emphasizing the need for more effective therapies.
The MLL fusion proteins, which result from the chromosomal translocations of the MLL gene, retain the N-terminal fragment of MLL (about 1400 amino acids)7,8 that is involved in the protein–protein interaction (PPI) with menin.1,9 Importantly, the menin–MLL interaction plays a crucial role in the MLL fusion protein mediated leukemogenic transformation.1,10 Menin is required by MLL fusion proteins for enhanced proliferation and differentiation block of leukemic cells.1,11 We previously demonstrated that the N-terminal fragment of MLL binds with a high affinity to the large central cavity on menin9,12,13 and validated that this protein–protein interaction is amenable to small molecule inhibition.12,14–16 All these findings imply that the menin–MLL protein–protein interaction represents an attractive molecular target to develop new targeted agents as a potential therapeutic strategy for MLL leukemia.
Despite an important role of protein–protein interactions (PPIs) in human diseases, including cancer,17 targeting PPIs with small molecules is still considered a challenging task due to large contact areas, lack of well-defined pockets, and flexibility of residues at PPI interfaces.18,19 Furthermore, development of cell-permeable and orally bioavailable PPI inhibitors with optimized druglike properties represents an additional challenge.20 Despite these limitations, there are a number of successful examples of small molecules blocking PPIs.21–25 More importantly, PPI inhibitors have been successfully advanced to clinical trials, including small molecules activating apoptotic pathways, such as Bcl-2 inhibitors and p53 activators,26,27 providing evidence that PPI inhibitors might serve as novel therapeutics.
Our own work led to the identification of two distinct classes of menin–MLL inhibitors, which demonstrated relatively potent activity in MLL leukemia cells14,15,28 but were not optimized for in vivo studies. More recently, we developed a new class of menin–MLL inhibitors by linking the cyanoindole ring with the thienopyrimidine core through the aminopiperidine linker, Figure 1a and compound 1 (MI-136) in Table 1.16 However, this class, represented by the lead compound 1 (IC50 = 31 nM, Table 1), requires further improvement of cellular activity and druglike properties, including metabolic stability, selectivity, and aqueous solubility, to develop compounds with potential therapeutic value. In this study we report structure-based optimization of compound 1 performed by systematic exploration of substituents introduced on the indole ring of 1, together with extensive characterization of their in vitro potency, cellular activity, selectivity, and microsomal stability to provide structure–activity and structure–property relationships. For several selected compounds we also determined the pharmacokinetic profile (PK) in mice. This work resulted in the identification of compound 27 (MI-538), which showed significantly improved activity, selectivity, and PK profile over 1 and demonstrated a pronounced effect in a mouse model of MLL leukemia. Interestingly, we discovered that simultaneous incorporation of multiple substituents optimized for each site on the scaffold does not always positively affect the activity or druglike properties of these compounds, proving an additional level of complexity in lead optimization. This study illustrates the difficulties and challenges in optimizing druglike properties of PPI inhibitors and emphasizes the need for extensive medicinal chemistry efforts combined with structural studies for successful optimization of such compounds.
Figure 1.
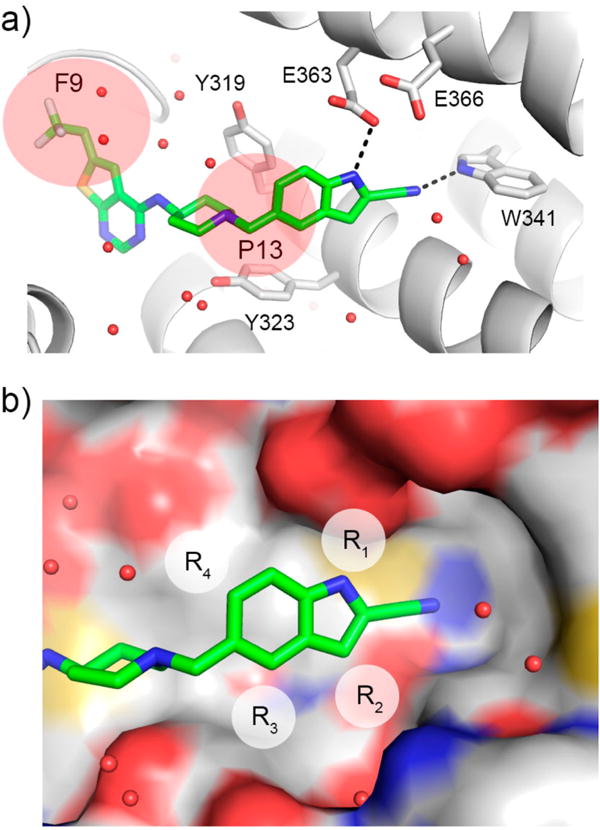
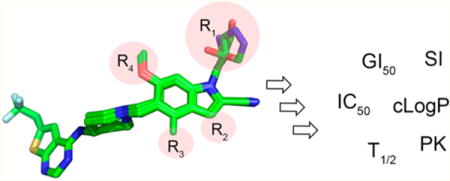
Compound 1 represents a good pharmacophore for development of menin–MLL inhibitors. (a) Binding mode of 1 to menin from the crystal structure of 1 bound to menin (4X5Z in PDB). Protein is shown in ribbon representation, and key menin residues involved in interactions with 1 are shown as sticks. Compound 1 is shown in stick representation with colors corresponding to the atom type (green, carbons; dark blue, nitrogens; yellow, sulfur; light blue, fluorine). Dashed lines represent hydrogen bonds between 1 and menin. Water molecules are represented as red spheres. P9 and P13 pockets are marked as pink spheres. (b) Crystal structure of menin–1 (green carbons) with substitution sites on indole of 1 explored for optimization marked as R1, R2, R3, and R4. Menin is shown in surface representation with atoms colored according to the atom type (red, oxygens; blue, nitrogens; yellow, sulfur; gray, carbons).
Table 1.
SAR and Properties of Analogues of Lead Compound 1 with Substitutions on Indole Nitrogen
| |||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | IC50(nM)a | GI50(μM)b
|
SIc | Tl/2 (min)d | cLogPe | |
| MLL-AF9 | HM-2 | ||||||
| 1 (MI-136) | H | 31 ± 3.5 | 0.55 | 3.0 | 5.5 | 6.8 | 4.3 |
| 2 |
|
35 ± 2.8f | 1.1 | 6.2 | 5.6 | 18 | 3.5 |
| 3-(RS) (MI-352) |
|
37 ± 2.8 | 3.4 | >12 | >3.5 | >60 | 3.1 |
| 3-(R) |
|
46 ± 2.8 | 1.7 | >40 | >24 | 48 | 3.1 |
| 3-(S) |
|
42 ± 0.7 | 3.8 | ND | ND | >60 | 3.1 |
| 4-(R) |
|
40 ± 1.4 | 0.36 | 9.7 | 27 | <3 | 3.9 |
| 4-(S) |
|
37 ± 6.0 | 0.66 | 6.9 | 11 | 5.2 | 3.9 |
| 5 |
|
101 ± 13 | 2.1 | ND | ND | <3 | 3.4 |
| 6 |
|
23 ± 1.4 | 0.18 | 3.5 | 19 | <3 | 4.6 |
| 7 (MI-389) |
|
25 ± 4.9f | 0.78 | 25 | 32 | 54 | 3.0 |
| 8 |
|
244 ± 57 | ND | ND | ND | ND | 3.3 |
| 9 |
|
27 ± 2.8 | 0.33 | 10 | 30 | 11 | 3.5 |
| 10 |
|
23 ± 4.2f | 0.26 | 6.9 | 27 | 8.3 | 3.9 |
IC50 values were measured by fluorescence polarization assay, average values from two to three independent measurements ± SD are provided.
Growth inhibition (GI50 values) measured in the MTT cell viability assay in MLL-AF9 or Hoxa9/Meis1 (HM-2) transformed murine bone marrow cells after 7 days of treatment with compounds. Activity in HM-2 cells was measured for compounds with best activity in MLL-AF9 cells.
SI, selectivity index calculated as a ratio of GI50 values measured in HM-2 cells (control cell line) and MLL-AF9 transformed cells.
Half-life of compounds in mouse liver microsomes.
Calculated with ChemBioDraw Ultra 14.0.
IC50 values reported before.16
ND: not determined.
RESULTS AND DISCUSSION
Compound 1 Represents a Valuable Pharmacophore for Development of Menin–MLL Inhibitors
We recently developed a new class of small molecule inhibitors of the menin–MLL interaction represented by compound 1, Figure 1a, Table 1.16 This compound strongly binds to menin (Kd = 24 nM), potently inhibits menin interaction with MLL (IC50 = 31 nM) (Table 1, Supporting Information Figure 1), and blocks proliferation of MLL leukemia cells at submicromolar concentrations (GI50 = 0.55 μM in the MLL-AF9 cells, Table 1), validating that it represents a valuable pharmacophore for menin–MLL inhibitors. In addition, compound 1 did not show substantial off-target activity when profiled on a panel of kinases and GPCRs, Supporting Information Tables 1 and 2. Compound 1, however, requires further improvement of druglike properties, including (i) potency and selectivity toward MLL leukemia cells (selectivity index for 1, SI < 6, Table 1), (ii) metabolic stability (T1/2 = 6.8 min in mouse liver microsomes), (iii) pharmacokinetic profile (PK), and (iv) polarity (clogP = 4.3), implying the need for additional medicinal chemistry efforts.
The crystal structure of the menin in complex with 1 revealed that it binds to the MLL binding site on menin and occupies the F9 and P13 pockets, with the 2-cyanoindole ring extending beyond the P13 pocket to form a hydrogen bond with the side chain of Trp341, Figure 1a. On the basis of the crystal structure, we concluded that additional substitutions on the 2-cyanoindole ring of 1 could be explored by introducing substituents at positions 1, 3, 4, and 6 (substituents R1–R4, Figure 1b). Substitutions at R2 and R4 were not tested before, while only a limited set of substituents at R1 and R3 were probed in our recent study,16 providing a good rationale for much more systematic structure–activity relationship (SAR) combined with structure–property relationship studies.
Development of Analogues with Substitutions at Indole Nitrogen
The crystal structure of the menin in complex with compound 1 revealed that the indole nitrogen in 1 is involved in a direct hydrogen bond with the side chain of Glu363 on menin (Figure 1a). This nitrogen atom is partly solvent exposed, suggesting that substituents of different size should be tolerable at this position. Furthermore, the close proximity of the side chains of Glu363 and Glu366 (Figure 1a) supports exploration of polar groups at R1 for hydrogen bond formation. To identify optimal R1 substituents, we focused on introducing a variety of substituents harboring hydrogen bond donor groups at R1, Table 1, with the goal to improve polarity while retaining or improving the potency of the new analogues.
First, we tested the hydroxyethyl group at R1 (compound 2), which was designed to form a hydrogen bond with one of the glutamic acid residues on menin, but no activity improvement was observed as compared to 1 (Table 1). We next designed compounds with 1,2-propanediol functional group or its derivatives (compounds 3-(RS) (MI-352), 3-(R), 3-(S), 4-(R), 4-(S), 5, 6), Table 1) to further increase capabilities for hydrogen bond formation with Glu363 and/or Glu366. We found that neither compound 3-(RS), which represents a racemic mixture, nor individual enantiomers of this compound (3-(R) and 3-(S)) demonstrate better inhibitory activity than 1, Table 1. To understand the molecular basis of inhibitory activity, we determined the crystal structure of menin in complex with 3-(RS), Figure 2a, Supporting Information Table 3. Interestingly, we observed that both enantiomers of 3-(RS) bind to menin in the same manner with the only difference being a distinct orientation of the secondary hydroxyl group, Figure 2a. This hydroxyl group is solvent exposed in both enantiomers, but only in one of them (3-R) it forms a hydrogen bond with Glu363, while the terminal hydroxyl group forms a hydrogen bond with Glu366 in both enantiomers of 3-(RS), Figure 2a. We next explored substitutions on the secondary hydroxyl group (compounds 4-(R), 4-(S), and 6) and found almost no effect on the inhibition of the menin–MLL interaction as compared to 3-(RS), Table 1, while alkylation of the terminal hydroxyl group (compound 5) resulted in ~3-fold reduced inhibitory activity. Overall, the series of R1 substitutions with the 1,2-propanediol group or its derivatives did not improve inhibitory activity over 1 despite the formation of additional hydrogen bonds with menin by some of these compounds.
Figure 2.
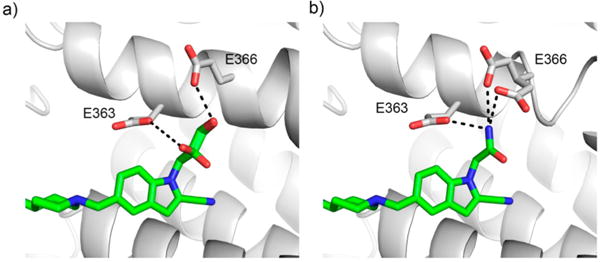
Structure-based optimization of polar interactions with menin. (a) Crystal structure of the menin–3-(RS) complex (PDB code 5DB0) demonstrating binding of both enantiomers of 3-(RS) to menin. Hydrogen bonds with Glu363 and Glu366 are marked. (b) Crystal structure of menin–7 complex (PDB code 5DB2) showing the interactions of the amide group of 7 with Glu363 and Glu366 on menin. The side chain of Glu366 exists in two conformations.
Incorporation of the acetamide group at R1 in compound 7 (MI-389), which results in formation of hydrogen bonds with both Glu363 and Glu366 as validated by the crystal structure of the complex (Figure 2b), also does not improve inhibition of the menin–MLL interaction (Table 1). We then introduced the α-methylated acetamide group at R1 (compound 8), which showed about 10-fold reduced inhibitory activity likely due to a limited space to accommodate an additional methyl group at this region of the binding site, Table 1.
Out of aromatic substituents at R1 (compounds 9 and 10, Table 1), we have recently explored a few five-member heterocycles harboring hydrogen bond donor groups, out of which the 4-methylpyrazole derivative (compound 10) showed best inhibitory activity (IC50 = 23 nM).16 Here, we followed up on these findings and introduced the 4-methyltriazole substituent at R1 (compound 9) to further increase polarity of these compounds. Importantly, we were able to retain strong inhibitory activity of compound 9 while improving polarity represented by a significantly lower clogP value when compared to 1, Table 1. Overall, despite the fact we did not improve substantially the in vitro inhibitory activity by substituting at R1 of 1, the majority of these compounds represent very potent inhibitors of the menin–MLL interactions with the IC50 values at the range of 25–45 nM, Table 1. Importantly, these new analogues are more polar than 1, reflected by the clogP values below 3.5 for most of them, which affects their cellular activity and other druglike properties (see below).
In the next step we assessed cellular activities and microsomal stabilities of the series of menin–MLL inhibitors with various R1 substituents. Interestingly, despite similar in vitro activity, these compounds demonstrate substantially different cellular activity in MLL leukemia cells (ranging from 0.2 to 4 μM), Table 1, likely associated with differences in their polarity (clogP values, Table 1) affecting cellular permeability, as assessed in the Caco-2 membrane permeability studies (Supporting Information Table 4). Likewise, compounds 1–10 exhibit different level of selectivity toward MLL-AF9 cells over HM-2 control cell line (reported as selectivity index, SI, ranging from 5 to 32), Table 1. Furthermore, the microsomal stability of these compounds in mouse liver microsomes also varied substantially, Table 1. For example, incorporation of the hydroxyethyl (2) or 1,2-propanediol (3-(RS), 3-(R), 3-(S)) substituents at R1 substantially increases polarity (clogP < 3.5) and metabolic stability (T1/2 = 18–60 min); however the effect of these compounds on proliferation of the MLL-AF9 cells (GI50 > 1 μM) is weaker than for 1, Table 1. Introduction of the methyl (4-(R)) or isopropyl (6) groups at the secondary hydroxyl substantially improves GI50 values (e.g., GI50 = 0.18 μM for 6); however these compounds are metabolically unstable (T1/2 < 3 min). Interestingly, 4-(R) has markedly improved cellular permeability over 3-(RS), Supporting Information Table 4, despite very small difference in their structures, Table 1. The efflux ratio was >20-fold lower for 4-(R) as compared to 3-(RS) (Supporting Information Table 4), with both apical to basolateral (A → B) and basolateral to apical (B → A) permeability markedly improved for 4-(R). Accordingly, cellular activity of 4-(R) is almost 10-fold increased over 3-(RS) despite similar IC50 values for these compounds, Table 1, emphasizing that increased cellular permeability correlates with cell growth inhibition. The analogue with the acetamide group at R1, compound 7, shows cellular activity (GI50 = 0.78 μM in MLL-AF9 cells) similar to 1; however this compound has substantially improved selectivity (SI = 34) and metabolic stability over 1, Table 1, and both are likely associated with its higher polarity. Compound 7 also showed low cellular permeability (efflux ratio of >90, Supporting Information Table 4) reflected by the modest cellular activity of this compound, Table 1. Finally, similar to the compound with 4-methylpyrazole at R1 (10), the analogue with 4-methyltriazole (9) has improved cellular activity (GI50 = 0.33 μM), selectivity (SI = 30), microsomal stability, and polarity when compared to 1, Table 1. Therefore, substitutions on the indole nitrogen are very valuable to modulate both cellular activity and druglike properties for this class of menin–MLL inhibitors. Overall, we concluded that hydroxyethyl (present in 2), acetamide (present in 7), 4-methylpyrazole (present in 10), and 4-methyltriazole (present in 9) represent the most valuable substituents at R1 due to either superior effect in MLL leukemia cells (9 and 10) or improved druglike properties (e.g., polarity for 2 and 7).
Activity and Property Optimization of Analogues Substituted at Positions 3, 4, or 6 of Indole
Analysis of the crystal structure of the menin–1 complex revealed that carbon at position 3 of the indole ring (position R2, Figure 1b) approaches the backbone of menin, which limits the possibility of substituting at this position. Nevertheless, since this position has not been explored before, we introduced a few small substituents (R2 substituents) at this position, namely, fluorine (compound 11), amino (compound 12), and methyl (compound 13), Table 2. The inhibitory activity of these compounds was, however, 3- to 4-fold reduced as compared to 1, Table 2, consistent with their relatively weak cellular activity in the MLL-AF9 cells. These results support that position 3 on the indole ring of 1 should remain unsubstituted.
Table 2.
SAR and Properties of Analogues with Substitutions at Position 3, 4, or 6 of Indole
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| compd | R2 | R3 | R4 | IC50(nM)a | GI50 (μM)b
|
SIc | T1/2 (min)d | clogPe | |
| MLL-AF9 | HM-2 | ||||||||
| 1 (MI-136) | H | H | H | 31 ± 3.5 | 0.55 | 3.0 | 5.5 | 6.8 | 4.3 |
| 11 | F | H | H | 96 ± 0.7 | 1.7 | ND | ND | 5.7 | 4.5 |
| 12 | NH2 | H | H | 124 ± 30 | 5.2 | ND | ND | 27 | 3.3 |
| 13 | CH3 | H | H | 111 ± 16 | 2.6 | ND | ND | 4.8 | 4.9 |
| 14 (MI-463) | H | CH3 | H | 15 ± 1.4f | 0.2 | 5.6 | 28 | 14 | 4.7 |
| 15 | H | Cl | H | 22 ± 3.5 | 1.1 | 11 | 10 | 28 | 5.0 |
| 16 | H | H | F | 109 ± 5.6 | 3.0 | >12 | >4.0 | 9 | 4.5 |
| 17 | H | H | OH | 20 ± 5.0 | 0.18 | >10 | >56 | 6.6 | 3.7 |
| 18 (MI-336) | H | H | OCH3 | 22 ± 0.7 | 0.34 | 16 | 47 | 3.7 | 4.3 |
| 19 | H | H | OEt | 47 ± 4.2 | 0.45 | 5.3 | 12 | 11 | 4.8 |
IC50 values were measured by fluorescence polarization assay, average values from two to three independent measurements ± SD are provided.
Growth inhibition (GI50 values) measured in the MTT cell viability assay in MLL-AF9 or Hoxa9/Meis1 (HM-2) transformed murine bone marrow cells after 7 days of treatment with compounds. Activity in HM-2 cells was measured for compounds with best activity in MLL-AF9 cells.
SI, selectivity index calculated as a ratio of GI50 values measured in HM-2 cells (control cell line) and MLL-AF9 transformed cells.
Half-life of compounds in mouse liver microsomes.
Calculated with ChemBioDraw Ultra 14.0.
IC50 values reported before.16
ND: not determined.
We have recently explored several substituents at position 4 of the indole ring (R3 substituent, Table 2) and concluded that small hydrophobic groups represent the best substituents at this position,16 with the methyl group resulting in ~2-fold improved activity (compound 14, MI-463, IC50 = 15 nM, Table 2), likely due to hydrophobic contacts with the aromatic ring of Tyr323, Figure 1a. Here, we completed the series of R3 substituted analogues by incorporating chlorine (compound 15, Table 2) at this position to assess whether it might represent an even more favorable substituent than a methyl group. The in vitro inhibitory activity of 15 (IC50 = 22 nM) was slightly improved over 1, but the effect on proliferation of the MLL leukemia cells was much weaker (GI50 > 1 μM), Table 2. Therefore, we concluded that methyl group represents the best substituent at position 4 of the indole ring.
Finally, we also probed position 6 on the indole ring (R4 substituents, Table 2), which was not explored previously for substitutions. The crystal structure of the menin–1 complex revealed that this position is partly solvent exposed and that relatively small polar groups could likely enhance inter- and intramolecular interactions (Figure 1a, Figure 1b). Indeed, incorporation of the hydroxyl in compound 17 or methoxy group in compound 18 (MI-336) at R4 resulted in about 1.5-fold improved inhibitory activity over 1, Table 2. More importantly, these two substituents have led to a significant improvement of growth inhibition in the MLL leukemia cells, in particular compound 17, which showed almost 3-fold better GI50 value (GI50 = 0.18 μM) than 1, Table 2. Further increase in the size of R4 (e.g., compound 19 with the ethoxy group) or introduction of fluorine (compound 16) resulted in the reduction of inhibitory activity, Table 2.
To understand the molecular basis of the activity of these compounds, we determined the crystal structure of 18 with menin, Supporting Information Table 3. The structure revealed an intramolecular hydrogen bond between the oxygen of the methoxy group and nitrogen in the piperidine ring, which likely stabilizes the conformation of 18 required for effective binding, Figure 3a. The same interaction is expected for compound 17 harboring a hydroxyl group at R4. In summary, on the basis of in vitro and cellular activity, we concluded that hydroxyl and methoxy groups represent optimal substituents at R4, and we selected both of them for preparation of multisubstituted derivatives of 1 (see below). The metabolic stability and polarity (particularly for 18) still need to be improved for compounds with R4 substituents (Table 2), but these liabilities may be compensated by the presence of other substituents on the indole ring (see below).
Figure 3.
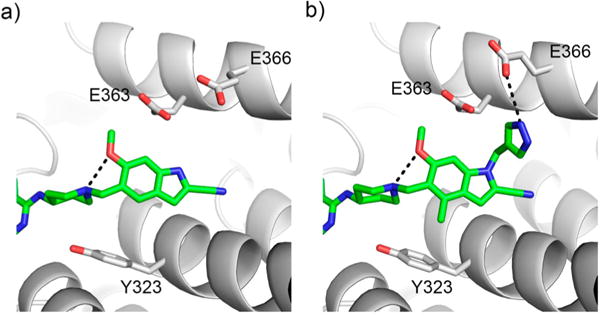
Structure-based optimization of R1 and R4 substituents. (a) Crystal structure of menin–18 complex (PDB code 5DB1) demonstrating intramolecular hydrogen bond between the methoxy group and nitrogen in the piperidine ring of 18. (b) Binding mode of triple substituted compound 31 to menin based on the crystal structure of the complex (PDB code 5DB3).
Activity of Analogues with Multiple Substitutions on Indole Ring
Having established the optimal substituents at different positions of the indole ring, we then assessed whether simultaneous incorporation of these substituents into the scaffold would provide further improvement of activity and druglike properties. For this purpose, we prepared a number of compounds to probe combinations of the best R1 substituents (hydroxyethyl, acetamide, 4-methylpyrazole, and 4-methyltriazole) with the methyl group as an optimal substituent at R3 and/or two best R4 substituents (hydroxyl or methoxy groups), Table 3. First, we prepared the R3, R4 disubstituted analogues (compounds 20 and 21), Table 3. Although neither compound showed improved in vitro inhibitory activity over 1, they demonstrate improved cellular activity in MLL-AF9 cells, Table 3, with compound 21 showing over 5-fold improved cell growth inhibition (GI50 = 100 nM) as compared to 1, Table 3. Interestingly, both 20 and 21 showed relatively high to moderate cellular permeability in Caco-2 cells, Supporting Information Table 4, which could contribute to strong growth inhibition in the MLL leukemia cells, Table 3.
Table 3.
SAR and Properties of Analogues with Multiple Substitutions on Indole
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R3 | R4 | IC50 (nM)a | GI50 (μM)b
|
SIc | Tl/2 (min)d | cLogPe | |
| MLL-AF9 | HM-2 | ||||||||
| 1 (MI-136) | H | H | H | 31 ± 3.5 | 0.55 | 3.0 | 5.5 | 6.8 | 4.3 |
| 20 | H | CH3 | OH | 27 ± 1.4 | 0.22 | 12 | 55 | 56 | 4.1 |
| 21 | H | CH3 | OCH3 | 40 ± 0.9 | 0.10 | 11 | 110 | 39 | 4.7 |
| 22 |
|
CH3 | H | 44 ± 8.4 | 0.44 | 5.6 | 13 | 23 | 3.9 |
| 23 |
|
CH3 | OH | 27 ± 0.5 | 0.40 | 10 | 25 | 11 | 3.7 |
| 24 |
|
CH3 | OCH3 | 16 ± 2.0 | 0.36 | 15 | 69 | 7.8 | 4.1 |
| 25 |
|
CH3 | H | 27 ± 5.6 | 0.47 | >25 | >53 | 59 | 3.4 |
| 26 |
|
CH3 | OCH3 | 37 ± 3.5 | 0.42 | >25 | >60 | >60 | 3.5 |
| 27 (MI-538) |
|
H | OH | 21 ± 1.4 | 0.083 | 9 | 108 | 14 | 3.7 |
| 28 |
|
H | OCH3 | 24 ± 3.0 | 0.29 | 6.7 | 23 | 35 | 4.1 |
| 29 (MI-503) |
|
CH3 | H | 14 ± 2.1f | 0.22 | >5 | >23 | 21 | 4.4 |
| 30 |
|
CH3 | OH | 18 ± 1.4 | 0.27 | 13 | 48 | 25 | 4.2 |
| 31 (MI-574) |
|
CH3 | OCH3 | 12 ± 2.0 | 0.18 | 4.2 | 23 | 55 | 4.5 |
| 32 |
|
CH3 | H | 23 ± 2.0 | 0.25 | 8 | 32 | 59 | 4.0 |
| 33 |
|
CH3 | OH | 17 ± 1.4 | 0.42 | 10 | 24 | 25 | 3.8 |
| 34 |
|
CH3 | OCH3 | 16 ± 1.4 | 0.41 | 9 | 22 | 32 | 4.1 |
IC50 values were measured by fluorescence polarization assay, average values from two to three independent measurements ± SD are provided.
Growth inhibition (GI50 values) measured in the MTT cell viability assay in MLL-AF9 or Hoxa9/Meis1 (HM-2) transformed murine bone marrow cells after 7 days of treatment with compounds.
SI, selectivity index calculated as a ratio of GI50 values measured in HM-2 cells (control cell line) and MLL-AF9 transformed cells.
Half-life of compounds in mouse liver microsomes.
Calculated with ChemBioDraw Ultra 14.0.
IC50 value reported before.16
The subsequent set of analogues (compounds 22, 23, 24) contained a hydroxyethyl substituent at R1, a methyl at R3 with R4 remaining either unsubstituted or substituted with hydroxyl or methoxy groups, Table 3. None of these compounds, however, showed substantially increased activity, either in vitro or in MLL leukemia cells, Table 3. Similarly, two analogues with an acetamide group at the indole nitrogen (compounds 25 and 26) did not demonstrate significantly improved in vitro or cellular activity over 1, Table 3. We next synthesized and tested compounds with aromatic substituents at the indole nitrogen, either 4-methylpyrazole (compounds 27–31) or 4-methyltriazole (compounds 32–34), Table 3. Notably, all these compounds showed strong inhibitory activity, with the IC50 values for inhibition of the menin–MLL interaction in the range of 12–24 nM, Table 3, representing an improvement over 1. We solved the crystal structure of menin in complex with the most potent 31 (MI-574), which harbors three substituents on the scaffold (Figure 3b). The structure shows that incorporation of several substituents on the indole ring does not affect the binding mode of 31 to menin as compared to 1. The structure also reveals two additional hydrogen bonds, one intramolecular formed by the methoxy group of 31 and an additional intermolecular between the pyrazole ring and Glu366, Figure 3b, which are likely responsible for ~2.6-fold improved activity of this compound over 1.
Importantly, simultaneous substitution at multiple sites of indole resulted in compounds with a pronounced effect on the proliferation of MLL leukemia cells, with the most potent compound 27 (MI-538), which showed GI50 = 83 nM in MLL-AF9 cells, representing almost 7-fold improvement over 1, Table 3. Compound 27 binds to menin with low nanomolar affinity (Kd = 6.5 nM), as assessed by isothermal titration calorimetry (Figure 4a), representing ~3.6-fold improvement in the binding affinity as compared to 1 (Kd = 23.6 nM).16 Therefore, potent cellular activity of 27 originates from the improved binding affinity to menin and possibly increased cell membrane permeability (e.g., improved permeability over 7 with a similar IC50 value), Supporting Information Table 4. Interestingly, 27, which is a double substituted analogue with methylpyrazole at R1 and hydroxyl at R4, has improved cellular activity by 2- to 3-fold over monosubstituted compound 10 or 17, which harbors only one of these substituents, Tables 1–3. Further addition of the methyl group at R3 resulting in compound 30 leads to reduced activity in MLL leukemia cells by more than 2-fold, Table 3. These results demonstrate that simultaneous incorporation of multiple substituents to the scaffold can result in improved cellular activity over monosubstituted analogues; however these substitutions are not fully additive, as cellular activity of triple substituted analogues is not improved over disubstituted analogues of 1, Table 3.
Figure 4.
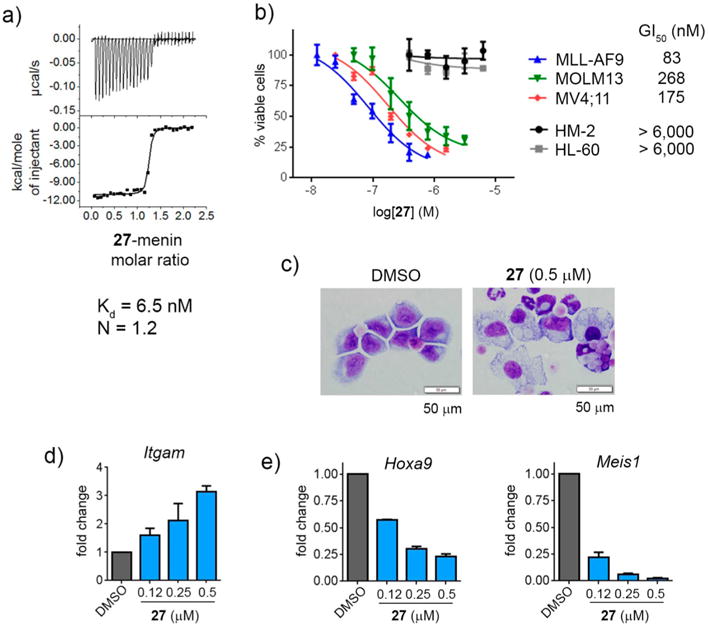
Binding to menin and cellular activity of compound 27. (a) Binding isotherm from the ITC experiment for 27 showing its direct binding to menin. (b) Titration curves from the MTT cell viability assay performed for 27 after 7 days of treatment of MLL-AF9 or Hoxa9/Meis1 (HM-2) transformed murine bone marrow cells as well as human MLL leukemia cell lines (MV4;11 and MOLM-13) and control cell line HL-60 (non-MLL leukemia cell line). Cell growth inhibition (GI50) values are provided for MLL leukemia cell lines. Data represent mean values of quadruplicates ± SD. The experiments were performed 3 times. (c) Wright–Giemsa stained cytospins for MLL-AF9 transformed murine bone marrow cells (BMCs) after 7 days of treatment with 27. Black line represents the scale bar (50 μm). (d) Quantification of Itgam expression in MLL-AF9 transformed murine bone marrow cells after 6 days of treatment with 27 as detected by quantitative real-time PCR (qRT-PCR), mean ± SD, n = 2. Expression of Itgam was normalized to 18S rRNA and referenced to DMSO-treated cells. (e) qRT-PCR performed in MLL-AF9 transformed BMCs after 6 days of treatment with 27. Expression of Hoxa9 and Meis1 was normalized to β-actin and referenced to DMSO-treated cells, mean ± SD, n = 2.
Multisubstituted Analogues Demonstrate Favorable Druglike Properties
In addition to activity, other properties of the analogues of compound 1 were affected by simultaneous introduction of multiple substituents. For example, substantial improvement in selectivity toward MLL leukemia cells, reflected by increase in the SI index, was observed for compounds with multiple substituents on the indole ring, Table 3. While 1 and the majority of monosubstituted analogues have SI < 30, Tables 1 and 2, additional substitutions on the indole led to increased selectivity, resulting in a number of compounds with SI > 50, Table 3. For instance, compounds with no substituent at R1 but substituted at R3 and R4 (20 and 21), analogues with acetamide at R1 (25 and 26) as well as 27 with 4-methylpyrazole at R1 and hydroxyl at R4 showed best selectivity, Table 3. Increase in the selectivity index can be attributed to both the increase in cellular activity toward MLL leukemia cells and a reduced effect in the control cell line HM-2 (e.g., for 25 and 26, Table 3). Although cellular selectivity was rather difficult to predict based upon a particular modification in the scaffold, in general the presence of an additional polar group improves selectivity, Table 3. Exceptions are compounds 33 and 34 due to their weaker effect in MLL-AF9 cells over 32.
Another important property we attempted to optimize was the polarity of these compounds, as the clogP value for the parent compound 1 is about 4.3, implying that it might need further improvement. Indeed, the majority of compounds with multiple substitutions on indole had lower clogP values than 1, Table 3, including the compound with the most potent activity in MLL leukemia cells, 27 (clogP = 3.7), which harbors the hydroxyl group at R4. This demonstrates that it is possible to simultaneously improve polarity, cellular activity, and selectivity for this class of menin–MLL inhibitors.
Finally, one liability of the lead compound 1 is its relatively poor metabolic stability in the murine liver microsomes (T1/2 = 6.8 min), Table 1, reflected by unsatisfactory PK profile (see below). By introducing multiple substituents to the indole ring, we were able to substantially improve metabolic stability of analogues of compound 1, with a number of compounds showing increase in T1/2 to over 30 min, Table 3. This includes compounds 20, 21, 31, which also demonstrated strong effect on cell proliferation (GI50 of ~200 nM or better in MLL-AF9 cells) and good selectivity (SI > 50). The analogue with the most potent cellular activity, compound 27, has about 2-fold increase in microsomal stability (T1/2 = 14 min) over 1, Table 3. Nevertheless, due to its strongest activity in MLL leukemia cells, high selectivity to MLL leukemia cells (SI > 100), and improved polarity (clogP = 3.7), 27 was chosen for pharmacokinetic studies in mice together with several other compounds.
Pharmacokinetic Studies of Menin–MLL Inhibitors in Mice
We selected 27 and several other compounds (20, 21, 31) that showed the best cellular activity in MLL-AF9 cells (GI50 < 0.2 μM), high level of selectivity to MLL leukemia cells (SI > 50), and good microsomal stability (T1/2 > 30 min) and performed in vivo pharmacokinetic (PK) studies in mice. The PK studies were also performed for the parent compound 1. Table 4 summarizes the PK parameters for all compounds tested using both intravenous (iv, 15 mg/kg dose) and oral (po, 30 mg/kg dose) administration. Importantly, these studies demonstrate substantially improved PK profile for 27 over 1 and other compounds tested, Table 4. Compound 27 demonstrated markedly improved exposure (area under the curve, AUC, values), Cmax (maximum compound concentration) in the blood plasma, and the lowest value of clearance (Clobs), Table 4. The half-life (T1/2) for 27 was reduced as compared to 1 but still remained relatively long (~1.6 h). To investigate the possible reason for reduced half-life for 27, we investigated phase II metabolism and found that this compound undergoes glucuronidation, consistent with the presence of the hydroxyl group in the structure (Supporting Information Figure 2). Importantly, 27 has also high oral bioavailability (~50%), similar to 1 and 21, Table 4. Out of the other compounds tested in PK studies, 31 with the trisubstituted 2-cyanoindole ring demonstrated good exposure, but this compound had very poor oral bioavailability, Table 4. Two other compounds, 20 and 21, with R3, R4 disubstitutions on the 2-cyanoindole ring did not show substantial improvement in the PK profile over parent compound 1, and 20 had even reduced exposure and increased clearance, Table 4. These results demonstrate that not all substitutions on the 2-cyanoindol ring of 1 result in additive effects on druglike properties, including PK profile, and multiple combinations of substituents need to be tested to identify compounds with the desired activity and properties. These studies also showed a lack of correlation between in vitro microsomal stability and in vivo PK profile, as exemplified for compound 20 when compared to 1 and for 27 with superior PK but relatively short T1/2 in mouse liver microsomes, Tables 3 and 4.
Table 4.
PK Parameters for Menin–MLL Inhibitorsa
| compd | route of admin | dose (mg/kg) | Tmax (h) | Cmax (ng/ml) | AUCobs (h·ng/mL) | T1/2 (h) | Clobs (mL min−1 kg−1) | Vss (mL/kg) | F (%) |
|---|---|---|---|---|---|---|---|---|---|
| 1b | iv | 15 | 6576 | 3.1 | 38.1 | 6516 | |||
| po | 30 | 1 | 1516 | 7509 | 3.1 | 57 | |||
| 20b | iv | 15 | 5666 | 1.7 | 44.1 | 4454 | |||
| po | 30 | 2 | 649 | 3846 | 4 | 34 | |||
| 21b | iv | 15 | 9397 | 1.8 | 26.6 | 3698 | |||
| po | 30 | 0.5 | 2113 | 8717 | 2 | 46 | |||
| 27b | iv | 15 | 17897 | 1.6 | 14.0 | 2347 | |||
| po | 30 | 2 | 4572 | 18090 | 1.6 | 51 | |||
| 31c | iv | 15 | 16235 | 1.4 | 15.0 | 1837 | |||
| po | 30 | 2 | 101 | 383 | NAd | 1 |
Cmax (maximum compound concentration) in blood plasma, AUCobs (area under the curve), Clobs (Clearance), T1/2 (half-life), Vss (volume of distribution), F (oral bioavailability).
24 h experiments.
8 h experiment.
Not available due to compound concentration below detection level;
Compound 27 Demonstrates On-Target Mechanism of Action in MLL Leukemia Cells
On the basis of strong effect on proliferation of the MLL-AF9 leukemia cells (GI50 = 83 nM), excellent selectivity, and best PK profile (Table 4), 27 was selected for further characterization in MLL leukemia models. First, we assessed the effect of this compound in human MLL leukemia cell lines, MV4;11 and MOLM13, harboring two different MLL translocations (MLL-AF4 and MLL-AF9, respectively), and observed a pronounced inhibition of cell growth (GI50 = 170–270 nM), Figure 4b. Importantly, 27 showed no effect (up to 6 μM) on growth of the control cell lines HL-60 and HM-2, which do not harbor MLL translocations, demonstrating good selectivity toward MLL fusion protein transformed cells (Figure 4b). Inhibition of the menin–MLL interaction is expected to result in the differentiation of MLL leukemia cells and downregulation of MLL fusion target genes.1,14 Indeed, we observed pronounced differentiation of MLL-AF9 cells upon treatment with 27 as reflected by cell morphology change (decreased nuclei to cytoplasm ratio, multilobed nuclei and highly vacuolated cytoplasm) as well as marked increase in the expression level of Itgam, a differentiation marker of myeloid cells (Figure 4c,d). Finally, treatment with 27 resulted in strong downregulation of expression of Hoxa9 and Meis1 genes, the downstream targets of MLL fusion proteins required for their leukemogenicity, Figure 4e. About 100 nM 27 was sufficient to reduce by ~50% Hoxa9 expression in MLL-AF9 cells, and even more pronounced effect was seen on Meis1 expression, consistent with the strong effect of this compound on proliferation of these cells, Figure 4b. All these findings further confirm on-target effects and specific mechanism of action of 27 menin–MLL inhibitor, suggesting that it represents a valid candidate for in vivo studies in a mouse model of MLL leukemia.
Compound 27 Demonstrates in Vivo Efficacy in a Xenograft Model of MLL Leukemia
Encouraged by the favorable PK profile as well as the pronounced and specific effect of 27 in MLL leukemia cells, we selected this compound for in vivo efficacy studies in a mouse xenograft model utilizing MV4;11 human MLL leukemia cells injected subcutaneously into CB17 SCID mice. Treatment with 27 (45 mg/kg, once daily via ip) was initiated when tumor size reached ~100 mm3 and was continued for 2 weeks, Figure 5. Importantly, treatment with 27 resulted in a pronounced, about 80%, reduction in the MV4;11 tumor volume (Figure 5), without causing substantial signs of toxicity reflected by less than 10% reduction of the body weight (not shown). These results validate that 27 not only is a potent inhibitor of MLL leukemia cell proliferation but can also effectively inhibit the growth of MV4;11 tumors in mice. These data demonstrate a strong potential for this class of menin–MLL inhibitors for further development toward therapeutically useful compounds.
Figure 5.
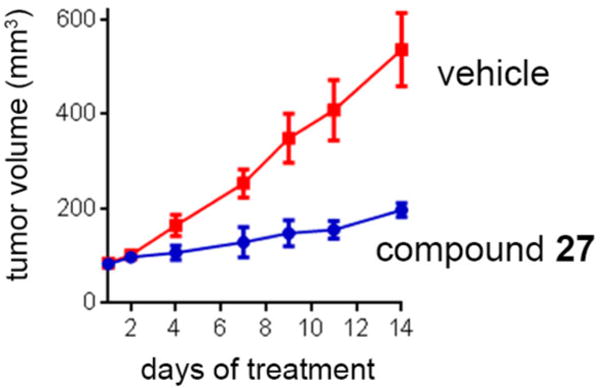
In vivo inhibition of tumor growth by compound 27. 27 inhibits tumor growth in MV4;11 subcutaneous model of MLL leukemia developed in CB17 SCID mice after injecting 0.5 × 107 MV4;11 cells, n = 9 per group. Mice were treated once daily with 27 (45 mg/kg) or vehicle via ip, resulting in substantial reduction of the tumor volume (p < 0.001). Error bars represent SEM.
CHEMISTRY
All inhibitors 1–34 contain the N-(piperidin-4-yl)-6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-amine moiety (39) that was synthesized in five steps starting from the 4-trifluorobuteraldehyde, Scheme 1, with the key step of Gewald thiophene synthesis.29 Condensation of thiophene with triethyl orthoformate gave pyrimidinone 37, which was then converted to 38. Nucleophilic substitution of 38 with Boc-protected aminopiperidine and subsequent deprotection provided 39 as hydrochloride salt. The process was optimized to provide high yields of 39 with no chromatography purification, resulting in overall yield of 45% over five steps after recrystallization of the final product.
Scheme 1. Synthesis of Compound 39a.
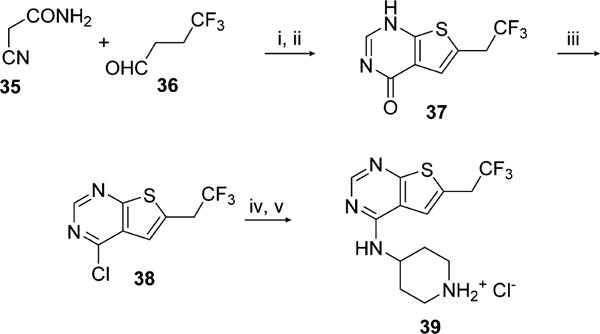
aReagents and conditions: (i) S, Et3N, DMF, rt, 24 h; (ii) AcOH, (EtO)3CH, reflux, 4 h, 76% (2 steps); (iii) POCl3, reflux, 3 h, 92%; (iv) tert-butyl 4-aminopiperidine-1-carboxylate, DIEA, IPA, reflux, 12 h; (v) HCl, dioxane, rt, overnight, 64% (2 steps).
Synthesis of 1 and 11 started from commercially available 5-methylindole-2-carboxylic acid, Scheme 2. The 3-substituted indole was obtained by reaction of 40a with 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate) as a source of electrophilic fluorine, providing 40b. The carboxylic acid functional group in 40a and 40b was then converted to nitrile by dehydratation of amide obtained from acid chloride. The Boc protection of indole nitrogen smoothly gave 42a,b that were subjected to free radical bromination. Bromo derivatives were used for coupling reaction with 39 followed by Boc group deprotection resulting in 1 and 11, Scheme 2.
Scheme 2. Synthesis of Compounds 1, 7, 8, and 11a.
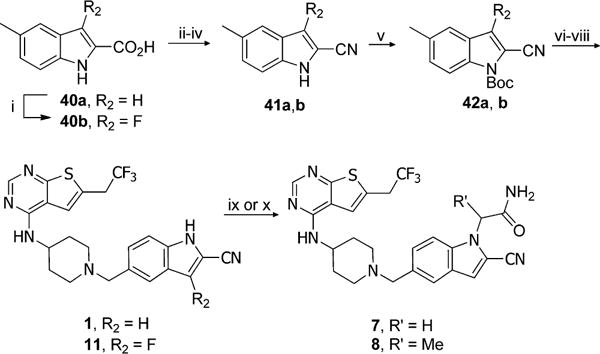
aReagents and conditions: (i) 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate), CH3CN, reflux, 12 h, 17%; (ii) SOCl2, CHCl3, reflux, 2 h; (iii) NH3·H2O, rt, 2 h; (iv) POCl3, CHCl3, reflux, 4 h, 44–55% (3 steps); (v) Boc2O, DMAP, CH3CN, rt, 1 h, 79–83%; (vi) NBS, AIBN, CCl4, reflux, 2 h; (vii) 39, DIEA, DCM, 18 h; (viii) SnCl4, CH3CN, 0 °C to rt, 0.5 h, 34–53% (3 steps); (ix) 2-bromoacetamide, Cs2CO3, DMF, rt, 4 h, 40% of 7; (x) 2-bromopropionamide, Cs2CO3, DMF, rt, 4 h, 27% of 8.
For preparation of 7 and 8, we explored the possibility of introducing substituents on indole nitrogen using compound 1 as a substrate, Scheme 2. Indeed, indole 1 was deprotonated using Cs2CO3 as a weak base and directly alkylated with bromoacetamide or 2-bromopropionamide to form 7 and 8, respectively, although in moderate to poor yields. Interestingly, similar conditions with other alkylating reagents did not yield the desired products. Therefore, we applied other methodologies to afford compounds 2–6, 9, 10, 23–34 as described below.
Synthesis of 2 was performed according to Scheme 3. Alkylation of indole 41a proceeded efficiently with NaH as a base producing the hydroxyethyloindole 44, which was then subjected to free radical bromination, followed by alkylation and deprotection reactions to afford final product 2. Similar approach was employed for synthesis of 1,2-propanediol derivatives of 3, where starting materials were either racemic mixture or enantiomerically pure alcohols 45, Scheme 4. After protection of hydroxyl groups in diol 45 with tert-butyldimethylsilyl ethers (TBS) the resulting intermediates reacted with indole 41a to obtain alkylated compound 46. Further bromination, alkylation, and deprotection reactions provided racemic compound 3 as well as its enantiomers, Scheme 4. Synthesis of individual enantiomers of 4 required selective methylation of secondary hydroxy group, which was accomplished by removal of both TBS protecting groups from 46 followed by exclusive protection of primary hydroxyl group with a bulky tert-butyldiphenylsilyl ether (TBDPS) and methylation of a secondary alcohol to afford 47. Subsequently, the bromination, alkylation, and deprotection reactions were performed to yield enantiomerically pure 4-(S) and 4-(R).
Scheme 3. Synthesis of Compound 2a.
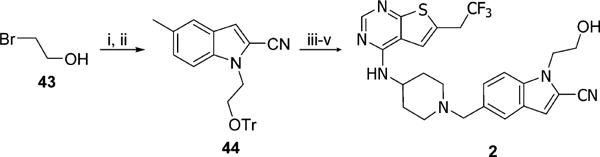
aReagents and conditions: (i) TrCl, DMAP, Et3N, DCM, rt, 24 h, 96%; (ii) 41a, NaH, DMF, rt to 60 °C, overnight, 81%; (iii) NBS, AIBN, CCl4, reflux, 2 h; (iv) 39, DIEA, DCM, 5 h; (v) HCl, MeOH, rt, 1 h, 42% (3 steps).
Scheme 4. Synthesis of Compounds 3-(RS), 3-(R), 3-(S), 4-(R), and 4-(S)a.
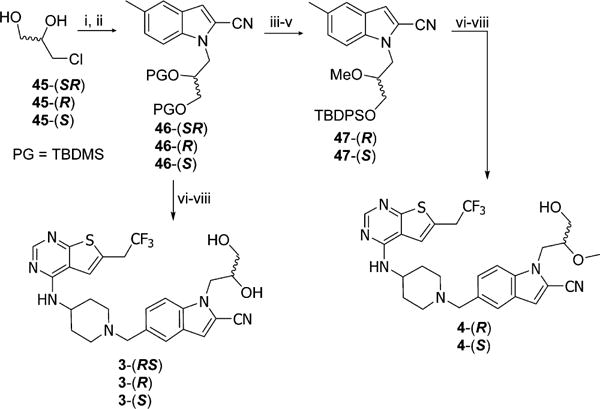
aReagents and conditions: (i) TBSCl, imidazole, DCM, rt, overnight, 84%; (ii) 41a, NaH, DMF, rt, 2 h, 49%; (iii) HCl, MeOH, rt, 4 h; (iv) TBDPSCl, imidazole, DCM, rt, 1 h; (v) NaH, MeI, THF, rt, 1 h, 11% (3 steps); (vi) NBS, AIBN, CCl4, reflux, 2 h; (vii) 39, DIEA, DCM, 5 h; (vii) HCl, MeOH, rt, 1 h, 24–58% (3 steps).
To synthesize compound 12, the carboxylic acid 48 was used as a starting material, Scheme 5. The carboxylic group of 48 was converted to nitrile via standard amidation and dehydration reactions, followed by reduction of the nitro group with tin chloride (II) to afford 49 in four steps. When 49 was heated with paraformaldehyde and KCN in the presence of zinc chloride (II), the N-cyanomethyl compound 50 was obtained, which was subjected to Boc protection with 4 equiv of Boc2O and simultaneous cyclization to afford 51. Free radical bromination followed by nucleophilic substitution with building block 39 and deprotection resulted in compound 12, Scheme 5.
Scheme 5. Synthesis of Compound 12a.
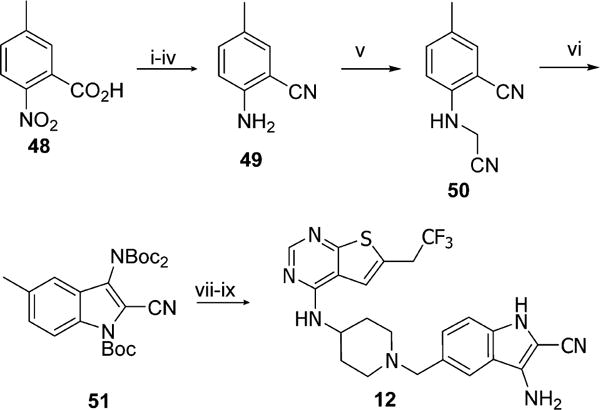
aReagents and conditions: (i) SOCl2, CHCl3, reflux, 3 h; (ii) NH3·H2O, rt, 2 h; (iii) POCl3, CHCl3, reflux, 4 h; (iv) SnCl2, EtOH, reflux, 1 h, 18% (4 steps); (v) paraformaldehyde, KCN, ZnCl2, AcOH, 60 °C, 6 h, 33%; (vi) Boc2O, DMAP, DMF, rt, 4 h, 25% (2 steps); (vii) NBS, AIBN, CCl4, reflux, 2 h; (viii) 39, DIEA, DCM, 5 h; (ix) SnCl4, CH3CN, 0 °C to rt, 0.5 h, 19% (3 steps).
Inhibitors bearing methyl groups at position 3 or 4 of indole required different synthetic procedure, in which reductive amination was employed to couple aldehydes 60a–d with 39, Scheme 6. Synthesis of 13 started from reaction of 2-oxobutyric acid and hydrazine 52 to produce 53, which after hydrolysis was converted to nitrile via amide to obtain indole derivative 59a. To synthesize aldehydes 60b–d, we used Hemetsberger indole synthesis.30 First, acids 56a,b were converted to azidoacrylates 57a,b. To obtain 57c, direct iodination of phenol 54 at para position was performed after protection of the hydroxyl group followed by conversion of the intermediate to aldehyde with simultaneous deprotection of phenol, which was then alkylated to yield 55 and further converted to afford 57c. Compounds 58a–c were obtained by refluxing of azidoacrylates 57a–c, resulting in products that precipitated out upon cooling. Subsequently, the 2-cyanoindole compounds 59a–d were obtained after conversion of esters 53 or 58a–c to nitriles, Scheme 6. Aldehydes 60a,c,d were synthesized by adapting the procedure described by Moyer et al.,31 while aldehyde 60b was prepared by free radical dibromination and hydrolysis of halo derivative in the presence of silver carbonate. Reductive amination with 39 went efficiently to produce 14, 15, 21 with good yields except for 13 (28% yield). Subsequently compound 20 was obtained from 21 by demethylation, while 25 and 26 were produced by direct alkylation of 14 and 21, respectively.
Scheme 6. Synthesis of Compounds 13–15, 20, 21, 25, and 26a.
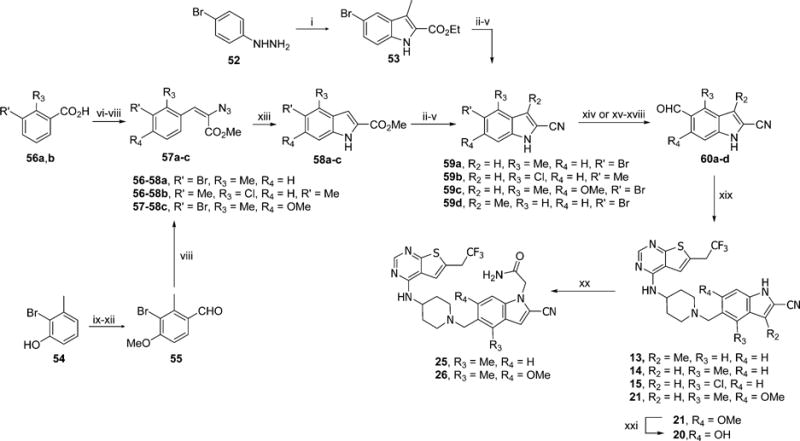
aReagents and conditions: (i) 2-oxobutyric acid, H2SO4, EtOH, reflux, 8 h, 58%; (ii) KOH, MeOH, reflux, 3 h; (iii) oxalyl chloride, DCM, rt, 1 h; (iv) NH3·H2O, rt, 2 h; (v) POCl3, CHCl3, reflux, 4 h, 56–96% (4 steps); (vi) LAH, THF, 0 °C to rt, 3 h, 80–97%; (vii) PCC, DCM, rt, 5 h, 78–86%; (viii) methyl azidoacetate, MeONa, MeOH, −15 to 4 °C, overnight 73–83%; (ix) TBDPSCl, imidazole, DCM, rt, 2 h; (x) NIS, TFA, CH3CN, 50 °C, 12 h, 92% (2 steps); (xi) BuLi, DMF, THF, −78 to 0 °C, 2 h; (xii) MeI, Me2CO, reflux, 2 h, 87% (2 steps); (xiii) (synthesis of 60a,c,d) xylene, reflux, 0.2 h, 50–81%; (xiv) KH, t-BuLi, DMF, THF, −90 to 0 °C, 2 h, 32–89%; (xv) (synthesis of 60b) Boc2O, DMAP, CH3CN, rt, 1 h, 96%; (xvi) 2.2 equiv of NBS, AIBN, CCl4, reflux, 5 h; (xvii) Ag2CO3, Me2CO/H2O, rt, 24 h, 60% (2 steps); (xviii) TFA, DCM, rt, 1 h, 81%; (ix) 39, Et3N, NaBH(OAc)3, DCM, rt, overnight, 28–93%; (xx) 2-bromoacetamide, Cs2CO3, DMF, rt, 4 h, 25–37%; (xxi) BBr3, DCM, rt, 24 h, 68%.
Compounds 16–19 were synthesized using the procedure shown in Scheme 7. First, the azidoacrylates 62a,b were synthesized from commercially available aldehydes 61a,b. Then, the rhodium trfluoroacetate catalyzed reaction was adapted as a cyclization method,32 resulting in 63a,b, which were subsequently converted to nitriles and protected with Boc group. Synthesis of compounds 16 and 18 was performed by bromination followed by nucleophilic substitution with 39 and removal of Boc protecting group. Compound 17 was obtained after demethylation of 18 with boron tribromide that was further alkylated with ethyl iodide to get final product 19.
Scheme 7. Synthesis of Compounds 16–19a.
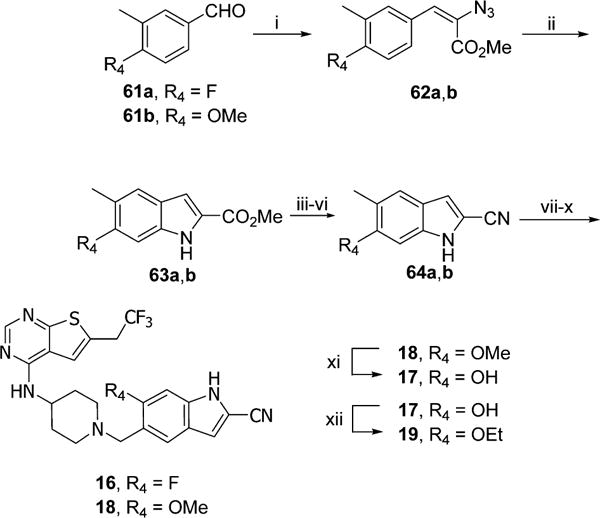
aReagents and conditions: (i) methyl azidoacetate, MeONa, MeOH, −15 to 4 °C, 60–65%; (ii) Rh2(CF3CO2)4, toluene, 50 °C, 24 h, 33–75%; (iii) KOH, MeOH, reflux, 3 h; (iv) oxalyl chloride, DCM, rt, 1 h; (v) NH3·H2O, rt, 2 h; (vi) POCl3, CHCl3, reflux, 4 h, 58–62% (4 steps); (vii) Boc2O, CH3CN, rt, 1 h; (viii) NBS, AIBN, CCl4, reflux, 2 h; (ix) 39, DIEA, DCM, 5 h; (x) SnCl4, CH3CN, 0 °C to rt, 0.5 h, 50–77% (4 steps); (xi) BBr3, DCM, rt, 24 h, 56%; (xii) EtI, K2CO3, CH3CN, rt, 24 h, 61%.
The synthesis of compounds 23, 24, 27–34, with multiple substituents on indole ring is depicted in Scheme 8. To prepare alcohol 24, the hydroxyethyl substituent was introduced by alkylation of 60c with trityl protected bromoethanol to afford 69. Pyrazole derivatives 27–31 were synthesized from commercially available ester 65, which was first protected with trityl group, then reduced to alcohol 66 and transformed to bromide 67 which was used immediately for alkylation of aldehyde 60a, 60c, or 68b, resulting in 70, 71, or 72. The triazol moiety was introduced through alkylation of 60c via click chemistry to obtain 73. Reductive amination and subsequent deprotection afforded the desired products 24, 28, 29, 31, and 34, Scheme 8. Finally demethylation of 24, 28, 31, and 34 resulted in 23, 27, 30, and 33, respectively.
Scheme 8. Synthesis of Compounds 23, 24, 27–31, 33, and 34a.
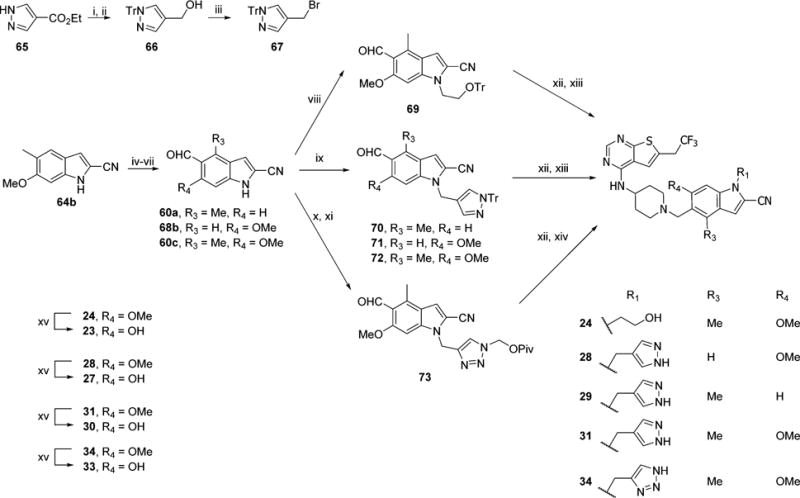
aReagents and conditions: (i) t-BuOK, TrCl, DMF, rt, 2 h; (ii) LAH, THF, 0 °C to rt, 2 h, 97% (2 steps); (iii) NBS, PPh3, DCM, rt, 0.5 h, 79%; (iv) Boc2O, DMAP, CH3CN, rt, 1 h; (v) 2.2 equiv of NBS, AIBN, CCl4, reflux, 5 h; (vi) Ag2CO3, Me2CO/H2O, rt, 24 h (2 steps); (vii) TFA, DCM, rt, 1 h, 38% (4 steps); (viii) BrCH2CH2OTr, Cs2CO3, DMF, rt, 2 h, 50%; (ix) 67, Cs2CO3, DMF, rt, 0.5 h, 82–94%; (x) propargyl bromide, Cs2CO3, DMF, rt, 4 h; (xi) t-BuCO2CH2N3, CuSO4, sodium ascorbate, t-BuOH/H2O, 60 °C, 4 h, 90%; (xii) 39, Et3N, NaBH(OAc)3, DCM, rt, 15 h; (xiii) HCl, MeOH, rt, 1 h, 50–72% (2 steps); (xiv) KOH, MeOH, rt, 1 h, 87% (2 steps); (xv) BBr3, DCM, rt, 24 h, 17–49%.
CONCLUSIONS
Development of very potent small molecule inhibitors of protein–protein interactions with optimized druglike properties, including cellular permeability and pharmacokinetic profile, is considered a challenging task due to the increased molecular weight of such compounds often required to achieve high potency.18,20 Here, we report optimization of a thienopyrimidine class of compounds, which block the protein–protein interaction between menin and MLL fusion proteins as a potential therapeutic strategy for MLL leukemia.1 The difficulty in developing menin–MLL inhibitors partly arises from the large size of the MLL binding site on menin (over 5000 Å3),12 implying that compounds with a relatively large molecular weight might be required to effectively block this PPI, complicating the optimization of their druglike properties.
In this study, we focused on structure-based optimization of our new class of menin–MLL inhibitors, represented by the lead compound 1,16 with the goal to identify compounds with improved activity and druglike properties and to select compound(s) for characterization in biological experiments. On the basis of the crystal structure of the menin–1 complex, we designed, synthesized, and extensively evaluated series of novel analogues by systematic exploration of substituents on the cyanoindole ring of 1. The most potent mono- and multisubstituted analogues demonstrated up to a few fold increase in in vitro activity over the lead compound 1. Furthermore, we discovered that incorporation of substituents at least at two positions of the 2-cyanoindole ring was required to substantially increase both cellular activity and metabolic stability (e.g., compounds 20, 21, 27, 29, 32, Table 3), with ~7-fold increased GI50 value for 27. Interestingly, favorable substituents introduced simultaneously at three positions of the cyanoindole ring did not have an additive effect on cellular activity, and in fact we observed a reduced effect on proliferation of the MLL leukemia cell for such compounds (e.g., trisubstituted compound 30 has weaker cellular activity than disubstituted compound 20 or 27) and on their metabolic stability (e.g., compound 30 as compared to 20). Furthermore, we found that poor cellular permeability (e.g., high efflux ratio) for some of the compounds reported here (e.g., for 7, 3-(RS), and 25) likely leads to the discrepancy in their in vitro and cellular activity. Other effects, including possible variations in the binding kinetics, stability in cells, and solubility of compounds as well as the need to compete with the full length MLL or MLL fusion proteins in cells, might also contribute to the observed differences between in vitro and cellular activity. Overall, these results highlight difficulties in lead optimization process, in particular for inhibitors targeting protein–protein interactions.
Our efforts led to the discovery of several menin–MLL inhibitors with greatly improved potency and druglike properties, out of which 27 displayed (1) strong in vitro inhibition of the menin–MLL interaction, IC50 = 21 nM, and the most potent binding affinity to menin (Kd = 6.5 nM) obtained for small molecule menin–MLL inhibitor reported to date; (2) high potency in inhibiting proliferation of MLL-AF9 transformed cells (GI50 = 83 nM), demonstrating the best cellular activity observed for any menin–MLL inhibitor reported to date; (3) specific mechanism of action in MLL leukemia cells; (4) superior, over 100-fold, selectivity to the MLL leukemia cells over control cell line; (5) improved polarity as compared to parent compound 1 (clogP = 3.7 for 27); (6) greatly improved PK profile, including high oral bioavailability; (7) pronounced efficacy in vivo in mice model of MLL leukemia. Overall, the development of 27 represents a great achievement for this new class of menin–MLL inhibitors. In fact this compound demonstrates a significant improvement over our recently reported MI-463 and MI-50316 in terms of cellular activity in MLL-AF9 cells, selectivity to MLL leukemia cells, and polarity (clogP value), Tables 2 and 3, making it an excellent candidate for development of a therapeutically useful compound. Furthermore, 27 has a favorable ligand efficiency index (LE = 0.28), which is above the average value of 0.24 reported for PPI inhibitors.33 Compound 27 discovered in this study constitutes a valuable addition to our menin–MLL inhibitor toolbox to investigate the role of menin–MLL interaction in physiological and disease state and for potential therapeutic applications. Overall, this study demonstrates that property optimization of inhibitors targeting protein–protein interactions represents a complex and multidimensional problem, with an outcome difficult to predict when a particular set of substituents is explored. The difficulties in optimizing PPI inhibitors are at least partly associated with the relatively large molecular weight of these compounds (e.g., most of the compounds reported here have Mw > 500 Da) and rather high hydrophobicity often required for potent binding to the PPI interfaces.25 Extensive medicinal chemistry efforts strongly supported by structure-based design are required to successfully conduct such studies to identify potent PPI inhibitors with optimized physicochemical properties and for advancing such compound to future potential therapeutics.
EXPERIMENTAL SECTION
Expression and Purification of Menin
The expression and purification of menin have been described previously.9,12
Biochemical Characterization of Menin–MLL Inhibitors
Inhibition of the menin–MLL interaction by small molecules was assessed by fluorescence polarization (FP) assay using the protocol described previously.9,14 Briefly, the fluorescein-labeled MLL (MBM1) peptide at 10 nM, menin at 100 nM, and varying concentrations of compounds were used for IC50 determination in the FP buffer (50 mM Tris, pH 7.5, 50 mM NaCl, 1 mM DTT). Compounds (5% final DMSO concentration) were added to the menin–MLL peptide complex and incubated for 1 h before changes in fluorescence polarization were measured using the PHERAstar microplate reader (BMG).
Isothermal Titration Calorimetry
Menin was extensively dialyzed at 4 °C against ITC buffer (50 mM phosphate, pH 7.5, 50 mM NaCl, 1 mM β-mercaptoethanol) and degassed prior to measurement. Compound 27 was dissolved in DMSO and diluted with the ITC buffer to final concentration (60 μM, 5% DMSO). Protein solution (6 μM) was adjusted to contain 5% DMSO final concentration. The titrations were performed using a VP-ITC titration calorimetric system (MicroCal) at 25 °C. The calorimetric cell, containing menin, was titrated with 27 injected in 10 μL aliquots. Data were analyzed using Origin 7.0 (OriginLab) to obtain Kd and stoichiometry.
Crystallization of Menin Complexes with Small Molecule Inhibitors
For cocrystallization experiments 2.5 mg/mL menin was incubated with small molecule inhibitors at 1:3 molar ratio. Crystals were obtained using the sitting drop technique at 10 °C by applying the procedure described previously.12 Prior to data collection, crystals were transferred into a cryo-solution containing 20% PEG550 MME and flash-frozen in liquid nitrogen.
Crystallographic Data Collection and Structure Determination
Diffraction data for menin and menin complexes were collected at the 21-ID-D and 21-ID-F beamlines at the Life Sciences Collaborative Access Team at the Advanced Photon Source. Data were processed with HKL-2000.34 Structures of the complexes were determined by molecular replacement using MOLREP with the apo-structure of human menin (PDB code 4GPQ) as a search model in molecular replacement. The model was refined using REFMAC,35 COOT,36 and the CCP4 package.37 Validation of the structures was performed using MOLPROBITY38 and ADIT.39 Details of data processing and refinement are summarized in Supporting Information Table 3. Coordinates and structure factors for menin–inhibitor complexes have been deposited in the Protein Data Bank under the following codes: 5DB0 (menin–3-(RS)), 5DB1 (menin–18), 5DB2 (menin–7), and 5DB3 (menin–31).
Compound Profiling
Profiling of compound 1 was outsourced to the Contract Research Organizations (CROs). Kinase profiling was performed by Nanosyn (http://www.nanosyn.com/productsandservices/screening-and-profiling/) and GPCR profiling was performed by Millipore (http://www.emdmillipore.com/).
Caco-2 Studies
The Caco-2 membrane permeability studies were performed at Cyprotex (www.cyprotex.com).
Viability Assays
The MLL-AF9 transformed murine bone marrow cells (BMCs) were prepared as described previously.40 MV4;11, MOLM-13, and HL-60 cells were cultured in RPMI-1640 medium with 10% FBS, 1% penicillin/streptomycin, and NEAA (nonessential amino acid) solution. For viability assay, MOLM-13 (1 × 105/mL), MV4;11 (1 × 105/mL), HL-60 (2 × 105/mL) human leukemia cells as well as MLL-AF9 and HM-2 murine bone marrow cells (2.5 × 104/mL) were plated (1 mL/well), treated with compounds or 0.25% DMSO, and cultured at 37 °C for 7 days. Media were changed at day 4 with viable cell number restored to the original concentration, and compounds were resupplied. An amount of 100 μL of cell suspension was transferred to 96-well plates for each sample in quadruplicates. A Vybrant MTT cell proliferation assay kit (Molecular Probes) was employed. Plates were read for absorbance at 570 nm using a PHERAstar BMG microplate reader. The experiments were performed 2–3 times in quadruplicate with calculation of mean and standard deviation for each condition.
Real-Time PCR
Effect of menin–MLL inhibitors on expression level of Hoxa9 and Meis1 was assessed by real-time quantitative PCR (qRT-PCR) using the protocol described previously.12
Cytospin/Wright–Giemsa Staining
Mouse bone marrow cells transduced with MLL-AF9 were plated in 12-well plates (1 mL/well) at an initial concentration of 3 × 105/mL cells, treated with compounds or 0.25% DMSO, and incubated at 37 °C in a 5% CO2 incubator. Cytospins were prepared as described previously14 at designated time points.
Microsomal Stability Studies
The metabolic stability was assessed using CD-1 mouse liver microsomes. 1 μM compounds were incubated with 1.3 mg/mL microsomes and 1.7 mM cofactor NADPH in 0.1 M phosphate buffer (pH = 7.4) containing 3.3 mM MgCl2 at 37 °C. The DMSO concentration was less than 0.1% in the final incubation system. At 0, 3, 5, 10, 15, 30, 45, and 60 min of incubation, the reactions were stopped by adding 3-fold excess of acetonitrile containing 100 ng/mL of internal standard for quantification. The collected fractions were centrifuged at 3000g for 20 min to collect the supernatant for LC–MS/MS analysis, from which the amount of compound remaining was determined. The natural log of the amount of compound remaining was plotted against time to determine the disappearance rate and the half-life of tested compounds.
Pharmacokinetic Studies in Mice
All animal experiments in this study were approved by the University of Michigan Committee on Use and Care of Animals and Unit for Laboratory Animal Medicine (ULAM).
The pharmacokinetics of menin–MLL inhibitors was determined in female C57BL/6 mice following intravenous (iv) dosing at 15 mg/kg and oral dosing (po) at 30 mg/kg. Compounds were dissolved in the vehicle containing 25% (v/v) DMSO, 25% (v/v) PEG-400, and 50% (v/v) PBS. Serial blood samples (50 μL) were collected over 24 h, centrifuged at 15 000 rpm for 10 min, and saved for analysis. Plasma concentrations of the compounds were determined by the LC–MS/MS method developed and validated for this study. The LC–MS/MS method consisted of an Agilent 1200 HPLC system, and chromatographic separation of tested compound was achieved using an Agilent Zorbax Extend-C18 column (5 cm × 2.1 mm, 3.5 μm; Waters). An AB Sciex QTrap 3200 mass spectrometer equipped with an electrospray ionization source (ABI-Sciex, Toronto, Canada) in the positive-ion multiple reaction monitoring (MRM) mode was used for detection. All pharmacokinetic parameters were calculated by noncompartmental methods using WinNonlin, version 3.2 (Pharsight Corporation, Mountain View, CA, USA). Similar procedure was applied for detection of glucuronidation of compound 27 in blood plasma after iv administration of the compound (15 mg/kg).
In Vivo Efficacy Studies
Freshly thawed MV4;11 cells (ATCC) were cultured in RPMI-1640 medium with 10% FBS and penicillin/streptomycin. On the day of injection, cells were resuspended in serum free medium at 1 × 108 cells/mL and mixed 1:1 with Matrigel. 5 ×106 cells (100 μL) were injected into the right flank of 4- to 6-week-old female BALB/c nude mice. Treatment started when tumor size has reached ~100 mm3. Mice were randomly grouped with each group containing eight mice. Vehicle or compounds were administrated once daily at designated doses using ip injections for 2 weeks. Body weight and tumor sizes were monitored three times a week. The tumor size was calculated as tumor volume = ab2/2 (a, long diameter; b, short diameter). All animal studies were performed under the Guidelines of the University of Michigan Committee for Use and Care of Animals.
General Chemistry
All solvents and reagents were used as obtained from commercial sources unless otherwise indicated. The 1H NMR data were taken on Bruker Avance III 600 MHz or Varian MR400. Chemical shifts are reported in ppm relative to tetramethylsilane or residual solvent signal. The mass measurements were determined on a Micromass LCT time-of-flight mass spectrometer using positive mode and electrospray ionization. The exact mass measurements were determined on Agilent Q-TOF time-of-flight mass spectrometer using positive ion mode and electrospray ionization. The melting points were determined on PerkinElmer DSC-7 differential scanning calorimeter and are not corrected. Analytical TLC was performed on Merck TLC aluminum plates precoated with F254 silica gel 60 (UV, 254 nm, and iodine). Infrared (IR) spectra were recorded on PerkinElmer FT-IR Spectrum BX on neat powder. The purity analysis of final compounds was determined on Shimadzu Prominence HPLC system (20 series: binary pump, UV/vis at 254 nm, heated column compartment 30 °C), using Restek Ultra C18 (5 μm) 150 mm × 4.6 mm column. The solvents were programmed to run at gradient starting 20% CH3CN in water to 80% during 8 min run. If not indicated, the purity of all final compounds was >95% as determined by HPLC via integration of UV spectra at 254 nm. All target compounds 1–34 were tested as hydrochloride salts unless otherwise stated. The standard procedure for HCl salt formation included dissolving in minimum volume of methanol, addition of 1 equiv of 1 M HCl in water, and thorough drying. Hydrochloride salts of inhibitors exist as a mixture of rotamers in solution of either DMSO or MeOH with approximate ratio of 10:1; NMR is reported for the major rotamer.
6-(2,2,2-Trifluoroethyl)thieno[2,3-d]pyrimidin-4(3H)-one (37)
4,4,4-Trifluorobuteraldehyde 20 g (158 mmol), cyanoacetamide 13.44 g (158 mmol), sulfur 5.12 g (158 mmol), and 27 mL (190 mmol) of Et3N were stirred in 160 mL of DMF for 24 h. Solvent was evaporated under reduced pressure. The 2-amino-5-(2,2,2-trifluoroethyl)-thiophene-3-carboxamide intermediate was obtained as an oil, which was dissolved in a mixture of 110 mL of triethyl orthoformate and 80 mL of acetic acid and refluxed for 4 h (monitored by TLC, EtOAc). Excess of volatiles was removed under reduced pressure to form an oily solid. 200 mL of hexane/EtOAc (1:1) was added. After stirring for 30 min the precipitate was filtered off, washed with 100 mL of hexane/EtOAc (1:1), and dried on filter affording 28 g of 37, 76% yield. 1H NMR CD3OD (600 MHz): δ 12.6 (br s, 1H), 8.14 (s, 1H), 7.42 (s, 1H), 4.07 (q, J = 11.0 Hz, 2H). 13C NMR CD3OD (150 MHz): δ 164.5, 157.01, 146.1, 128.4, 124.6, 123.5, 33.6 (q, J = 31.5 Hz).
4-Chloro-6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidine (38)
28 g (119 mmol) of 37 was added to 70 mL of POCl3 with 2 drops of DMF. The heterogeneous mixture was refluxed until all solid dissolved and no starting material was observed on TLC (3 h) and then evaporated to remove the excess of phosphorus oxychloride. The residue was quenched carefully with ice and saturated ammonia solution with additional external cooling (CAUTION: highly exothermic reaction). 300 mL of chloroform was added, and the mixture was transferred in a separatory funnel. Water phase was extracted with additional 100 mL of chloroform. Combined organic extracts were evaporated (no drying), and the residual material was used in the next step without further purification, 28 g of 38, 92% yield. 1H NMR CDCl3 (600 MHz): δ 8.86 (s, 1H), 7.39 (s, 1H), 3.76 (q, J = 9.9 Hz, 2H). 13C NMR CDCl3 (150 MHz): δ 169.0, 154.7, 153.2, 129.9, 125.3, 123.5, 121.3, 35.9 (q, J = 33.0 Hz).
N-(Piperidin-4-yl)-6-(2,2,2-trifluoroethyl)thieno[2,3-d]-pyrimidin-4-amine Hydrochloride (39)
28 g (111 mmol) of 4-chloro-6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidine 38, 26.64 g (133 mmol) of 1-Boc-4-aminopiperidine, and 40 mL (228 mmol) of N,N-diisopropylethylamine were stirred under reflux in 400 mL of IPA for 24 h. Reaction mixture was evaporated to dryness, and then 500 mL of water was added resulting in precipitation of the crude product. After stirring for 12 h product was filtered off, washed with 1 L of water, and dried on filter overnight. Solid was transferred to a flask and stirred with 200 mL of diethyl ether for 12 h. The product was filtered off, washed with 50 mL of hexane/EtOAc (1:1) and 50 mL of hexane/EtOAc (3:1). Thorough drying afforded 31 g of Boc-protected intermediate, 64% yield. 1H NMR CD3CN (600 MHz): δ 8.34 (s, 1H), 7.35 (s, 1H), 6.16 (br s, 1H), 4.29 (m, 1H), 4.05 (m, 2H), 3.82 (q, J = 9.9 Hz, 2H), 2.91 (m, 2H), 1.97 (m, 2H), 1.47 (m, 2H), 1.44 (s, 9H). Boc protected intermediate was dissolved in 60 mL of mixture DCM/MeOH 10:1. 100 mL of 4 M HCl solution in dioxane was added, and biphasic mixture was stirred overnight (no staring material was observed on TLC). The reaction mixture was evaporated to dryness, dissolved in 200 mL of MeOH, and evaporated again forming an oily residue. Oil was dissolved in 100 mL of MeOH and stored in freezer overnight. The precipitated product was filtered off, dried on funnel for several hours to afford 27 g of 39. Free base 1H NMR CDCl3 (600 MHz): δ 8.47 (s, 1H), 7.13 (s, 1H), 5.32 (d, J = 7.7 Hz, 1H), 4.32 (m, 1H), 3.64 (q, J = 10 Hz, 2H), 3.19 (m, 2H), 2.83 (m, 2H), 2.57 (br s, 1H), 2.14 (m, 2H), 1.55 (m, 2H). 13C NMR CDCl3 (150 MHz): δ 166.9, 156.0, 154.3, 128.1, 126.6 (q, J = 276 Hz), 118.7, 116.5, 48.0, 45.3, 35.6 (q, J = 31.5 Hz), 33.1. ESI MS [M + H+] m/z: 317.2.
3-Fluoro-5-methyl-1H-indole-2-carboxylic Acid (40b)
The mixture of 790 mg (4.5 mmol) of 40a and 1.76 g (5 mmol) of 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis-(tetrafluoroborate) was refluxed in 10 mL of CH3CN for 12 h. Mixture was diluted with saturated NH4Cl and extracted with chloroform. Organic phase was concentrated and purified using preparative TLC (silica gel, hexane/EtOAc/AcOH (10:1:1) to afford 150 mg of 40b, 17% yield. 1H NMR CDCl3 (400 MHz): δ 10.03 (br s, 1H), 7.29 (s, 1H), 7.22 (d, J = 8.4 Hz, 1H), 7.02 (d, J = 8.4 Hz, 1H), 6.35 (br s, 1H), 2.33 (s, 3H).
5-Methyl-1H-indole-2-carbonitrile (41a)
A mixture of 0.5 g (2.9 mmol) of 5-methylindole-2-carboxylic acid, 0.25 mL of thionyl chloride (3.4 mmol), 5 mL of chloroform, and small drop of DMF was refluxed for 2 h. The reaction mixture was cooled to rt, poured into a mixture of 5 g of ice and 5 mL of 25% ammonia solution, and then stirred for 2 h. The precipitated product was filtered off, washed with water, and dried to yield 350 mg of 5-methylindole-2-carboxamide. It was added to 1.5 g (9.75 mmol) of phosphorus oxychloride and 8 mL of chloroform refluxed for 2 h. Then cooled solution was poured into 20 mL of water and stirred for 1 h. After separation the organic layer was dried over sodium sulfate and concentrated. The residue was purified by column chromatography (silica gel, hexane/EtOAc 5:1) to afford 245 mg of 5-methyl-1H-indole-2-carbonitrile (41a), 55% yield. 1H NMR CDCl3 (600 MHz): δ 8.61 (br s, 1H), 7.44 (s, 1H), 7.30 (d, J = 8.4 Hz, 1H), 7.21 (d, J = 8.4 Hz, 1H), 7.11 (s, 1H), 2.44 (s, 3H). 13C NMR CDCl3 (150 MHz): δ 135.3, 131.3, 128.3, 126.5, 121.3, 114.4, 114.0, 111.4, 106.1, 21.4.
3-Fluoro-5-methyl-1H-indole-2-carbonitrile (41b)
To obtain 41b, a similar procedure as described for 41b was used but starting with 150 mg (0.78 mmol) of 40b. Isolated 60 mg, 44% yield. 1H NMR CDCl3 (600 MHz): δ 9.02 (br s, 1H), 7.56 (s, 1H), 7.37 (d, J = 8.4 Hz, 1H), 7.24 (d, J = 8.4 Hz, 1H), 7.15 (s, 1H), 2.46 (s, 3H).
tert-Butyl 2-Cyano-5-methyl-1H-indole-1-carboxylate (42a)
To a solution of 245 mg (1.6 mmol) of 5-methyl-1H-indole-2-carbonitrile in 5 mL of acetonitrile, 0.434 mL (1.9 mmol) of Boc2O and 29 mg (0.24 mmol) of DMAP were added and stirred at room temperature for 1 h. The solvent was removed in vacuo, and the resultant crude product was purified by column chromatography (silica gel, hexane/EtOAc, 10:1) to afford 334 mg of 42a, 83% yield. 1H NMR CDCl3 (600 MHz): δ 8.10 (d, J = 8.8 Hz, 1H), 7.39 (s, 1H), 7.31 (d, J = 8.8 Hz, 1H), 7.26 (s, 1H), 2.45 (s, 3H), 1.72, (s, 9H). 13C NMR CDCl3 (150 MHz): δ 148.2, 134.9, 133.8, 129.9, 121.6, 121.2, 115.5, 113.5, 108.8, 85.6, 28.1, 21.2.
tert-Butyl 2-Cyano-3-fluoro-5-methyl-1H-indole-1-carboxylate (42b)
To obtain 42b, a similar procedure as described for 41b was used but starting with 60 mg (0.34 mmol) of 41b. Isolated 74 mg, 79% yield. 1H NMR CDCl3 (600 MHz): δ 9.02 (br s, 1H), 7.56 (s, 1H), 7.37 (d, J = 8.4 Hz, 1H), 7.24 (d, J = 8.4 Hz, 1H), 7.15 (s, 1H), 2.46 (s, 3H).
5-((4-((6-(2,2,2-Trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)-amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (1)
To a stirred solution of 334 mg (1.3 mmol) of tert-butyl 2-cyano-5-methyl-1H-indole-1-carboxylate in carbon tetrachloride (5 mL), 232 mg (1.3 mmol) of N-bromosuccinimide and 11 mg (0.065 mmol) of AIBN were added. The mixture was refluxed for 1 h, then cooled, concentrated, and the residue was filtered through silica gel column, using hexane/EtOAc (10:1) to give crude tert-butyl 2-cyano-5-bromomethyl-1H-indole-1-carboxylate. 340 mg (1.06 mmol) of tert-butyl 2-cyano-5-bromomethyl-1H-indole-1-carboxylate and 375 mg (1.06 mmol) of N-(piperidin-4-yl)-6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-amine hydrochloride were dissolved in 12 mL of DCM. 820 mg (3.2 mmol) of DIEA was added to that solution, and reaction mixture was stirred for 18 h. Then reaction mixture was directly loaded on silica gel column and the product was eluted with DCM–MeOH (30:1). After evaporation of solvent, the Boc-protected intermediate was dissolved in 10 mL of CH3CN and 1.27 mL (11 mmol) of SnCl4 was added. The homogeneous reaction mixture was stirred for 1 h, and then all volatiles were removed in vacuo. The residue was quenched with ammonia solution and extracted with ethyl acetate. Combined organic fractions were dried over MgSO4 and concentrated. The residue was purified by column chromatography (silica gel, hexane/EtOAc/MeOH, 1:1:0.1) to afford 331 mg of 5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]-pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile, 1. Its monohydrochloride salt was obtained by adding 1 equiv of 1 M HCl solution in diethyl ether to a solution of compound in ethanol and thorough drying to afford 1 in 53% yield. 1H NMR DMSO-d6 (600 MHz): δ 12.62 (s, 1H), 10.74 (br s, 1H), 8.33 (s, 1H), 8.07 (d, J = 7 Hz, 1H), 7.93 (s, 1H), 7.70 (s, 1H), 7.62 (d, J = 12 Hz, 1H), 7.56 (d, J = 12 Hz, 1H), 7.45 (s, 1H), 4.36 (s, 2H), 4.30 (m, 1H), 4.03 (q, J = 11 Hz, 2H), 3.41 (m, 2H), 3.11 (m, 2H), 2.12 (m, 2H), 1.98 (m, 2H). 13C NMR DMSO-d6 (150 MHz): δ 165.9, 155.7, 153.8, 137.2, 128.4, 127.0, 125.8, 125.4 (q, J = 276 Hz), 125.4, 122.2, 121.3, 116.1, 114.2, 113.4, 112.5, 106.8, 59.3, 50.4, 45.5, 33.7 (q, J = 33 Hz), 28.2. HRMS (ESI): [M + H+] calculated 471.1573; found 471.1579.
3-Fluoro-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]-pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (11)
To prepare 11, a similar procedure as for 1 but starting was applied using 74 mg (0.26 mmol) of 42b. Isolated 43 mg, 34% yield. 1H NMR CD3OD (600 MHz): δ 8.71 (s, 1H), 7.95 (s, 1H), 7.84 (s, 1H), 7.65 (d, J = 7 Hz, 1H), 7.53 (d, J = 7 Hz, 1H), 4.65 (m, 1H), 4.53 (s, 2H), 4.00 (q, J = 11 Hz, 2H), 3.65 (m, 2H), 3.34 (m, 2H), 2.37 (m, 2H), 2.15 (m, 2H). 13C NMR CD3OD (150 MHz): δ 157.4, 153.0, 151.2, 149.6, 136.0, 133.0, 130.7, 125.3 (q, J = 276 Hz), 123.3, 122.8, 118.7, 115.8, 115.8, 114.7, 111.7, 94.0, 61.6, 48.0, 35.2(q, J = 33 Hz), 29.5. HRMS (ESI): [M + H+] calculated 489.1479; found 489.1484.
2-(2-Cyano-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]-pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indol-1-yl)-acetamide (7)
760 mg (1.5 mmol) of 1 (free base) and 207 mg (1.5 mmol) of bromoacetamide were dissolved in 3.6 mL of dry DMF. Then 1.96 g (6 mmol) of cesium carbonate was added and reaction mixture was stirred for 4 h. The reaction was quenched with 50 mL of water and extracted with DCM/MeOH (10:1). Combined organic extracts were evaporated with silica gel and purified by column chromatography (silica gel, DCM/MeOH 10:1). The solvent was evaporated and recrystallized from MeOH to produce 319 mg of 2-(2-cyano-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)-methyl)-1H-indol-1-yl)acetamide (7), which was converted to hydrochloride salt by dissolving in 5 mL of MeOH and adding 1 equiv of 1 M HCl in water. After evaporation and drying hydrochloride product was obtained as white crystalline material (7), 40% yield. 1H NMR CD3OD (600 MHz): δ 8.44 (s, 1H), 7.91 (s, 1H), 7.57 (m, 2H), 7.41 (s, 1H), 5.10 (s, 2H), 4.53 (m, 2H), 4.47 (s, 1H), 3.89 (q, J = 10.3 Hz, 2H), 3.62 (m, 2H), 3.24 (m, 2H), 2.35 (m, 2H), 1.93 (m, 2H). 13C NMR CD3OD (150 MHz): δ 171.1, 157.7, 153.6, 140.7, 130.8, 129.6, 128.00, 127.1, 126.6 (q, J = 276 Hz), 123.5, 121.9, 118.3, 114.8, 113.7, 113.5, 112.6, 62.0, 52.7, 49.6, 48.00, 47.4, 35.5 (q, J = 33 Hz), 30.0. HRMS (ESI): [M + H+] calculated 528.1788; found 528.1783.
2-(2-Cyano-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]-pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indol-1-yl)-propanamide (8)
Similar procedure as for 7 but starting with 50 mg (0.11 mmol) of 1. Isolated 16 mg, 27% yield. 1H NMR CD3OD (600 MHz): δ 8.33 (s, 1H), 8.04 (s, 1H), 7.74 (m, 1H), 7.62 (m, 2H), 7.53 (s, 1H), 5.22 (q, J = 9.8 Hz, 1H), 4.50 (m, 3H), 3.85 (q, J = 10.3 Hz, 2H), 3.62 (m, 2H), 3.26 (m, 2H), 2.33 (m, 2H), 2.02 (m, 2H), 1.56 (d, J = 9.8 Hz, 3H). 13C NMR CD3OD (150 MHz): δ 167.0, 161.6, 157.7, 154.8, 140.27, 139.1, 130.0, 129.2, 128.6, 128.2, 126.6 (q, J = 276 Hz), 123.9, 122.5, 121.8, 118.2, 113.0, 111.0, 107.01, 54.1, 52.8, 47.1, 35.6 (q, J = 33 Hz), 30.0, 15.8. HRMS (ESI): [M + H+] calculated 542.1944; found 542.1937.
5-Methyl-1-(2-(trityloxy)ethyl)-1H-indole-2-carbonitrile (44)
2.5 g (19.5 mmol) of 2-bromoethanol, 6 g (21.5 mmol) of trityl chloride, 95 mg (0.78 mmol) of DMAP, and 4.6 mL (33 mmol) of triethylamine were stirred in 20 mL of DCM for 24 h. Reaction mixture was diluted with water, washed with brine, dried over sodium sulfate, and concentrated to produce 6.9 g of crude 1-tritoxy-2-bromoethane that was used without purification. 468 mg (3 mmol) of 41a was dissolved in 6 mL of DMF, and 240 mg (6 mmol) of sodium hydride (60% suspension in oil) was added. After stirring for 5 min 1.32 g (3.6 mmol) of 1-tritoxy-2-bromoethane was added and reaction mixture was heated in a sand bath at 60 °C overnight. Reaction mixture was carefully quenched with ice and extracted with ethyl acetate. Organic fractions were evaporated with silica gel purified by column chromatography (silica gel, hexane/EtOAc 5:1) to afford 1.07 g of 44, 81% yield. 1H NMR CDCl3 (600 MHz): δ 8.02 (d, J = 8.8 Hz, 1H), 7.23 (m, 16H), 7.12 (d, J = 8.8 Hz, 1H), 7.03 (s, 1H), 4.32 (t, J = 5.3 Hz, 2H), 3.45 (t, J = 5.3 Hz, 2H), 2.43 (s, 3H).
1-(2-Hydroxyethyl)-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (2)
Bromination and alkylation were carried out as described for 1, starting with 1 g (2.4 mmol) of 44 to yield 1.6 g of trityl protected intermediate, which was dissolved in 2 mL of MeOH, and 0.5 mL of 12 M HCl was added. After 30 min the mixture was quenched with 1 mL of 30% NH3·H2O, diluted with 50 mL of DCM, and evaporated with silica gel. The product was purified by column chromatography (silica gel, hexane/EtOAc/MeOH 0.5:1:0.1) to afford 580 mg, 42% of pure 2, that was converted to hydrochloride salt. 1H NMR CD3OD (600 MHz): δ 8.75 (s, 1H), 7.96 (s, 1H), 7.88 (d, 1H, J = 8.8 Hz), 7.74 (d, 1H, J = 8.8 Hz), 7.64 (s, 1H), 7.37 (s, 1H), 4.67 (m, 1H), 4.51 (m, 3H), 4.03 (q, 2H, J = 10.6 Hz), 3.93 (m, 2H), 3.65 (m, 2H), 3.30 (m, 2H), 2.36 (m, 2H), 2.18 (m, 2H). 13C NMR CD3OD (150 MHz): δ 164.9, 139.7, 133.3, 129.2, 128.0, 127.1, 127.1, 126.4 (q, J = 275 Hz), 125.5, 123.1, 122.8, 122.8, 118.7, 114.2, 113.1, 113.0, 63.0, 62.0, 52.3, 49.6, 48.5, 35.1 (q, J = 33 Hz), 29.5. HRMS (ESI): [M + H+] calculated 515.1835; found 515.1828.
(S)-1-(2,3-Bis((tert-butyldimethylsilyl)oxy)propyl)-5-methyl-1H-indole-2-carbonitrile (46-(S))
331 mg (3 mmol) of (R)-3-chloropropane-1,2-diol and 530 mg (7.8 mmol) of imidazole were dissolved in 5 mL of dry DCM. Then 7.2 mL of 1 M TBSCl in DCM was added. Reaction mixture was stirred overnight and then diluted with 20 mL of water. After separation the organic layer was dried over sodium sulfate and concentrated to produce 850 mg of (R)-5-(chloromethyl)-2,2,3,3,8,8,9,9-octamethyl-4,7-dioxa-3,8-disiladecane, 84% yield, which was used in the next step. To a solution of 39 mg (0.25 mmol) of 5-methyl-1H-indole-2-carbonitrile in 0.5 mL of DMF, 15 mg (0.375 mmol) of NaH (60% suspension in oil) was added, and mixture was stirred for 30 min. Then 170 mg (0.5 mmol) of (R)-5-(chloromethyl)-2,2,3,3,8,8,9,9-octamethyl-4,7-dioxa-3,8-disiladecane was added and stirring continued for 24 h. The reaction mixture was diluted with 10 mL of water and extracted with DCM. Combined organic extracts were dried over sodium sulfate, concentrated, and purified by column chromatography (silica gel, hexane/EtOAc 50:1) to afford 56 mg of (S)-1-(2,3-bis((tert-butyldimethylsilyl)oxy)propyl)-5-methyl-1H-indole-2-carbonitrile (46-(S)), 49% yield. 1H NMR CDCl3 (600 MHz): δ 8.09 (d, 1H, J = 8.8 Hz), 7.35 (s, 1H), 7.24 (d, 1H, J = 8.8 Hz), 7.18 (s, 1H), 4.04 (m, 1H), 3.82 (m, 1H), 3.77 (m, 2H), 3.68 (m, 1H), 2.43 (s, 3H), 1.08 (m, 18H), 0.26 (m, 12H).
(S)-1-(3-((tert-Butyldiphenylsilyl)oxy)-2-methoxypropyl)-5-methyl-1H-indole-2-carbonitrile (47-(S))
824 mg (1.8 mmol) of 46-(S) was dissolved in 2 mL of MeOH, and 1 mL of 4 M HCl in dioxane was added. After 4 h TLC indicated no starting material, and reaction mixture was evaporated to dryness. The residue was dissolved in 20 mL of DCM, and 0.78 mL (3 mmol) of TBDPSCl was added followed by 218 mg (3.2 mmol) of imidazole. After 1 h reaction was washed with saturated sodium bicarbonate solution, dried over sodium sulfate, and concentrated to dryness. Crude mixture was dissolved in 4 mL of THF, and 220 mg (5.5 mmol) of NaH (60% suspension in mineral oil) was added. After stirring for 5 min, 0.34 mL (5.5 mmol) of MeI was added. Reaction mixture was carefully quenched with ice after 4 h and diluted with water. The product was extracted with diethyl ether, organic fractions were evaporated with silica gel, and the product was purified by column chromatography (silica gel, hexane/EtOAc). The product containing fractions were concentrated to afford 95 mg of 47-(S), 11% yield. 1H NMR CDCl3 (600 MHz): δ 8.11 (d, J = 8.8 Hz, 1H), 7.71(m, 4H), 7.45 (m, 2H), 7.38 (m, 4H), 7.34 (s, 1H), 7.24 (d, J = 8.8 Hz, 1H), 7.19 (s, 1H), 4.04 (m, 1H), 3.82 (m, 1H), 3.77 (m, 2H), 3.68 (m, 1H), 3.43 (s, 3H), 1.13 (s, 9H).
(S)-1-(2,3-Dihydroxypropyl)-5-((4-((6-(2,2,2-trifluoroethyl)-thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (3-(S))
To a stirred solution of 55 mg (0.12 mmol) of 47-(S) in 0.5 mL of carbon tetrachloride were added 21.3 mg of (0.12 mmol) NBS and 1.1 mg (0.0065 mmol) of AIBN. The mixture was refluxed for 1 h, then cooled, concentrated, and filtered through short silica gel plug using hexane/EtOAc (10:1) to give crude bromide. Bromide was then dissolved in 0.2 mL of DCM; 36 mg (0.12 mmol) of 39 and 52 mg (0.4 mmol) of DIEA were added to that solution, and reaction mixture was stirred for 18 h. Then reaction mixture was directly purified by column chromatography (silica gel, DCM/MeOH, 30:1). After evaporation of solvent TBS-protected intermediate was dissolved in 0.2 mL of MeOH and 0.02 mL of 12 M HCl was added. The homogeneous reaction mixture was stirred overnight, and then all volatiles were removed in vacuo. The residue was quenched with concentrated ammonia solution and extracted with ethyl acetate. Combined organic fractions were dried over MgSO4 and concentrated. The residue was purified by column chromatography (silica gel, DCM/MeOH, 20:1) to afford 15.9 mg of 5-((4-((6-(2,2,2-trifluoroethyl)-thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile, 3-(S), 24% yield. Its monohydrochloride salt was obtained by adding 1 equiv of 1 M HCl solution in diethyl ether to a solution of compound in ethanol. 3-(RS) and 3-(R) were synthesized in the same manner but starting with 45-(RS) and 45-(S), respectively, and analytical data matched those reported for 3-(S). 1H NMR CD3OD (600 MHz): δ 8.63 (s, 1H), 7.95 (s, 1H), 7.78 (s, 1H), 7.77 (d, J = 8.6 Hz, 1H), 7.63 (d, J = 8.6 Hz, 1H), 7.37 (s, 1H), 4.57 (m, 1H), 4.56 (m, 1H), 4.50 (s, 2H) 4.37 (m, 1H), 4.04 (m, 1H), 3.99 (q, J = 10.3 Hz, 2H), 3.65 (m, 2H), 3.60 (d, J = 5.5 Hz, 2H), 3.29 (m, 2H), 2.37 (m, 2H), 2.12 (m, 2H). 13C NMR CD3OD (150 MHz): δ 157.5, 151.0, 139.9, 132.3, 129.2, 127.9, 127.4, 127.0, 126.4 (q, J 275 Hz), 125.5, 123.1, 122.5, 118.6, 114.3, 114.3, 113.4, 113.3, 72.4, 64.9, 62.0, 52.4, 49.8, 48.1, 35.3 (q, J = 33 Hz), 29.6. HRMS (ESI): [M + H+] calculated 545.1941; found 545.1950.
(S)-1-(3-Hydroxy-2-methoxypropyl)-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (4-(S))
To obtain 4-(S), a similar procedure was used as for 3-(S) but starting with 95 mg (0.2 mmol) of 47-(S), and 65 mg of 4-(S) was isolated, 58% yield. 4-(R) was synthesized in the same manner but starting with 45-(S). Analytical data matched those reported for 4-(S). 1H NMR CD3OD (600 MHz): δ 8.44 (s, 1H), 7.92 (s, 1H), 7.74 (d, J = 8.6 Hz, 1H), 7.62 (s, 1H), 7.59 (d, J = 8.6 Hz, 1H), 7.36 (s, 1H), 4.54 (m, 2H), 4.47 (s, 2H), 4.44 (m, 1H), 3.89 (q, J = 10.3 Hz, 2H), 3.61 (m, 5H), 3.24 (m, 2H), 3.21 (s, 3H), 2.36 (m, 2H), 2.02 (m, 2H). 13C NMR CD3OD (150 MHz): δ 157.6, 153.4, 139.8, 130.9, 129.3, 127.9, 126.4 (q, J = 275 Hz), 123.3, 122.0, 118.34, 114.44, 114.2, 113.4, 113.3, 82.3, 62.0, 61.8, 58.6, 54.8, 52.7, 47.8, 47.5, 35.5 (q, J = 33 Hz), 29.9. HRMS (ESI): [M + H+] calculated 559.2098; found 559.2091.
2-Amino-5-methylbenzonitrile (49)
A mixture of 905 mg (5 mmol) of 48, 1 mL of thionyl chloride (12 mmol), 8 mL of chloroform, and small drop of DMF was refluxed for 2 h. The reaction mixture was cooled to rt, poured into a mixture of 10 g of ice and 10 mL of 25% NH3·H2O, and then stirred for 2 h. The precipitated product was filtered off, washed with water, and dried. It was added to 2.6 g (17 mmol) of phosphorus oxychloride and 8 mL of chloroform and then refluxed for 2 h. Cooled solution was poured into 20 mL of water and stirred for 1 h. After separation the organic layer was dried over sodium sulfate and concentrated. The residue was dissolved in 7 mL of ethanol, and 4.1 g (22 mmol) SnCl2 was added. The mixture was refluxed for 1 h and the silica gel was added followed by 2 mL of 25% NH3·H2O. All volatiles were removed and residue was purified by column chromatography (silica gel, hexane/EtOAc, 1:1) to afford 120 mg of 49, 18% yield. HRMS (ESI): [M + H+] calculated 133.0760; found 133.0769.
2-((Cyanomethyl)amino)-5-methylbenzonitrile (50)
120 mg (0.93 mmol) of 49, 42 mg (1.4 mmol) of paraformaldehyde, 506 mg (3.7 mmol) of ZnCl2, 90 mg (1.4 mmol) of potassium cyanide, and 5 μL of conc H2SO4 were dissolved in 2.8 mL of acetic acid heated to 60 °C for 6 h. Reaction mixture was diluted with water, neutralized with solid sodium bicarbonate, and extracted with EtOAc. Organic phase was dried over sodium sulfate and evaporated. The residue was purified by column chromatography (silica gel, hexane/EtOAc) to afford 63 mg of 50, 33% yield. 1H NMR CDCl3 (600 MHz): δ 7.30 (m, 2H), 6.73 (d, J = 8.6 Hz, 1H), 4.95 (br s, 1H), 4.19 (d, J = 6.6 Hz, 2H), 2.27 (s, 3H).
tert-Butyl 3-((1-Bis(tert-butoxycarbonyl)amino)-2-cyano-5-methyl-1H-indole-1-carboxylate (51)
126 mg (0.46 mmol) of 50, 22 mg (0.16 mmol) of DMAP were dissolved in 2 mL of CH3CN, and 0.42 mL (1.84 mmol) of Boc2O was added. After stirring for 4 h the reaction mixture was subjected to purification by preparative TLC (silica gel, hexane/EtOAc, 7:1), obtaining 53 mg (25% yield) of 51, along with 87 mg (25% yield) of di-Boc intermediate. 1H NMR CDCl3 (600 MHz): δ 8.11 (d, J = 8.6 Hz, 1H), 7.32 (d, J = 8.6 Hz, 1H), 7.22 (s, 1H), 2.45 (s, 3H), 1.72 (s, 9H), 1.45 (s, 18H).
3-Amino-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]-pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (12)
Similar procedure was used as for 1 but starting with 47 mg (0.1 mmol) of 51. Isolated 9.2 mg, 19% yield. 1H NMR CD3OD (600 MHz): δ 8.55 (s, 1H), 7.91 (s, 1H), 7.72 (s, 1H), 7.50 (d, J = 9 Hz, 1H), 7.39 (d, J = 9 Hz, 1H), 4.55 (m, 1H), 4.44 (s, 2H), 3.94 (q, J = 11 Hz, 2H), 3.62 (m, 2H), 3.25 (m, 2H), 2.34 (m, 2H), 2.08 (m, 2H). 13C NMR CD3OD (150 MHz): δ 157.4, 151.6, 138.7, 136.4, 131.9, 130.1, 127.4 (q, J = 276 Hz), 124.6, 122.4, 120.9, 120.4, 118.5, 115.2, 113.9, 93.0, 62.2, 52.4, 35.4 (q, J = 33 Hz), 29.7. HRMS (ESI): [M + H+] calculated 486.1682; found 486.1678.
5-Bromo-3-methyl-1H-indole-2-carboxylic Acid (53)
892 mg (4 mmol) of 53 hydrochloride and 408 mg (4 mmol) of 2-oxobutyric acid were refluxed in 6.5 mL of absolute ethanol in the presence of 2.4 g of H2SO4. After 8 h the reaction mixture was cooled in the freezer and the precipitate was filtered off to afford 655 mg of 53, 58% yield. 1H NMR CDCl3 (600 MHz): δ 9.19 (br s, 1H), 7.79 (s, 1H), 7.38 (d, J = 9.0 Hz, 1H), 7.29 (d, J = 9.0 Hz, 1H), 4.41 (q, J = 7.3 Hz, 2H), 2.57 (s, 3H), 1.43 (t, J = 7.3 Hz, 3H).
3-Bromo-4-methoxy-2-methylbenzaldehyde (55)
5 g (26 mmol) of 54 was dissolved in 200 mL of DCM, and 10.4 mL (40 mmol) of TBDPSCl was added followed by 3.5 g (52 mmol) of imidazole. After 1 h reaction was evaporated to dryness and the product was purified by column chromatography (silica gel, hexane/EtOAc, 50:1). Crude intermediate was dissolved in 130 mL of CH3CN/DCM 1:1, and 9.65 g (39 mmol) of NIS and 1.99 mL of TFA were added. The reaction mixture was heated at 50 °C for 12 h. Then it was diluted with 100 mL of DCM, washed with saturated NaHCO3 and Na2S2O3, evaporated with silica gel, and purified by column chromatography (silica gel, hexane/EtOAc, 50:1) to afford 13.5 g (2-bromo-4-iodo-3-methylphenoxy)(tert-butyl)diphenylsilane. 1H NMR CDCl3 (600 MHz): δ 7.71 (m, 4H), 7.45 (m 2H), 7.38 (m, 4H), 7.27 (d, overlaps with solvent residual signal, 1H), 6.02 (d, J = 8.8 Hz, 1H), 2.68 (s, 3H), 1.13 (s, 9H).
11.9 g (21.5 mmol) of (2-bromo-4-iodo-3-methylphenoxy)(tert-butyl)diphenylsilane was dissolved in dry 86 mL of THF, the flask was flushed with argon cooled to −78 °C (internal temperature, cooling bath EtOAc/N2(liq)), and 9.5 mL of 1.6 M butyllithium (26 mmol) was slowly added to maintain the temperature. After stirring for 0.5 h, 5 mL of DMF (75 mmol) was added dropwise and stirring was continued for 30 min at the same temperature. Then 100 mL of 1 M HCl was added and the reaction mixture was allowed to warm to rt. Organic fraction was evaporated with silica gel and the product was purified by column chromatography (silica gel, hexane/EtOAc, 1:1) to afford 4 g of 3-bromo-4-hydroxy-2-methylbenzaldehyde. It was dissolved in 60 mL of acetone, and 15.5 g (112 mmol) of K2CO3 and 13.7 mL (222 mmol) of methyl iodide were added. The reaction was refluxed overnight, then filtered off and concentrated to afford 4.3 g of 55, 88% yield over four steps. 1H NMR CDCl3 (600 MHz): δ 10.08 (s, 1H), 7.81 (s, 1H), 7.68 (d, J = 8.3 Hz, 1H), 6.96 (d, J 8.3 = Hz, 1H), 3.22 (s, 3H), 2.75 (s, 3H).
Methyl 2-Azido-3-(3-bromo-2-methylphenyl)acrylate (57a)
30 g (139 mmol) of 3-bromo-2-methylbenzoic acid was dissolved in 180 mL of THF, cooled to 0 °C, and 9.5 g (250 mmol) of LAH was added in small portions. After stirring for 3 h no starting material was observed by TLC. Reaction mixture was carefully quenched with 20 mL of ethyl acetate and 20 mL of water. Silica gel was added and mixture was evaporated to dryness and the product was purified by column chromatography (silica gel, hexane/EtOAc, 1:1), resulting in 24 g of pure alcohol intermediate. 1H NMR CDCl3 (600 MHz): δ 7.50 (d, J = 8.1 Hz, 1H), 7.29 (d, J = 8.1 Hz, 1H), 7.04 (t, J = 8.1 Hz, 1H), 4.68 (s, 2H), 2.40 (s, 3H), 1.90 (br s, 1H). 13C NMR CDCl3 (150 MHz): 140.6, 135.8, 132.0, 127.1, 126.7, 126.1.
24 g (119 mmol) of (3-bromo-2-methylphenyl)methanol was dissolved in 240 mL of dichloromethane, and 103 g (1.2 mol) of manganese(IV) oxide was added. After stirring overnight TLC showed no starting material. Reaction mixture was evaporated with silica gel and the product was purified by column chromatography (silica gel, hexane/EtOAc, 10:1) to afford 18.6 g of pure aldehyde intermediate. 1H NMR CDCl3 (600 MHz): δ 10.25 (s, 1H), 7.78 (m, 2H), 7.23 (t, J = 7.7 Hz, 1H), 2.75 (s, 3H). 13C NMR CDCl3 (150 MHz): 191.8, 137.7, 130.9, 130.2, 127.6, 127.4, 126.8, 18.1.
To the mixture of 18.6 g (93 mmol) of 3-bromo-2-methylbenzaldehyde and 26.8 g (233 mmol) of methyl azidoacetate in 130 mL of MeOH was added 43 mL of 5.4 M MeONa over 30 min at −10 °C. After addition the mixture was stirred for additional hour at the same temperature and then transferred to cold room (+4 °C) and stirred overnight. The reaction mixture was then poured in 1 L mixture of ice and saturated ammonium chloride solution, stirred for 10 min, and filtered off. The solid was washed with plenty of ice cold water and then moved to ambient temperature. After air drying for 1 h the solid was dissolved in 100 mL of DCM, dried over magnesium sulfate, and passed through short silica gel plug. Evaporation of solvent gave 21.1 g of 57a, 76% yield, which was used in the next step without further purification. 1H NMR CDCl3 (600 MHz): δ 7.72 (d, J = 7.7 Hz, 1H), 7.53 (d, J = 7.7 Hz, 1H), 7.10 (s, 1H), 7.07 (t, J = 7.7 Hz, 1H), 3.93 (s, 3H), 2.42 (s, 3H). 13C NMR CDCl3 (150 MHz): 163.6, 136.7, 133.9, 133.1, 129.3, 128.8, 126.8, 125.8, 123.6, 53.1, 20.0.
Methyl 2-Azido-3-(2-chloro-3-methylphenyl)acrylate (57b)
Similar procedure was used as for 57a but starting with 10 g (57 mmol) of 56b. Isolated 4.6 g, 63% yield (3 steps). 1H NMR CDCl3 (600 MHz): δ 7.95 (m, 1H), 7.35 (s, 1H), 7.21 (m, 2H), 3.94 (s, 3H), 2.40 (s, 3H).
Methyl 2-Azido-3-(3-bromo-4-methoxy-2-methylphenyl)-acrylate (57c)
Similar procedure was used as the last step for 57a but starting with 4.89 g (21 mmol) of 55. Isolated 5.72 g, 83% yield (3 steps). 1H NMR CDCl3 (600 MHz): δ 7.87 (d, J = 8.8 Hz, 1H), 7.11 (s, 1H), 6.80 (d, J = 8.8 Hz, 1H), 3.93 (s, 3H), 3.92 (s, 3H), 2.48 (s, 3H).
Methyl 5-Bromo-4-methyl-1H-indole-2-carboxylate (58a)
21.1 g (71 mmol) of 57a was dissolved in 700 mL of xylene. The mixture was refluxed for 10 min. Reaction mixture was cooled to rt and kept at −20 °C overnight. The precipitated product was filtered off and dried on funnel to produce 10.0 g of 58a, 50% yield. 1H NMR CDCl3 (600 MHz): δ 8.98 (br s, 1H), 7.44 (d, J = 8.4 Hz, 1H), 7.24 (s, 1H), 7.14 (d, J = 8.4 Hz, 1H), 3.96 (s, 3H), 2.60 (s, 3H). 13C NMR CDCl3 (150 MHz): δ 162.1, 135.4, 131.6, 129.5, 129.0, 127.4, 115.9, 110.8, 107.7, 52.2, 19.0.
Methyl 4-Chloro-5-methyl-1H-indole-2-carboxylate (58b)
Similar procedure was used as for 57a but starting with 4.6 g (18 mmol) of 57b. Isolated 3.0 g, 74% yield. 1H NMR CDCl3 (600 MHz): δ 9.01 (br s, 1H), 7.28 (s, 1H), 7.23 (d, J = 8.4 Hz, 1H), 7.17 (d, J = 8.4 Hz, 1H), 3.96 (s, 3H), 2.46 (s, 3H).
Methyl 5-Bromo-6-methoxy-4-methyl-1H-indole-2-carboxylate (58c)
Similar procedure was used as for 57a but starting with 5.7 g (17 mmol) of 57c. Isolated 4.2 g, 81% yield. 1H NMR CDCl3 (600 MHz): δ 8.83 (br, 1H), 7.20 (s, 1H), 6.74 (s, 1H), 3.93 (s, 6H), 2.62 (s, 3H).
5-Bromo-4-methyl-1H-indole-2-carbonitrile (59a)
10.0 g (38 mmol) of 58a was refluxed in a solution of 10.6 g (190 mmol) of KOH in 130 mL of methanol for 1 h. Reaction mixture was then concentrated and acidified with 12 M HCl in water. Precipitated product was filtered off. After drying the 5-bromo-4-methyl-1H-indole-2-carboxylic acid was added to solution of 6.5 mL (76 mmol) of oxalyl chloride in 200 mL of dichloromethane with 0.6 mL of DMF. After stirring for 1 h reaction mixture was cooled in ice–water bath and 40 mL of conc ammonia solution in water was added dropwise. The heterogeneous mixture was stirred for another 3 h and filtered off to obtain amide. A mixture of 5-bromo-4-methyl-1H-indole-2-carboxamide from previous step, 36 mL (380 mmol) of phosphorus oxychloride, and 120 mL of chloroform was refluxed for 5 h. Then reaction mixture was evaporated to dryness and quenched with ice and conc ammonia solution in water (about 40 mL). The formed precipitate was filtered off, washed with water, and dissolved in 100 mL of THF. The solution was evaporated with silica gel and the product was purified by column chromatography (silica gel, hexane/EtOAc, 2:1), affording 7.4 g of pure nitrile 59a, 82% yield. 1H NMR (CD3)2CO (600 MHz): δ 11.42 (br s, 1H), 7.49 (d, J = 8.8 Hz, 1H), 7.42 (s, 1H), 7.32 (d, J = 8.8 Hz, 1H), 2.59 (s, 3H). 13C NMR (CD3)2CO (150 MHz): δ 137.5, 132.0, 131.0, 129.3, 117.2, 114.9, 113.7, 113.0, 108.5, 19.6.
4-Chloro-5-methyl-1H-indole-2-carbonitrile (59b)
Similar procedure was used as for 59a but starting with 3.0 g (13 mmol) of 58b. Isolated 1.5 g, 60% yield. 1H NMR CDCl3 (600 MHz): δ 8.79 (br s, 1H), 7.42 (d, 1H, J 8.4 Hz), 7.34 (s, 1H), 7.28 (d, 1H, J 8.4 Hz), 2.61 (s, 3H).
5-Bromo-6-methoxy-4-methyl-1H-indole-2-carbonitrile (59c)
Similar procedure was used as for 59a but starting with 4.2 g (13 mmol) of 58c. Isolated 3.3 g, 96% yield. 1H NMR (CD3)2CO (600 MHz): δ 10.24 (br s, 1H), 7.13 (s, 1H), 6.80 (s, 1H), 3.93 (s, 3H), 2.61 (s, 3H).
5-Bromo-3-methyl-1H-indole-2-carbonitrile (59d)
Similar procedure was used as for 59a but starting with 655 mg (2.32 mmol) of 53. Isolated 300 mg, 56% yield. 1H NMR CDCl3 (600 MHz): δ 9.35 (br s, 1H), 7.81 (s, 1H), 7.39 (d, J = 9.0 Hz, 1H), 7.32 (d, J 9.0 = Hz, 1H), 2.62 (s, 3H).
5-Formyl-4-methyl-1H-indole-2-carbonitrile (60a)
2.35 g (10 mmol) of 5-bromo-4-methyl-1H-indole-2-carbonitrile 59a was dissolved in 100 mL of THF. The flask was flushed with argon, and 3.2 g (24 mmol) of potassium hydride was added (30% suspension in oil). After stirring for 5 min the reaction mixture was cooled to −90 °C (internal temperature with probe, ethanol/N2(liq)) and 11.8 mL of tert-butyllithium (20 mmol) was slowly added to maintain the temperature at the range −95 to −90 °C. After stirring for 1 h 3.8 mL (50 mmol) of DMF was added dropwise and reaction mixture was allowed to warm to −70 °C and kept 30 min at this temperature. The reaction mixture was quenched with 2.9 mL (50 mmol) of acetic acid and warmed to rt. After addition of 100 mL of brine organic phase was separated, evaporated with silica gel and the product was purified by column chromatography (silica gel, hexane/THF, 1:1). After evaporation, 1.11 g of pure aldehyde 60a was obtained, 60% yield. 1H NMR CD3CN (600 MHz): δ 10.39 (s, 1H), 7.82 (d, J = 8.4 Hz, 1H), 7.51 (s, 1H), 7.43 (d, J = 8.4 Hz, 1H), 2.86 (s, 3H). 13C NMR CD3CN (150 MHz): δ 191.2, 138.9, 137.2, 127.3, 126.9, 126.6, 113.7, 113.1, 110.0, 107.2, 13.4.
4-Chloro-5-formyl-1H-indole-2-carbonitrile (60b)
To a solution of 702 mg (3.7 mmol) of 59b in 5 mL of acetonitrile, 1.0 mL (4.4 mmol) of Boc2O and 45 mg (0.37 mmol) of DMAP were added and stirred at room temperature for 1 h. The solvent was removed in vacuo, and the crude product was filtered through silica gel column using hexane/EtOAc (10:1) as an eluent to afford 1.03 g of Boc-protected intermediate, 96% yield. The intermediate (3.5 mmol) was dissolved in 35 mL of carbon tetrachloride, and 1.45 g (8.2 mmol) of N-bromosuccinimide and 60 mg (0.37 mmol) of AIBN were added. The mixture was refluxed for 2 h, then cooled to room temperature and evaporated to dryness. Then it was dissolved in 70 mL of acetone/water mixture (1:1) and 1.17 g (4.2 mmol) of silver carbonate was added. The mixture was stirred for 24 h, then concentrated and diluted with 50 mL of EtOAc. Mixture was filtered off through Celite, organic phase was evaporated with silica gel, and the product was purified by column chromatography (silica gel, hexane/DCM, 1:2) to afford 641 mg of Boc-protected intermediate 60b, 60% yield. It was then dissolved in 4 mL of DCM/TFA (1:1) and stirred for 1 h. The mixture was evaporated and partitioned in EtOAc and saturated with NaHCO3 solution. Organic phase was evaporated with silica gel and the product was purified by column chromatography (silica gel, DCM:acetone, 40:1) to afford 330 mg of 60b, 81%. 1H NMR CD3CN (600 MHz): δ 11.63 (br s, 1H), 10.56 (s, 1H), 7.92 (d, J = 8.6 Hz, 1H), 7.47 (d, J = 8.6 Hz, 1H), 7.43 (s, 1H).
5-Formyl-6-methoxy-4-methyl-1H-indole-2-carbonitrile (60c)
Similar procedure was used as for 60a but starting with 1.06 g (4 mmol) of 59c. Isolated 766 mg, 89% yield. 1H NMR CD3CN (600 MHz): δ 10.57 (s, 1H), 9.89 (br s, 1H), 7.24 (s, 1H), 6.92 (s, 1H), 3.90 (s, 3H), 2.71 (s, 3H).
5-Formyl-3-methyl-1H-indole-2-carbonitrile (60d)
Similar procedure was used as for 60a but starting with 93 mg (0.4 mmol) of 59d. Isolated 23 mg, 32% yield. 1H NMR CD3CN (600 MHz): δ 10.96 (br s, 1H), 10.06 (s, 1H), 8.19 (s, 1H), 7.90 (d, J = 8.8 Hz, 1H), 7.53 (d, J = 8.8 Hz, 1H), 2.57 (s, 3H).
4-Methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]-pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (14)
800 mg (4.35 mmol) of 5-formyl-4-methyl-1H-indole-2-carbonitrile, 1.83 g (5.22 mmol) of N-(piperidin-4-yl)-6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-amine hydrochloride, and 1.2 mL (8.7 mmol) of triethylamine were mixed in 43 mL of dry dichloromethane. 1.84 g (8.7 mmol) of sodium triacetoxyborohydride was added to it in one portion. After stirring overnight TLC showed no presence of starting material. The reaction mixture was transferred to a separatory funnel and washed with 50 mL of 1 M NaOH. Organic phase was evaporated with silica gel and the product was purified by column chromatography (silica gel, DCM/MeOH/NH3(aq) 40:1:0.1 to 15:1:0.1). Evaporation of solvent gave 2.08 g of oily product, which was dissolved in 5 mL of diethyl ether and crystallized upon standing. The product was filtered off, washed with additional 10 mL of ether, and thoroughly dried to afford 1.95 g of product, 93% yield. The compound was converted to hydrochloride salt by dissolving in 5 mL of MeOH followed by addition of 4.4 mL of 1 M HCl in water (4.4 mmol) and drying to afford 14. 1H NMR CD3OD (600 MHz): δ 8.35 (s, 1H), 7.53 (br s, 1H), 7.48 (d, J = 8.4 Hz, 1H), 7.41 (d, J = 8.4 Hz, 1H), 7.40 (m, 2H), 4.51 (s, 2H), 4.46 (m, 1H), 3.86 (q, J = 10.5 Hz, 2H), 3.60 (m, 2H), 3.33 (m, 2H), 2.70 (s, 3H), 2.33 (m, 2H), 1.95 (m, 2H). 13C NMR CD3OD (150 MHz): δ 167.1, 157.7, 154.9, 138.9, 134.5, 130.4, 130.0, 128.9, 126.6 (q, J = 275 Hz), 121.7, 118.2, 114.9, 113.6, 111.7, 108.4, 59.1, 52.9, 49.6, 47.2, 35.7 (q, J = 32 Hz), 30.1, 15.9. HRMS (ESI): [M + H+] calculated 485.1730; found 485.1733.
3-Methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]-pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (13)
Similar procedure was used as for 14 but starting with 29 mg (0.16 mmol) of 60d. Isolated 21 mg, 28% yield. 1H NMR CD3OD (600 MHz): δ 8.36 (s, 1H), 7.89 (s, 1H), 7.55 (s, 1H), 7.52 (m, 2H), 4.47 (s, 2H), 4.45 (m, 1H), 3.86 (q, J = 10.5 Hz, 2H), 3.61 (m, 2H), 3.24 (m, 2H), 2.51 (s, 3H), 2.35 (m, 2H), 1.98 (m, 2H). 13C NMR CD3OD (150 MHz): δ 164.8, 156.1, 153.0, 137.7, 128.7, 127.8, 126.5, 125.0 (q, J = 275 Hz), 123.7, 123.5, 120.7, 120.3, 116.7, 113.0, 112.4, 106.0, 60.7, 51.2, 45.7, 35.4 (q, J = 32 Hz), 28.5, 7.9. HRMS (ESI): [M + H+] calculated 485.1730; found 485.1724.
4-Chloro-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]-pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (15)
Similar procedure was used as for 14 but starting with 29 mg (0.16 mmol) of 60d. Isolated 21 mg, 28% yield. 1H NMR CD3OD (600 MHz): δ 8.42 (s, 1H), 7.86 (s, 1H), 7.59 (s, 1H), 7.55 (m, 2H), 4.48 (s, 2H), 4.44 (m, 1H), 3.87 (q, J = 10.5 Hz, 2H), 3.62 (m, 2H), 3.26 (m, 2H), 2.54 (s, 3H), 2.31 (m, 2H), 1.99 (m, 2H). 13C NMR CD3OD (150 MHz): δ 164.3, 155.9, 152.7, 138.1, 129.1, 127.6, 126.3, 125.4 (q, J = 275 Hz), 123.4, 123.2, 121.0, 120.4, 115.9, 113.6, 112.7, 106.6, 60.4, 51.1, 45.3, 35.4 (q, J = 32 Hz), 28.9. HRMS (ESI): [M + H+] calculated 505.1184; found 505.1183.
6-Methoxy-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno-[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (21)
Similar procedure was used as for 14 but starting with 107 mg (0.5 mmol) of 60c. Isolated 227 mg, 87% yield. 1H NMR CD3OD (600 MHz): δ 8.70 (s, 1H), 7.86 (s, 1H), 7.32 (s, 1H), 6.96 (s, 1H), 4.64 (m, 1H), 4.54 (s, 2H), 4.00 (s, 3H), 3.99 (q, J = 10.5 Hz, 2H), 3.68 (m, 2H), 3.43 (m, 2H), 2.68 (s, 3H), 2.32 (m, 2H), 2.21 (m, 2H). 13C NMR CD3OD (150 MHz): δ 159.3, 159.1, 150.0, 139.90, 139.85, 135.8, 135.6, 133.0, 126.0 (q, J = 275 Hz), 122.9, 118.8, 115.4, 114.2, 112.9, 106.6, 92.5, 56.8, 55.0, 53.5, 50.4, 35.3 (q, J = 32 Hz), 29.6, 16.5. HRMS (ESI): [M + H+] calculated 515.1835; found 515.1833.
6-Hydroxy-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno-[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (20)
26 mg (0.05 mmol) of 21 was added to 0.5 mL of 1 M BBr3 in DCM at 0 °C, and the reaction mixture was brought to rt. After 24 h ice was added to the reaction mixture in the presence of sodium bicarbonate. Volatile organic was evaporated and the residue was partitioned between water and EtOAc/MeOH (10:1). Organic layer was evaporated with silica gel and the product was purified by column chromatography (silica gel, hexane/EtOAc/MeOH, 1:1:0.1) to afford 17 mg of 20, 68% yield. 1H NMR CD3OD (600 MHz): δ 8.45 (s, 1H), 7.64 (s, 1H), 7.28 (s, 1H), 6.84 (s, 1H), 4.55 (m, 1H), 4.53 (s, 2H), 4.00 (s, 3H), 3.90 (q, J = 10.5 Hz, 2H), 3.67 (m, 2H), 3.40 (m, 2H), 2.65 (s, 3H), 2.31 (m, 2H), 2.05 (m, 2H). 13C NMR CD3OD (150 MHz): δ 157.6, 157.0, 153.4, 140.1, 135.3, 135.1, 130.8, 126.2 (q, J = 275 Hz), 122.6, 122.1, 118.3, 115.4, 114.2, 114.1, 112.3, 106.2, 95.2, 55.1, 53.4, 47.5, 35.0 (q, J = 32 Hz), 29.8, 16.1. HRMS (ESI): [M + H+] calculated 501.1679; found 501.1674.
2-(2-Cyano-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno-[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indol-1-yl)acetamide (25)
Similar procedure was used as for 7 but starting with 13 mg (0.025 mmol) of 14. Isolated 5 mg, 37% yield. 1H NMR CD3OD (600 MHz): δ 8.37 (s, 1H), 7.57 (d, J = 8.4 Hz, 1H), 7.52 (s, 1H), 7.45 (d, J = 8.4 Hz, 1H), 5.09 (s, 2H), 4.62 (m, 1H), 4.53 (s, 2H), 3.88 (q, J = 10.5 Hz, 2H), 3.61 (m, 2H), 2.72 (s, 3H), 2.34 (m, 2H), 1.96 (m, 2H). 13C NMR CD3OD (150 MHz): 171.2, 167.1, 157.7, 154.9, 139.8, 130.0, 129.4, 128.7, 126.6 (q, J = 275 Hz), 121.8, 118.2, 114.0, 113.9, 112.6, 110.2, 58.9, 54.8, 52.9, 48.0, 35.6 (q, J = 32 Hz), 30.1, 15.8. HRMS (ESI): [M + H+] calculated 542.1944; found 542.1949.
2-(2-Cyano-6-methoxy-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indol-1-yl)acetamide (26)
Similar procedure was used as for 7 but starting with 26 mg (0.05 mmol) of 21. Isolated 7 mg, 25% yield. 1H NMR CD3OD (600 MHz): δ 8.64 (s, 1H), 7.79 (s, 1H), 7.43 (s, 1H), 7.00 (s, 1H), 5.05 (s, 2H), 4.62 (m, 1H), 4.53 (s, 2H), 4.02 (s, 3H), 3.98 (q, J = 10.5 Hz, 2H), 3.66 (m, 2H), 3.41 (m, 2H), 2.67 (s, 3H), 2.32 (m, 2H), 2.15 (m, 2H). 13C NMR CD3OD (150 MHz): δ 171.4, 159.7, 150.8, 141.0, 136.3, 132.5, 126.2 (q, J = 275 Hz), 123.2, 122.7, 122.6, 118.7, 114.6, 114.3, 113.4, 110.9, 110.8, 91.5, 57.0, 54.8, 53.4, 48.03, 35.4, (q, J = 32 Hz), 29.6, 16.2. HRMS (ESI): [M + H+] calculated 572.2050; found 572.2055.
Methyl 2-Azido-3-(4-fluoro-3-methylphenyl)acrylate (62a)
Similar procedure was used as for 57a but starting with 0.83 mL (5 mmol) of 61a. Isolated 723 mg, 60% yield. 1H NMR CDCl3 (400 MHz): δ 7.66 (m, 2H), 7.01 (m, 1H), 6.85 (s, 1H), 3.91 (s, 3H), 2.30 (s, 3H).
Methyl 2-Azido-3-(4-methoxy-3-methylphenyl)acrylate (62b)
Similar procedure was used as for 57a but starting with 1.46 mL (10 mmol) of 61a. Isolated 1.6 g, 65% yield. 1H NMR CDCl3 (400 MHz): δ 7.65 (d, J = 8.8 Hz, 1H), 7.05 (s, 1H), 7.14 (s, 1H), 6.92 (d, J = 8.8 Hz, 1H), 3.91 (s, 3H), 3.89 (s, 3H), 2.45 (s, 3H).
Methyl 6-Fluoro-5-methyl-1H-indole-2-carboxylate (63a)
376 mg (1.6 mmol) of 62a was dissolved in 1.5 mL of toluene. 45 mg (0.075 mmol) of rhodium(II) trifluoroacetate dimer was added, and the reaction mixture was heated at 50 °C for 24 h. Then solvent was evaporated, and the residue product was purified by column chromatography (silica gel, hexane/EtOAc, 10:1). 110 mg (33% yield) of compound 63a was obtained. 1H NMR (600 MHz, CDCl3): δ 8.81 (br s, 1H), 7.56 (s, 1H), 7.34 (s, 1H), 7.14 (s, 1H), 3.92 (s, 3H), 2.56 (s, 3H).
Methyl 6-Methoxy-5-methyl-1H-indole-2-carboxylate (63b)
Similar procedure was used as for 63a but starting with 250 mg (1.05 mmol) of 62a. Isolated 156 mg, 75% yield. 1H NMR CDCl3 (400 MHz): δ 8.73 (br s, 1H), 7.39 (s, 1H), 7.10 (s, 1H), 6.77 (s, 1H), 3.92 (s, 3H), 3.88 (s, 3H), 2.28 (s, 3H).
6-Fluoro-5-methyl-1H-indole-2-carbonitrile (64a)
Similar procedure was used as for 59a but starting with 65 mg (0.31 mmol) of 63a. Isolated 31 mg, 58% yield. 1H NMR CDCl3 (400 MHz): δ 7.56 (s, 1H), 7.34 (s, 1H), 7.15 (s, 1H), 3.90 (s, 1H), 2.52 (s, 3H).
6-Methoxy-5-methyl-1H-indole-2-carbonitrile (64b)
Similar procedure was used as for 59a but starting with 203 mg (1 mmol) of 63b. Isolated 116 mg, 62% yield. 1H NMR CDCl3 (400 MHz): δ 7.37 (s, 1H), 7.06 (s, 1H), 6.76 (s, 1H), 3.88 (s, 1H), 2.35 (s, 3H).
6-Fluoro-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]-pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (16)
Similar procedure was used as for 1 but starting with 16 mg (0.1 mmol) of 64a. Isolated 25 mg, 50% yield. 1H NMR CD3OD (600 MHz): δ 8.70 (s, 1H), 8.04 (s, 1H), 7.87 (s, 1H), 7.36 (s, 1H), 7.32 (s, 1H), 4.65 (m, 1H), 4.55 (s, 2H), 4.01 (q, J = 10.5 Hz, 2H), 3.70 (m, 2H), 3.35 (m, 2H), 2.36 (m, 2H), 2.17 (m, 2H). 13C NMR CD3OD (150 MHz): δ 162.5, 160.9, 157.3, 149.5, 139.8, 133.2, 128.9, 126.5 (q, J = 275 Hz), 124.8, 123.0, 118.8, 114.9, 114.6, 112.4, 109.9, 99.7, 55.6, 52.5, 35.3 (q, J = 32 Hz), 29.6. HRMS (ESI): [M + H+] calculated 489.1479; found 489.1484.
6-Methoxy-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]-pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (18)
Similar procedure was used as for 1 but starting with 116 mg (0.62 mmol) of 64b. Isolated 287 mg, 77% yield. 1H NMR CD3OD (600 MHz): δ 8.71 (s, 1H), 7.87 (s, 1H), 7.86 (s, 1H), 7.24 (s, 1H), 7.12 (s, 1H), 4.66 (m, 1H), 4.49 (s, 2H), 4.03 (m, 5H), 3.69 (m, 2H), 3.34 (m, 2H), 2.36 (m, 2H), 2.18 (m, 2H). 13C NMR CD3OD (150 MHz): δ 158.7, 149.7, 140.9, 133.1, 128.2, 127.8, 127.3, 126.4 (q, J = 275 Hz), 123.4, 122.8, 121.7, 118.7, 115.1, 114.8, 114.6, 107.5, 94.6, 57.7, 56.6, 52.7, 35.2 (q, J = 33 Hz), 29.5. HRMS (ESI): [M + H+] calculated 501.1679; found 501.1674.
6-Hydroxy-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]-pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (17)
Similar procedure was used as for 20 but starting with 50 mg (0.1 mmol) of 18. Isolated 27 mg, 56% yield. 1H NMR CD3OD (600 MHz): δ 8.70 (s, 1H), 7.81 (s, 1H), 7.77 (s, 1H), 7.21 (s, 1H), 6.98 (s, 1H), 4.64 (m, 1H), 4.48 (s, 2H), 4.01 (q, J = 10.3 Hz, 2H), 3.66 (m, 2H), 3.35 (m, 2H), 2.37 (m, 2H), 2.12 (m, 2H). 13C NMR CD3OD (150 MHz): δ 157.6, 156.6, 153.9, 141.1, 130.5, 127.8, 127.5, 126.4 (q, J = 275 Hz), 125.7, 122.0, 121.6, 118.3, 115.2, 114.9, 114.0, 107.1, 97.5, 57.8, 52.9, 47.9, 35.5 (q, J = 33 Hz), 30.0. HRMS (ESI): [M + H+] calculated 487.1522; found 487.1519.
6-Ethoxy-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]-pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (19)
14 mg (0.029 mmol) of 17 was dissolved in 0.1 mL of CH3CN. 12 mg (0.087 mmol) of K2CO3 and 4.7 μL (0.058 mmol) of EtI were added, and the mixture was stirred for 24 h. Then reaction mixture was directly subjected to purification by preparative TLC (silica gel, hexane/EtOAc/MeOH, 1:1:0.2) and the product was extracted from silica gel with methanol to afford 9 mg of 19, 61% yield. 1H NMR CD3OD (600 MHz): δ 8.65 (s, 1H), 7.84 (s, 1H), 7.80 (s, 1H), 7.28 (s, 1H), 7.25 (s, 1H), 4.60 (m, 1H), 4.47 (s, 2H), 4.29 (m, 2H), 3.99 (q, J = 10.3 Hz, 2H), 3.67 (m, 2H), 3.58 (m, 2H), 2.35 (m, 2H), 2.10 (m, 2H), 1.55 (t, J = 7 Hz, 3H). 13C NMR CD3OD (150 MHz): δ 157.9, 157.4, 150.4, 132.6, 128.3, 126.2 (q, J = 275 Hz), 122.6, 121.3, 118.6, 114.8, 114.6, 111.5, 94.8, 65.7, 57.4, 52.8, 43.8, 35.5 (q, J = 33 Hz), 29.6, 15.0. HRMS (ESI): [M + H+] calculated 515.1835; found 515.1834.
(1-Trityl-1H-pyrazol-4-yl)methanol (66)
5 g of ethyl 1H-pyrazole-4-carboxylate (35.7 mmol) was dissolved in 70 mL of DMF, and 4.8 g (42.8 mmol) of potassium tert-butoxide was added. After stirring for 30 min 13.9 g (50 mmol) of trityl chloride was added in small portions. TLC indicated no starting material after 1 h of stirring, and the reaction mixture was diluted with 300 mL of water and extracted with 3 × 50 mL of DCM. Organic extracts were evaporated and then dissolved in 300 mL of THF. 2.03 g (53.6 mmol) of lithium aluminohydride was added in small portions. After 1 h the reaction mixture was carefully quenched with 10 mL of ethyl acetate followed by addition of 5 mL of water. 15 g of silica gel was added, and the solvent was evaporated. The product was purified by column chromatography (silica gel, hexane/EtOAc, 1:1), resulting in 11.75 g of 66, 97% yield. 1H NMR CDCl3 (600 MHz): δ 7.67 (s, 1H), 7.38 (s, 1H), 7.31 (m, 9H), 7.14 (m, 6H), 4.55 (s, 2H), 1.47 (s, 1H). 13C NMR CDCl3 (150 MHz): δ 143.2, 139.1, 131.4, 130.1, 127.8, 120.0, 78.7, 56.3.
4-(Bromomethyl)-1-trityl-1H-pyrazole (67)
To a solution of 11.75 g (34.5 mmol) of 66 and 9.04 g (34.5 mmol) of triphenylphosphine in 90 mL of DCM was added 6.14 g (34.5 mmol) of N-bromosuccinimide at 0 °C. Then mixture was stirred for 30 min and transferred to a separatory funnel, washed with saturated sodium bicarbonate solution and brine, dried over sodium sulfate. After evaporation, the product was purified by column chromatography (neutralized silica gel, hexane/EtOAc, 10:1). Silica gel neutralization (for 50 g of silica gel): washed with 500 mL of 0.05% of ammonia (25% in water) in EtOAc, 500 mL of pure EtOAc, and 500 mL of hexane. 11.09 g of 67 was obtained and it was used in subsequent step immediately (decomposes upon standing), 79% yield.
5-Formyl-6-methoxy-1H-indole-2-carbonitrile (68b)
Similar procedure was used as for 60b but starting with 744 mg (4 mmol) of 64b. Isolated 300 mg, 38% yield. 1H NMR CD3CN (600 MHz): δ 10.46 (s, 1H), 9.78 (br s, 1H), 7.33 (s, 1H), 7.15 (s, 1H), 6.99 (s, 1H), 3.92 (s, 3H).
5-Formyl-6-methoxy-4-methyl-1-(2-(trityloxy)ethyl)-1H-in-dole-2-carbonitrile (69)
A mixture of 44 mg (0.21 mmol) of 60c, 113 mg (0.31 mmol) of 1-tritoxy-2-bromoethane (see 44 for preparation), and 197 mg (0.6 mmol) of Cs2CO3 in 0.4 mL of DMF was heated at 40 °C for 3 h. The reaction mixture was diluted with water and extracted with DCM. Organic phase was evaporated with silica gel and loaded on silica gel column. The product was purified with hexane/EtOAc 1:1 to afford 50 mg of 69, 50% yield. 1H NMR CD3CN (600 MHz): 10.65 (s, 1H), 7.32 (s, 1H), 7.17 (m, 15H), 6.65 (s, 1H), 4.41 (t, 5.3 Hz, 2H), 3.90 (s, 3H), 3.50 (t, 5.3 Hz, 2H), 2.81 (s, 3H).
5-Formyl-4-methyl-1-((1-trityl-1H-pyrazol-4-yl)methyl)-1H-indole-2-carbonitrile (70)
11.09 g (27.5 mmol) of 67, 2.53 g (13.8 mmol) of 5-formyl-4-methyl-1H-indole-2-carbonitrile, and 13.6 g (41.4 mmol) of cesium carbonate were stirred in 28 mL of DMF for 30 min. Then the reaction mixture was diluted with 300 mL of water and 200 mL of diethyl ether. After stirring for 4 h the precipitate was filtered off, washed with additional 100 mL of water and 100 mL of diethyl ether, and dried to afford 6.4 g of 70, 92% yield. 1H NMR DMSO-d6 (600 MHz): δ 10.02 (s, 1H), 7.85 (s, 1H), 7.82 (d, J = 8.4 Hz, 1H), 7.77 (d, J = 8.4 Hz, 1H), 7.58 (s, 1H), 7.50 (s, 1H), 7.36 (m, 9H), 7.00 (m, 6H), 5.47 (s, 2H), 2.83 (s, 3H)). 13C NMR DMSO-d6 (150 MHz): δ 191.1, 162.3, 142.7, 142.6, 138.5, 132.1, 129.7, 129.6, 127.9, 127.8, 127.1, 126.6, 115.6, 114.9, 109.8, 109.4, 78.5, 78.1, 14.1.
5-Formyl-6-methoxy-1-((1-trityl-1H-pyrazol-4-yl)methyl)-1H-indole-2-carbonitrile (71)
Similar procedure was used as for 70 but starting with 300 mg (1.5 mmol) of 68b. Isolated 642 mg of 71, 82% yield. 1H NMR DMSO-d6 (600 MHz): δ 9.89 (s, 1H), 7.56 (s, 1H), 7.51 (s, 1H), 7.44(s, 1H), 7.34 (m, 9H), 7.23 (s, 1H), 7.15 (s, 1H), 7.02 (m, 6H), 5.43 (s, 2H), 2.72 (s, 3H).
5-Formyl-6-methoxy-4-methyl-1-((1-trityl-1H-pyrazol-4-yl)-methyl)-1H-indole-2-carbonitrile (72)
Similar procedure was used as for 70 but starting with 535 mg (2.5 mmol) of 60c. Isolated 1.30 g, 94% yield. 1H NMR CDCl3 (600 MHz): 10.63 (s, 1H), 7.57 (s, 1H), 7.42 (s, 1H), 7.30 (m, 9H), 7.10 (m, 6H), 6.57 (s, 1H), 5.24 (s, 2H), 3.84 (s, 3H), 2.76 (s, 3H).
(4-((2-Cyano-5-formyl-6-methoxy-4-methyl-1H-indol-1-yl)-methyl)-1H-1,2,3-triazol-1-yl)methyl Pivalate (73)
Similar procedure was used as for 69 but starting with 180 mg (0.84 mmol) of 60c and 0.14 mL (1.26 mmol) of propargyl bromide (80% in toluene). Crude intermediate, 0.13 mL (0.84 mmol) of t-BuCO2CH2N3, 50 mg (0.25 mmol) of sodium ascorbate, and 6.7 mg (0.042 mmol) of CuSO4 were dissolved in 2.5 mL of t-BuOH/H2O (1:1), and the reaction mixture was heated at 60 °C for 4 h. The reaction mixture was then diluted with water and extracted with DCM. Organic phase was evaporated with silica gel and loaded on silica gel column. The product was purified by column chromatography (silica gel, hexane/EtOAc, 1:1) to afford 307 mg of 73, 90% yield. 1H NMR CDCl3 (600 MHz): δ 10.56 (s, 1H), 7.78 (s, 1H), 7.39 (s, 1H), 6.58 (s, 1H), 5.27 (s, 2H), 4.98 (s, 2H), 3.83 (s, 3H), 2.74 (s, 3H), 1.89 (s, 9H).
1-(2-Hydroxyethyl)-6-methoxy-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (24)
Similar procedure was used as for 14 for reductive amination and for 2 for deprotection but starting with 100 mg (0.2 mmol) of 69. Isolated 80 mg, 72% yield. 1H NMR CD3OD (600 MHz): δ 8.53 (s, 1H), 7.70 (s, 1H), 7.39 (s, 1H), 7.08 (s, 1H), 4.56 (m, 1H), 4.43 (s, 2H), 4.43 (t, J = 5.1 Hz, 2H), 4.03 (s, 3H), 3.92 (m, 4H), 3.65 (m, 2H), 3.40 (m, 2H), 2.67 (s, 1H), 2.33 (m, 2H), 2.09 (m, 2H). 13C NMR CD3OD (150 MHz): δ 159.4, 157.7, 152.5, 140.5, 136.1, 135.9, 131.6, 129.4, 126.6 (q, J = 275 Hz), 122.7, 122.6, 122.4, 118.6, 114.8, 114.1, 113.2, 110.4, 91.9, 61.9, 56.9, 54.9, 53.5, 50,4, 47.9, 35.5 (q, J = 33 Hz), 29.8, 16.2. HRMS (ESI): [M + H+] calculated 559.2098; found 559.2092.
6-Hydroxy-1-(2-hydroxyethyl)-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (23)
Similar procedure was used as for 20 but starting with 50 mg (0.089 mmol) of 24. Isolated 22 mg, 46% yield. 1H NMR CD3OD (600 MHz): δ 8.38 (s, 1H), 7.58 (s, 1H), 7.34 (s, 1), 6.88 (s, 1H), 4.61 (m, 1H), 4.53 (s, 2H), 4.32 (t, J = 5.1 Hz, 2H), 3.87 (m, 4H), 3.66 (m, 2H), 3.39 (m, 2H), 2.64 (s, 3H), 2.32 (m, 2H), 2.04 (m, 2H). 13C NMR CD3OD (150 MHz): δ 166.6, 158.2, 157.7, 154.9, 141.1, 136.4, 136.3, 130.8, 128.1, 126.5 (q, J = 275 Hz), 122.8, 122.5, 118.8, 115.4, 114.8, 114.6, 113.1, 110.5, 94.8, 63.5, 62.1, 55.5, 54.0, 47.7, 36.3 (q, J = 33 Hz), 30.4, 16.5. HRMS (ESI): [M + H+] calculated 545.1941; found 545.1941.
1-((1H-Pyrazol-4-yl)methyl)-6-methoxy-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)-piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (28)
Similar procedure was used as for 14 for reductive amination and 2 for deprotection but starting with 642 mg (1.23 mmol) of 71. Isolated 430 mg, 60% yield. 1H NMR CD3OD (600 MHz): δ 8.46 (s, 1H), 7.83 (s, 1H), 7.66 (s, 1H), 7.64 (s, 2H), 7.30 (s, H), 7.28 (s, 1H), 5.47 (s, 2H), 4.49 (m, 1H), 4.44 (s, 2H), 4.06 (s, 3H), 3.90 (q, J = 10.3 Hz, 2H), 3.62 (m, 2H), 3.28 (m, 2H), 2.32 (m, 2H), 2.04 (m, 2H). 13C NMR CD3OD (150 MHz): δ 164.5, 159.0, 157.5, 152.9, 140.6, 134.2, 131.0, 128.7, 126.4 (q, J = 275 Hz), 122.4, 122.2, 121.4, 121.2, 118.3, 117.7, 115.2, 115.0, 114.6, 110.2, 93.6, 61.0, 57.4, 56.8, 56.2, 52.9, 52.7, 47.5, 40.3, 35.5 (q, J = 33 Hz), 29.8. HRMS (ESI): [M + H+] calculated 581.2053; found 581.2059.
1-((1H-Pyrazol-4-yl)methyl)-6-hydroxy-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (27)
Similar procedure was used as for 20 but starting with 580 mg (1 mmol) of 28. Isolated 253 mg, 45% yield. 1H NMR CD3OD (600 MHz): δ 8.35 (s, 1H), 7.77 (s, 1H), 7.58 (s, 2H), 7.35 (s, 1H), 7.24 (s, 1H), 7.07 (s, 1H), 5.35 (s, 2H), 4.44 (m, 3H), 3.86 (m, 2H), 3.64 (m, 2H), 3.27 (m, 2H), 2.32 (m, 2H), 1.98 (m, 2H). 13C NMR CD3OD (150 MHz): δ 176.4, 166.6, 157.7, 156.9, 154.6, 140.8, 130.0, 128.5, 126.5 (q, J 275 Hz), 121.8, 121.4, 118.2, 117.6, 151.2, 114.6, 114.4, 110.0, 96.4, 57.5, 52.9, 47.0, 40.4, 35.6 (q, J = 33 Hz), 30.0. HRMS (ESI): [M + H+] calculated 567.1897; found 567.1892.
1-((1H-Pyrazol-4-yl)methyl)-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (29)
Similar procedure was used as for 14 for reductive amination and 2 for deprotection but starting with 5.5 g (10.8 mmol) of 70. Isolated 3.7 g, 58% yield. 1H NMR CD3OD (600 MHz): 1H NMR CD3OD (600 MHz): δ 8.36 (s, 1H), 7.63 (m, 5H), 7.43 (s, 1H), 5.45 (s, 2H), 4.53 (s, 2H), 4.46 (m, 1H), 3.86 (q, J = 10.5 Hz, 2H), 3.63 (m, 2H), 3.34 (m, 2H), 2.67 (s, 3H), 2.32 (m, 2H), 2.03 (m, 2H). 13C NMR CD3OD (150 MHz): δ 165.72, 157.61, 154.23, 138.70, 135.18, 134.22, 130.91, 130.30, 128.73, 126.60 (q, J = 289.5 Hz), 122.71, 121.99, 121.12, 118.22, 114.39, 114.04, 111.04, 110.61, 58.83, 52.87, 47.25, 40.50, 35.61 (q, J = 32 Hz), 29.90, 27.34, 15.89. HRMS (ESI): [M + H+] calculated 565.2104; found 565.2107.
1-((1H-Pyrazol-4-yl)methyl)-6-methoxy-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)-piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (31)
Similar procedure was used as for 14 for reductive amination and for 2 for deprotection but starting with 1053 mg (3 mmol) of 72. Isolated 890 mg, 50% yield. 1H NMR CD3OD (600 MHz): δ 8.39 (s, 1H), 7.62 (s, 2H), 7.60 (s, 1H), 7.39 (s, 1H), 7.17 (s, 1H), 5.46 (s, 1H), 4.52 (s, 2H), 4.48 (m, 1H), 4.05 (s, 3H), 3.87 (q, 2H, J = 10.3 Hz), 3.64 (m, 2H), 3.39 (m, 2H), 2.65 (s, 3H), 2.32 (m, 2H), 2.05 (m, 2H). 13C NMR CD3OD (150 MHz): 165.7, 159.8, 157.9, 154.4, 139.9, 136.5, 136.3, 134.4, 130.7, 126.8 (q, J = 275 Hz), 122.3, 118.5, 118.0, 115.0, 114.8, 113.6, 109.6, 91.8, 57.2, 54.9, 53.8, 47.5, 40.6, 35.8 (q, J = 33 Hz), 30.07, 16.4. HRMS (ESI): [M + H+] calculated 595.2210; found 595.2217.
1-((1H-Pyrazol-4-yl)methyl)-6-hydroxy-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)-piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (30)
Similar procedure was used as for 20 but starting with 36 mg (0.06 mmol) of 31. Isolated 17 mg, 49% yield. 1H NMR CD3OD (600 MHz): 8.36 (s, 1H), 7.56 (s, 2H), 7.49 (s, 1H), 7.36 (s, 1H), 6.94 (s, 1H), 5.34 (s, 2H), 4.52 (s, 2H), 4.45 (m, 1H), 3.86 (m, 2H), 3.65 (m, 2H), 3.40 (m, 2H), 2.32 (m, 2H), 2.01 (m, 2H). 13C NMR CD3OD (150 MHz): 166.8, 157.8, 154.8, 154.2, 140.0, 135.9, 130.2, 126.4 (q, J = 275 Hz), 125.1, 122.5, 122.0, 118.8, 117.8, 115.6, 114.6, 112.9, 112.3, 109.2, 94.3, 55.1, 53.7, 47.2, 42.2, 40.5, 35.7 (q, J = 33 Hz), 30.0, 16.1. HRMS (ESI): [M + H+] calculated 581.2053; found 581.2046.
1-((1H-1,2,3-Triazol-4-yl)methyl)-6-methoxy-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)-piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (34)
Reductive amination was completed using a similar procedure as for synthesis of 13, starting with 307 mg (0.75 mmol) of 73. Protected intermediate was dissolved in 2 mL of MeOH, and 530 mg (9.3 mmol) of KOH was added. After 30 min the reaction mixture was diluted with water and the product was extracted with EtOAc. Organic phase was evaporated with silica gel and the product was purified by column chromatography (silica gel, DCM/MeOH, 10:1) to afford 390 mg of 34, 87% yield. 1H NMR CD3OD (600 MHz): δ 8.35 (s, 1H), 7.57 (s, 1H), 7.40 (s, 1H), 7.18 (s, 1H), 5.63 (s, 2H), 4.51 (s, 2H), 4.45 (m, 1H), 4.04 (s, 3H), 3.86 (q, J = 10.3 Hz, 2H), 3.63 (m, 2H), 3.36 (m, 2H), 2.65 (s, 3H), 2.31 (m, 2H), 2.04 (m, 2H). 13C NMR CD3OD (150 MHz): δ 166.6, 159.7, 157.8, 154.7, 140.1, 136.3, 130.2, 126.5 (q, J = 275 Hz), 122.7, 122.0, 118.3, 114.9, 114.6, 113.6, 109.7, 91.9, 57.0, 54.8, 53.7, 47.2, 41.2, 35.7 (q, J = 33 Hz), 30.0, 16.2. HRMS (ESI): [M + H+] calculated 596.2162; found 596.2157.
1-((1H-1,2,3-Triazol-4-yl)methyl)-6-hydroxy-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)-piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (33)
Similar procedure was used as for 20 but starting with 100 mg (0.17 mmol) of 34. Isolated 17 mg, 17% yield. 1H NMR CD3OD (600 MHz): δ 8.35 (s, 1H), 7.72 (br s, 1H), 7.56 (s, 1H), 7.38 (s, 1H), 6.95 (s, 1H), 5.52 (s, 2H), 4.51 (s, 2H), 4.45 (m, 1H), 3.86 (q, J = 10.5 Hz, 2H), 3.64 (m, 2H), 3.37 (m, 2H), 2.63 (s, 3H), 2.31 (m, 2H), 2.01 (m, 2H). 13C NMR CD3OD (150 MHz): δ 166.7, 157.8, 157.5, 154.8, 140.3, 136.2, 130.2, 126.7 (q, J = 277.3 Hz), 122.4, 122.0, 118.3, 114.9, 114.7, 113.1, 109.5, 94.5, 55.0, 53.6, 47.2, 41.2, 35.8 (q, J = 31.9 Hz), 30.0, 16.1. HRMS (ESI): [M + H+] calculated 582.2006; found 582.2011.
Compounds 5, 6, 9, 10, 22, and 32 were obtained from synthesis on demand, with all compounds provided with the certificate of analysis and in addition fully characterized in house by HRMS and NMR. Characterization results of compounds 5, 6, 9, 10, 22, and 32 are the following.
1-(2-Hydroxy-3-methoxypropyl)-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (5)
1H NMR DMSO-d6 (600 MHz): δ 10.90 (br s, 1H), 8.48 (br s, 1H), 8.39 (s, 1H), 7.89 (s, 1H), 7.73 (s, 1H), 7.65 (m, 1H), 7.47 (s, 1H), 4.38 (m, 1H), 4.32 (s, 2H), 4.27 (m, 1H), 4.21 (m, 1H), 4.04 (q, J = 10.5 Hz, 2H), 3.92 (m, 1H), 3.36 (m, 2H), 3.29 (m, 1H), 3.26 (s, 3H), 3.06 (m, 2H), 2.07 (m, 2H), 1.98 (m, 2H). 13C NMR DMSO-d6 (150 MHz): δ 155.4, 137.8, 128.4, 127.2 (q, J = 277.3 Hz), 125.6, 125.5, 124.6, 122.4, 121.6, 116.3, 113.6, 113.2, 111.8, 111.1, 74.1, 68.2, 59.1, 58.5, 50.3, 48.8, 45.9, 33.6 (q, J = 31.9 Hz), 31.3, 29.0, 28.1, 22.1, 14.0. HRMS (ESI): [M + H+] calculated 559.2098; found 559.2093.
1-(3-Hydroxy-2-isopropoxypropyl)-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (6)
1H NMR DMSO-d6 (600 MHz): δ 11.09 (br, 1H), 8.76 (br, 1H), 8.48 (s, 1H), 7.94 (s, 1H), 7.82 (s, 1H), 7.71 (m, 2H), 7.51 (s, 1H), 4.52 (m, 1H), 4.37 (s, 2H), 4.34 (m, 1H), 4.22 (m, 1H), 4.08 (q, J = 10.5 Hz, 2H), 3.61 (m, 1H), 3.50 (m, 1H), 3.39 (m, 2H), 3.24 (m, 1H), 3.10 (m, 1H), 2.10 (m, 4H), 0.95 (d, J = 6.2 Hz, 3H), 0.53 (d, J = 6.2 Hz, 3H). 13C NMR DMSO-d6 (150 MHz): δ 151.8, 137.6, 128.4, 125.4 (q, J = 277.3 Hz), 125.7, 125.6, 122.5, 121.7, 116.5, 113.6, 113.1, 111.7, 111.3, 76.5, 70.9, 61.6, 59.1, 50.3, 47.6, 47.0, 46.2, 33.6 (q, J = 31.9 Hz), 28.0, 22.8, 21.4. HRMS (ESI): [M + H+] calculated 587.2411; found 587.2416.
1-((1H-1,2,3-Triazol-4-yl)methyl)-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (9)
1H NMR DMSO-d6 (600 MHz): δ 11.1 (br s, 1H), 8.83 (br s, 1H), 8.50 (s, 1H), 7.94 (s, 1H), 7.92 (br s, 1H), 7.87 (s, 1H), 7.85 (s, 1H), 7.83 (br s, 1H), 7.72 (m, 2H), 7.57 (s, 1H), 5.68 (s, 2H), 4.36 (s, 2H), 4.10 (q, J = 10.5 Hz, 2H), 3.41 (m, 2H), 3.08 (m, 2H), 2.08 (m, 2H), 2.04 (m, 2H). 13C NMR DMSO-d6 (150 MHz): δ 137.3, 128.9, 125.3 (q, J = 277.3 Hz), 125.8, 125.6, 122.8, 121.7, 116.5, 114.0, 113.3, 111.7, 110.1, 59.0, 50.2, 46.2, 33.7 (q, J = 31.9 Hz), 28.0. HRMS (ESI): [M + H+] calculated 552.1900; found 552.1909.
1-((1H-Pyrazol-4-yl)methyl)-5-((4-((6-(2,2,2-trifluoroethyl)-thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (10)
1H NMR DMSO-d6 (600 MHz): δ 10.89 (br s, 1H), 8.61 (br s, 1H), 8.45 (s, 1H), 7.93 (s, 1H), 7.79 (s, 1H), 7.70 (m, 1H), 7.64 (m, 2H), 7.55 (s, 1H), 5.46 (s, 2H), 4.36 (s, 2H), 4.32 (m, 1H), 4.90 (q, J = 10.5 Hz, 2H), 3.40 (m, 2H), 3.08 (m, 2H), 2.10 (m, 2H), 2.02 (m, 2H). 13C NMR DMSO-d6 (150 MHz): δ 137.0, 128.8, 125.8, 125.6, 125.2 (q, J = 277.3 Hz), 122.7, 121.6, 116.4, 115.7, 113.9, 113.5, 111.7, 109.5, 59.1, 50.3, 46.0, 33.6 (q, J = 31.9 Hz), 30.7, 28.1. HRMS (ESI): [M + H+] calculated 551.1948; found 551.1957.
1-(2-Hydroxyethyl)-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)-thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (22)
1H NMR DMSO-d6 (600 MHz): δ 10.90 (br s, 1H), 9.10 (br s, 1H), 8.57 (s, 1H), 7.90 (m, 1H), 7.76 (d, J = 8.2 Hz, 1H), 7.60 (s, 1H), 7.56 (d, J = 8.2 Hz, 1H), 4.40 (m, 3H), 4.12 (q, J = 10.5 Hz, 2H), 3.72 (m, 2H), 3.42 (m, 2H), 3.25 (m, 2H), 2.64 (s, 3H), 2.16 (m, 2H), 2.09 (m, 2H). 13C NMR DMSO-d6 (150 MHz): δ 137.2, 133.3, 129.8, 126.7, 125.3 (q, J = 277.3 Hz), 121.9, 120.1, 116.6, 113.8, 112.6, 110.0, 109.3, 59.8, 56.1, 50.4, 48.0, 46.5, 33.6 (q, J = 31.9 Hz), 27.9, 22.5, 15.4. HRMS (ESI): [M + H+] calculated 515.1835; found 515.1829.
1-((1H-1,2,3-Triazol-4-yl)methyl)-4-methyl-5-((4-((6-(2,2,2-trifluoroethyl)thieno[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)methyl)-1H-indole-2-carbonitrile (32)
1H NMR DMSO-d6 (600 MHz): δ 10.61 (br s, 1H), 8.70 (br s, 1H), 8.48 (s, 1H), 7.90 (br s, 1H), 7.82 (m, 1H), 7.76 (m, 1H), 7.68 (m, 2H), 5.66 (s, 2H), 4.39 (s, 2H), 4.37 (m, 2H), 4.09 (q, J = 10.5 Hz, 2H), 3.41 (m, 2H), 3.32 (m, 2H), 2.62 (s, 3H), 2.08 (m, 4H). 13C NMR DMSO-d6 (150 MHz): δ 137.0, 133.5, 130.2, 126.7, 125.3 (q, J = 277.3 Hz), 121.7, 120.5, 116.4, 113.6, 113.4, 109.3, 109.2, 56.1, 50.5, 46.1, 33.6 (q, J = 31.9 Hz), 30.7, 28.1, 15.5. HRMS (ESI): [M + H+] calculated 515.1835; found 515.1829.
Supplementary Material
Acknowledgments
The authors thank Drs. Szymon Klossowski and Felicia Gray for critical reading of this manuscript. This work was funded by the National Institutes of Health (NIH) Grant R01 (Grant 1R01CA160467) to J.G., a Leukemia and Lymphoma Society (LLS) TRP Grant 6116-12 to J.G., LLS Scholar (Grant 1215-14) to J.G., LLS TAP (Therapy Acceleration Program) to J.G., American Cancer Society Grant RSG-11-082-01-DMC to T.C. and Grant RSG-13-130-01-CDD to J.G., and the Department of Pathology, University of Michigan. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (Grant 085P1000817). The mouse work was performed under oversight of UCUCA at the University of Michigan.
ABBREVIATIONS
- ALL
acute lymphoblastic leukemia
- AML
acute myeloid leukemia
- DIEA
N,N-diisopropylethylamine
- FP
fluorescence polarization
- MLL
mixed lineage leukemia
- PPI
protein–protein interaction
- SI
selectivity index
- TBDPS
tert-butyldiphenylsilyl
- NEAA
nonessential amino acid
- FBS
fetal bovine serum
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.5b01305.
Table 1 with profiling of compound 1 on a panel of kinases, Table 2 with profiling of compound 1 on a panel of GPCRs, Table 3 with crystallography data, Table 4 with Caco-2 membrane permeability data, Figure 1 with selected titration curves from the FP assay, and Figure 2 with phase II metabolism data (PDF)
Accession Codes
The atomic coordinate files have been deposited in PDB with the PDB codes 5DB0, 5DB1, 5DB2, and 5DB3.
Notes
The authors declare the following competing financial interest(s): Dmitry Borkin, Jolanta Grembecka, Tomasz Cierpicki, Duxin Sun, and Xiaoqin Li are inventors in the patent filed by the University of Michigan, which covers the menin–MLL inhibitors disclosed in the current study. The patent has been licensed by Kura Oncology, Inc. Drs. Grembecka and Cierpicki receive research support from Kura Oncology. They are also receiving compensation as members of the scientific advisory board of Kura Oncology, and they have an equity ownership in the company. Other co-authors declare no potential conflict of interest.
References
- 1.Yokoyama A, Somervaille TC, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005;123:207–218. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 2.Marschalek R. Mechanisms of leukemogenesis by MLL fusion proteins. Br J Haematol. 2011;152:141–154. doi: 10.1111/j.1365-2141.2010.08459.x. [DOI] [PubMed] [Google Scholar]
- 3.Tomizawa D, Koh K, Sato T, Kinukawa N, Morimoto A, Isoyama K, Kosaka Y, Oda T, Oda M, Hayashi Y, Eguchi M, Horibe K, Nakahata T, Mizutani S, Ishii E. Outcome of risk-based therapy for infant acute lymphoblastic leukemia with or without an MLL gene rearrangement, with emphasis on late effects: a final report of two consecutive studies, MLL96 and MLL98, of the Japan Infant Leukemia Study Group. Leukemia. 2007;21:2258–2263. doi: 10.1038/sj.leu.2404903. [DOI] [PubMed] [Google Scholar]
- 4.Slany RK. When epigenetics kills: MLL fusion proteins in leukemia. Hematol Oncol. 2005;23:1–9. doi: 10.1002/hon.739. [DOI] [PubMed] [Google Scholar]
- 5.Popovic R, Zeleznik-Le NJ. MLL: how complex does it get? J Cell Biochem. 2005;95:234–242. doi: 10.1002/jcb.20430. [DOI] [PubMed] [Google Scholar]
- 6.Dimartino JF, Cleary ML. Mll rearrangements in haematological malignancies: lessons from clinical and biological studies. Br J Haematol. 1999;106:614–626. doi: 10.1046/j.1365-2141.1999.01439.x. [DOI] [PubMed] [Google Scholar]
- 7.Hess JL. MLL: a histone methyltransferase disrupted in leukemia. Trends Mol Med. 2004;10:500–507. doi: 10.1016/j.molmed.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 8.Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, Golub TR, Armstrong SA. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- 9.Grembecka J, Belcher AM, Hartley T, Cierpicki T. Molecular basis of the mixed lineage leukemia-menin interaction: implications for targeting mixed lineage leukemias. J Biol Chem. 2010;285:40690–40698. doi: 10.1074/jbc.M110.172783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yokoyama A, Cleary ML. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell. 2008;14:36–46. doi: 10.1016/j.ccr.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caslini C, Yang Z, El-Osta M, Milne TA, Slany RK, Hess JL. Interaction of MLL amino terminal sequences with menin is required for transformation. Cancer Res. 2007;67:7275–7283. doi: 10.1158/0008-5472.CAN-06-2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi A, Murai MJ, He S, Lund G, Hartley T, Purohit T, Reddy G, Chruszcz M, Grembecka J, Cierpicki T. Structural insights into inhibition of the bivalent menin-MLL interaction by small molecules in leukemia. Blood. 2012;120:4461–4469. doi: 10.1182/blood-2012-05-429274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murai MJ, Chruszcz M, Reddy G, Grembecka J, Cierpicki T. Crystal structure of menin reveals binding site for mixed lineage leukemia (MLL) protein. J Biol Chem. 2011;286:31742–31748. doi: 10.1074/jbc.M111.258186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grembecka J, He S, Shi A, Purohit T, Muntean AG, Sorenson RJ, Showalter HD, Murai MJ, Belcher AM, Hartley T, Hess JL, Cierpicki T. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol. 2012;8:277–284. doi: 10.1038/nchembio.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He S, Senter TJ, Pollock J, Han C, Upadhyay SK, Purohit T, Gogliotti RD, Lindsley CW, Cierpicki T, Stauffer SR, Grembecka J. High-affinity small-molecule inhibitors of the menin-mixed lineage leukemia (MLL) interaction closely mimic a natural protein-protein interaction. J Med Chem. 2014;57:1543–1556. doi: 10.1021/jm401868d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borkin D, He S, Miao H, Kempinska K, Pollock J, Chase J, Purohit T, Malik B, Zhao T, Wang J, Wen B, Zong H, Jones M, Danet-Desnoyers G, Guzman ML, Talpaz M, Bixby DL, Sun D, Hess JL, Muntean AG, Maillard I, Cierpicki T, Grembecka J. Pharmacologic inhibition of the Menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell. 2015;27:589–602. doi: 10.1016/j.ccell.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kar G, Gursoy A, Keskin O. Human cancer protein-protein interaction network: a structural perspective. PLoS Comput Biol. 2009;5:e1000601. doi: 10.1371/journal.pcbi.1000601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buchwald P. Small-molecule protein-protein interaction inhibitors: therapeutic potential in light of molecular size, chemical space, and ligand binding efficiency considerations. IUBMB Life. 2010;62:724–731. doi: 10.1002/iub.383. [DOI] [PubMed] [Google Scholar]
- 19.Fry DC. Protein-protein interactions as targets for small molecule drug discovery. Biopolymers. 2006;84:535–552. doi: 10.1002/bip.20608. [DOI] [PubMed] [Google Scholar]
- 20.Azzarito V, Long K, Murphy NS, Wilson AJ. Inhibition of alpha-helix-mediated protein-protein interactions using designed molecules. Nat Chem. 2013;5:161–173. doi: 10.1038/nchem.1568. [DOI] [PubMed] [Google Scholar]
- 21.Arkin MR, Whitty A. The road less traveled: modulating signal transduction enzymes by inhibiting their protein-protein interactions. Curr Opin Chem Biol. 2009;13:284–290. doi: 10.1016/j.cbpa.2009.05.125. [DOI] [PubMed] [Google Scholar]
- 22.Morelli X, Bourgeas R, Roche P. Chemical and structural lessons from recent successes in protein-protein interaction inhibition (2P2I) Curr Opin Chem Biol. 2011;15:475–481. doi: 10.1016/j.cbpa.2011.05.024. [DOI] [PubMed] [Google Scholar]
- 23.Smith MC, Gestwicki JE. Features of protein-protein interactions that translate into potent inhibitors: topology, surface area and affinity. Expert Rev Mol Med. 2012;14:e16. doi: 10.1017/erm.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 25.Cierpicki T, Grembecka J. Targeting protein-protein interactions in hematologic malignancies: still a challenge or a great opportunity for future therapies? Immunol Rev. 2015;263:279–301. doi: 10.1111/imr.12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nero TL, Morton CJ, Holien JK, Wielens J, Parker MW. Oncogenic protein interfaces: small molecules, big challenges. Nat Rev Cancer. 2014;14:248–262. doi: 10.1038/nrc3690. [DOI] [PubMed] [Google Scholar]
- 27.Bai L, Wang S. Targeting apoptosis pathways for new cancer therapeutics. Annu Rev Med. 2014;65:139–155. doi: 10.1146/annurev-med-010713-141310. [DOI] [PubMed] [Google Scholar]
- 28.He S, Malik B, Borkin D, Miao H, Shukla S, Kempinska K, Purohit T, Wang J, Chen L, Parkin B, Malek SN, Danet-Desnoyers G, Muntean AG, Cierpicki T, Grembecka J. Menin-MLL inhibitors block oncogenic transformation by MLL-fusion proteins in a fusion partner-independent manner. Leukemia. 2015 doi: 10.1038/leu.2015.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Puterova Z, Krutosíkova A, Veghc D. Gewald reaction: synthesis, properties and applications of substituted 2-aminothiophenes. ARKIVOC. 2010;(i):209–246. [Google Scholar]
- 30.Gribble GW. Recent developments in indole ring synthesis-methodology and applications. J Chem Soc, Perkin Trans. 2000;1:1045–1075. [Google Scholar]
- 31.Moyer MP, Shiurba JF, Rapoport H. Metal-Halogen Exchange of Bromoindoles. A Route to Substituted Indoles. J Org Chem. 1986;51:5106–5110. [Google Scholar]
- 32.Stokes BJ, Dong H, Leslie BE, Pumphrey AL, Driver TG. Intramolecular C-H amination reactions: exploitation of the Rh(2)(II)-catalyzed decomposition of azidoacrylates. J Am Chem Soc. 2007;129:7500–7501. doi: 10.1021/ja072219k. [DOI] [PubMed] [Google Scholar]
- 33.Higueruelo AP, Schreyer A, Bickerton GR, Pitt WR, Groom CR, Blundell TL. Atomic interactions and profile of small molecules disrupting protein-protein interfaces: the TIMBAL database. Chem Biol Drug Des. 2009;74:457–467. doi: 10.1111/j.1747-0285.2009.00889.x. [DOI] [PubMed] [Google Scholar]
- 34.Otwinowski Z, Minor W. Processing of X-ray diffraction data. In: Carter CW Jr, Sweet RM, editors. Macromolecular Crystallography, Part A. Vol. 276. Academic Press; San Diego, CA: 1997. pp. 307–326. (Methods in Enzymology). [DOI] [PubMed] [Google Scholar]
- 35.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr, Sect D: Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.The CCP4 suite: programs for protein crystallography. Acta Crystallogr, Sect D: Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 38.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang H, Guranovic V, Dutta S, Feng Z, Berman HM, Westbrook JD. Automated and accurate deposition of structures solved by X-ray diffraction to the Protein Data Bank. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:1833–1839. doi: 10.1107/S0907444904019419. [DOI] [PubMed] [Google Scholar]
- 40.Muntean AG, Tan J, Sitwala K, Huang Y, Bronstein J, Connelly JA, Basrur V, Elenitoba-Johnson KS, Hess JL. The PAF complex synergizes with MLL fusion proteins at HOX loci to promote leukemogenesis. Cancer Cell. 2010;17:609–621. doi: 10.1016/j.ccr.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.