
Figure 2.
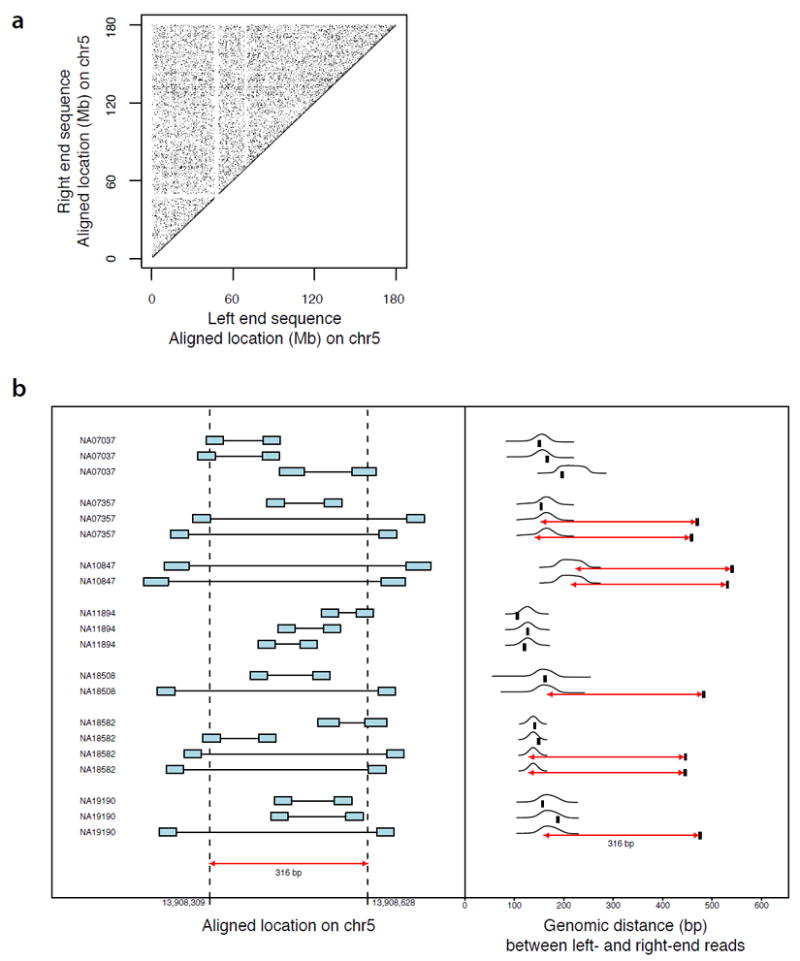
Identifying coherent sets of aberrantly mapping reads from a population of genomes.
(a) Millions of end-sequence pairs from sequencing libraries show aberrant alignment locations, appearing to span vast genomic distances. Almost all of these observations derive not from true structural variants but from chimeric inserts in molecular sequencing libraries. Data shown: paired-end alignments on chromosome 5, from 41 initial genome sequencing libraries from the 1000 Genomes Project.
(b) A set of “coherently aberrant” end-sequence pairs from many genomes. At this genomic locus, paired-end sequences (sequences of the two ends of the inserts in a molecular library) fall into two classes: (i) end-sequence pairs that show the genomic spacing expected given the insert size distribution of each sequencing library, such as the three read-pair alignments for genome NA07037; and (ii) end-sequence pairs that align to genomic locations unexpectedly far apart, but which relate to their expected insert size distributions by a shared correction factor (red arrows). A unifying model in which these eight read pairs from five genomes arise from a shared deletion allele (size of red arrows) converts all of these aberrant read pairs to likely observations. (In right panel, black tick marks indicate genomic distance between left and right end sequences; black curves indicate insert size distributions of the molecular library from which each sequence-pair is drawn.)