

Abstract
Sustained autophagy contributes to the metabolic adaptation of cancer cells to hypoxic and acidic microenvironments. Since cells in such environments are resistant to conventional cytotoxic drugs, inhibition of autophagy represents a promising therapeutic strategy in clinical oncology. We previously reported that the efficacy of hydroxychloroquine (HCQ), an autophagy inhibitor under clinical investigation is strongly impaired in acidic tumor environments, due to poor uptake of the drug, a phenomenon widely associated with drug resistance towards many weak bases. In this study we identified salinomycin (SAL) as a potent inhibitor of autophagy and cytotoxic agent effective on several cancer cell lines under conditions of transient and chronic acidosis. Since SAL has been reported to specifically target cancer-stem cells (CSC), we used an established model of breast CSC and CSC derived from breast cancer patients to examine whether this specificity may be associated with autophagy inhibition. We indeed found that CSC-like cells are more sensitive to autophagy inhibition compared to cells not expressing CSC markers. We also report that the ability of SAL to inhibit mammosphere formation from CSC-like cells was dramatically enhanced in acidic conditions. We propose that the development and use of clinically suitable SAL derivatives may result in improved autophagy inhibition in cancer cells and CSC in the acidic tumor microenvironment and lead to clinical benefits.
Keywords: Autophagy, chloroquine, tumor acidosis, cancer therapy, pH
INTRODUCTION
Malignant cells develop under the selective pressure of metabolic stress conditions including chronic and intermittent hypoxia and limited nutrients availability [1–3]. Metabolic adaptation to such environment allows rapid and continuous growth of cancer cells, leading to developing of abnormal blood vessels, which arrange irregularly and are structurally heterogeneous [4]. The vascular endothelium in tumors is defective and leaky [5] and defective tissue perfusion coupled with high metabolic rates contribute to extracellular acidosis in the tumor microenvironment [6, 7]. It has been shown in different animal models that the tumor extracellular pH (pHe) ranges between 5.9 and 7.2 [8–10], with average tumor pHe often reported ~6.5 [11–13]. Tumor acidosis represents an important selective force affecting cancer progression and therapeutic resistance [1, 14–18]. Indeed, efforts and strategies to counteract tumor acidosis have shown promising results in different preclinical models [3, 13, 18–20].
Macroautophagy (autophagy) is an evolutionary conserved process that contributes to cellular homeostasis by degrading damaged or redundant organelles, and misfolded proteins [21]. The process begins with the formation of a double membrane structure, which engulfs parts of the cytoplasm creating a double membrane vesicle, which fuses with lysosome (autolysosome). Lysosomal enzymes active in the acidic environment of autolysosomes degrade the sequestered cargo and provide hydrocarbons, fatty acids, amino acids and nucleotides to cells. Autophagy has a context-dependent role in initiation, development and progression of cancer [22, 23]. Autophagy suppresses malignant transformation by different mechanisms such as protection from genomic instability through elimination of dysfunctional mitochondria and decreased reactive oxygen species (ROS) production, degradation of onco-proteins, induction of senescence and cell death following oncogene activation, anti-inflammatory function and helping the immune system to eliminate the malignant cells [24]. After establishment of a neoplastic lesion, autophagy is believed to promote and sustain tumorigenesis and to mediate resistance to different forms of anticancer therapy [25]. In fact, proficient autophagy contributes to metabolic reprogramming and adaptation to different metabolic stresses, including hypoxia, acidosis and nutrient deprivation. About seventy clinical trials are registered in clinicaltrials.gov testing the use of HCQ or chloroquine (CQ) as autophagy inhibitors in combination anticancer therapies [26] but the use of CQ derivatives for this purpose may require further investigation. In fact, results from the first phase I/II trials support autophagy inhibition as a promising therapeutic strategy but also indicate limitation in the efficacy of HCQ to inhibit autophagy at the tumor site [27–32], underlining the need to identify better and more specific autophagy inhibitors [33]. Recently, others and we reported that autophagy contributes to the survival and adaptation of cancer cells to an acidic environment [34, 35]. However, CQ and HCQ fail to inhibit autophagy in acidic conditions because of a reduction in cellular uptake of the drugs [36]. Interestingly, the new CQ-derivative Lys-01, which displays lower pKa values, has a better activity both as cytotoxic agent and autophagy inhibitor in acidic conditions [36, 37].
CSC represent a subpopulation of cells with tumor initiating ability and they are believed to contribute to resistance to chemotherapy, metastatic spread and disease relapse [38]. It has also been suggested that tumor development is characterized by a dynamic transition between CSC and differentiated tumor cells and that both these subpopulations should be considered as target for therapy [39]. Gupta et al. reported that SAL, a polyether antibiotic widely used in veterinary medicine is a potent agent able to selectively target breast CSC and to inhibit mammary tumor growth in vivo [40]. It has been reported that autophagy promotes maintenance of breast CSC and tumorigenicity [41, 42] and that SAL can inhibit autophagy and lysosomal proteolytic activity in both breast CSC and cancer cells [43]. SAL has been described as a potassium ionophore inhibiting Wnt signaling and interfering with the proton gradient within lysosomes [44], although no effect on lysosomal pH have been reported in SAL-treated breast cancer cells [43].
In this study we analysed the pH-dependent cytotoxic and autophagy inhibiting activities of SAL towards cancer cell lines and CSC. We found that SAL is a potent inhibitor of the autophagic flux and cytotoxic agent showing increased efficacy towards cancer cells under low pH conditions.
RESULTS
Salinomycin is a potent autophagy inhibitor in acidic conditions
We recently showed that the clinically used autophagy inhibitors CQ and HCQ are not effective in blocking autophagy in the acidic environment of human tumors [36]. This effect was associated with a complete lack of cytotoxicity in acidic conditions in several cancer cell lines. In search of new autophagy inhibitors active also in acidic conditions we focused on SAL, an acidic ionophore compound used as anticoccidiosis in veterinary medicine. SAL was reported to induce cell death via autophagy upregulation in some experimental models [45, 46]. However, it was recently reported that 2 μM SAL inhibits the autophagic flux in breast and hepatocellular carcinomas [43, 47]. In order to establish the activity of SAL on autophagic flux, we started our investigation by using HOS cells stably transfected with a GFP-LC3 vector, which allows the analysis of the autophagic flux by flow cytometry by monitoring the accumulation of GFP-LC3-positive autophagosomes in the presence of lysosomal inhibitors [48]. BafA1 acts as inhibitor of the V-ATPase and raises lysosomal pH, thus inhibiting autolysosomes formation and leading to accumulation of GFP-LC3-positive autophagosomes. The autophagic flux here represents the ratio of GFP-LC3 fluorescence between presence and absence of saturating concentration of Bafilomycin A1 (BafA1). First, we observed that HOS-GFP-LC3 cells treated with 2 μM SAL for 6 hours accumulate a large number of intracellular vacuoles, with cells cultured at pH 6.8 showing an increased vacuolization with respect to cells kept at pH 7.4 (Figure 1A). As expected, autophagosomes-associated LC3-GFP fluorescence was increased in control cells treated with BafA1 at both pH conditions, indicating the presence of proficient autophagy in both pH conditions (Figure 1B), with autophagic flux being 2.2±0.23 and 2.2±0.36, respectively at pH 7.4 and 6.8. A significant increase in GFP-LC3 fluorescence was observed also in cells treated only with SAL in both pH conditions. The combined treatment with BafA1 showed only a minor increase in cells at pH 7.4, indicating that SAL reduces the autophagic flux without blocking it (1.5±0.1). Conversely, in cells kept at pH 6.8 and treated with SAL the GFP-LC3 signal intensity was similar in presence or absence of BafA1, suggesting that in HOS cells in acidic conditions SAL totally blocks the autophagic flux (1±0.1, Figure 1B). To further test the dose-dependent effects of SAL in these cells we used high-content fluorescence microscopy to quantify the number of GFP-LC3-positive vesicles in cells treated with different doses of SAL in absence or presence of BafA1. The data show that SAL induces a dose-dependent accumulation of GFP-LC3-positive vesicles (Figure 1C-1D) and that at 1-2 μM SAL the number of GFP-LC3-positive vesicles in BafA1 treated and untreated cells is similar, thus suggesting that SAL inhibits autophagic flux (Figure 1E).
Figure 1. Effects of SAL on accumulation of GFP-LC3+ vesicles in HOS cells at pH 7.4 and 6.8.
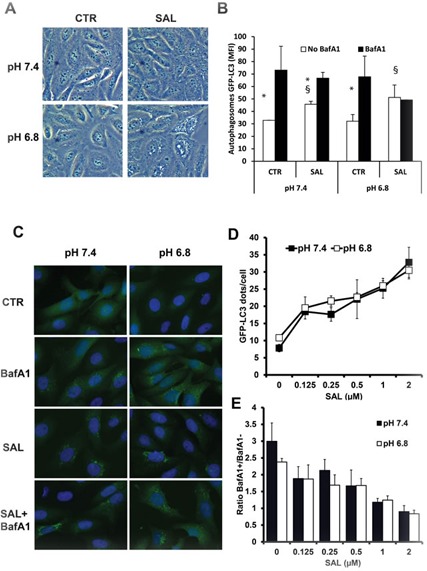
A. Phase contrast microscopy pictures of HOS-GFP-LC3 cells cultured with medium buffered at pH 7.4 or pH 6.8 overnight and treated with SAL (2 μM) for 6 hours. B. In the same settings as in A, BafA1 (50 nM) was added for 2 hours before flow cytometric analysis of saponin-permeabilised cells to quantify autophagosomes-associated GFP-LC3 (data are from three independent experiments run in duplicate samples). Differences between MFI in the presence or absence of BafA1 is indicated by *, showing a P < 0.05. Differences between MFI in the presence or absence of SAL is indicated by §, showing a P < 0.05. C. Fluorescence microscopy images of GFP-LC3-HOS cells under different treatments showing the presence of GFP-LC3+ vesicles. D. Quantitative analysis of the dose-dependent activity of SAL on the number of GFP-LC3+ vesicles run in triplicates. E. The ratio between the number of GFP-LC3+ vesicles in presence and absence of BafA1 is shown. Ratio of 1 indicates no additive effects of BafA1 on the SAL-mediated inhibition of the autophagic flux.
In order to properly demonstrate the pH-dependent activity of SAL, we extended our analysis to different cell lines representing different tumor hystotypes and in conditions of both transient and chronic acidosis. First, we used HCT116 (colon carcinoma) and Me30966 (melanoma) cell lines from which we developed sub-lines chronically adapted to grow at pH 6.8 [36]. As indicated above, BafA1 as a lysosomal inhibitor blocks the degradation and turnover of LC3-II and other substrates such as SQSTM1. The autophagic flux was then defined as the ratio between the normalised LC3-II levels in presence and absence of BafA1 at saturating concentrations (100 nM) (LC3-II AF). In these settings, a ratio of 1 indicates total blockage of autophagic flux. As observed in the HOS-GFP-LC3 cells, SAL induced a dose-dependent inhibition of the turnover of both LC3-II and SQSTM1 in all cell lines tested, with best activity detected at 2 μM (Figure S1A-B). As shown in Figure 2A and 2C, SAL treatment induced a strong time-dependent increase in LC3-II expression in both parental and pH 6.8-adapted HCT116 and Me30966 cells. In all cell lines, autophagic flux was strongly inhibited already after 4 hours treatment. Despite some variability likely dependent on different substrate-specific kinetics, the turnover of both LC3-II and to a lesser degree SQSTM1 was reduced in all cell lines independently of the pH culture conditions (Figure 2B and 2D).
Figure 2. Effects of SAL on autophagic flux in cancer cell lines at pH 7.4 and 6.8.
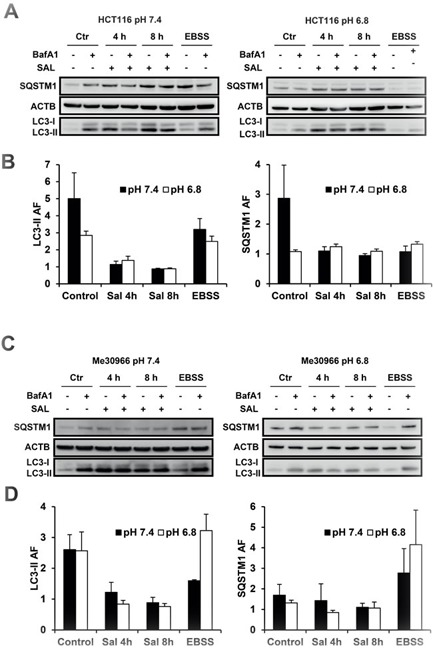
Parental and low pH-adapted HCT116 A. and Me30966 C. cells were exposed to SAL (2 μM) for 4-8 hours, with or without BafA1 (100 nM) during the last 2 hours. WB analysis of LC3-II and SQSTM1 was performed to measure the autophagic flux. EBSS-treated cells were used as controls. Data in panels A and C are representative of four independent experiments. Quantification of LC3-II and SQSTM1 are shown for HCT116 B. and Me30966 D. cells with data expressed as mean and SE.
The data shown so far suggest that SAL efficiently inhibits the autophagic flux in cancer cells also at pH 6.8. However, tumor tissues may be characterised by regions with even lower pH and the average pH in tumors is reported to be around 6.5 [10, 12, 16, 49]. We have reported that CQ failed to inhibit autophagy at pH 6.8 while the CQ dimer Lys-01 had still a detectable activity at pH 6.8. However, when tested at pH 6.5, Lys-01 did not induce LC3-II increase in HCT116 cells, suggesting that its activity is also inhibited at very acidic conditions (Figure S1C). Thus, we tested the activity of SAL on the autophagic flux in cells after exposing them to medium buffered at pH as low as 6.5. We exposed HCT116 cells to media buffered at different pH overnight and added 2 μM SAL for 8 hours, with BafA1 added during the last 2 hours incubation. We observed that SAL maintained a strong autophagy blocking activity even at pH 6.5 as shown by LC3-II and SQSTM1 expression (Figure 3A) and a similar observation was made using Me30966 cells cultured at pH 6.5 (Figure S1D).
Figure 3. Effects of SAL on HCT116 cells at pH 6.5 and in multicellular spheroids.
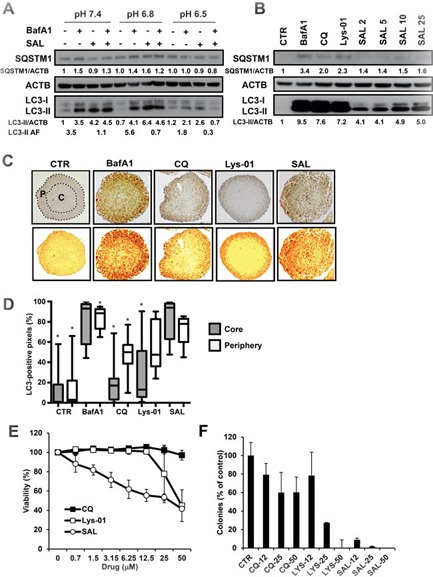
A. HCT116 cells were exposed to medium at pH 7.4, pH 6.8 and pH 6.5 overnight. SAL (2 μM) was added to cells for 6 hours, with or without BafA1 (100 nM) during the last 2 hours. The picture shows one representative WB analysis of three independent experiments. B. MCS derived from HCT116 cells were treated for 24 hours with different autophagy inhibitors (50 nM BafA1, 50 μM CQ and Lys-01) and SAL at different concentrations (μM). WB analysis on cell lysates was performed to analyse the expression of LC3-II and a representative WB from three different experiments is shown. C.-D. HCT116 MCS (n = 24) were treated with different autophagy inhibitors for 24 hours and analysed by IHC to detect the distribution of the augmented LC3 expression. LC3 positive pixel analysis was performed in the peripheral (P) and core (C) regions of the MCS as indicated for one representative untreated MCS. Data are shown as box plots, normalised and expressed as percentage of the total number of pixels in the specific area. The * indicates a P < 0.05 for differences between SAL and all treatment groups, calculated by the Mann-Whitney test. E. HCT116 MCS were treated for 3 days with different concentrations of CQ, Lys-01 and SAL. Cell viability was determined with the acid phosphatase assay. F. HCT116 MCS were treated for 3 days with different concentrations of CQ, Lys-01 and SAL. Single cell suspensions were obtained from MCS and clonogenic survival assay was performed. Experiments in E-F were repeated three times and means and SD are shown.
The activity of SAL is detectable in the acidic core of multicellular spheroids
To gain further insight into the capacity of SAL to block autophagy in a more complex environment we took advantage of the multicellular spheroid (MCS) culture system, where cells grow in a 3D structure with a central core characterised by acidic pH in the range 6.6-6.8 [50]. MCS from HCT116 cells were treated with CQ, Lys-01 and SAL and the accumulation of LC3-II was analysed. First, we observed by Western Blot (WB) that SAL induced a dose-dependent accumulation of LC3-II and SQSTM1 after 24 hours treatment (Figure 3B). The accumulation of LC3-II was even higher in MCS treated with Lys-01 (50 μM), BafA1 (50 nM) and CQ (50 μM).
Compounds blocking the autophagic turnover of LC3-II are expected to induce an increase in total LC3 positivity in tissues due to lack of LC3-II degradation. Interestingly, when analyzing the distribution pattern of LC3 signal intensity in relation to MCS areas by immunohistochemistry (IHC), we observed that MCS treated with CQ and Lys-01 showed a strong LC3 signal localised in the outer peripheral layer but almost completely absent in the MCS core, suggesting a failure to block autophagy in the acidic/hypoxic core of the MCS (Figure 3C). Conversely, low concentrations of SAL (2 μM) were able to increase LC3 intensity throughout the whole MCS area, similarly to the effects observed after treatment with BafA1 (Figure 3C). A quantitative image analysis of LC3 positive pixels normalised to the total number of pixels in each area showed that CQ and Lys-01 mostly induced accumulation of LC3 in the peripheral layer of MCS while BafA1 and SAL induced a significantly higher increase in LC3 positive pixels both in the peripheral region and in the core region of MCS with respect to CQ and Lys-01 (Figure 3C-3D).
In line with a more effective autophagy blocking activity in this 3D culture model, SAL showed a much stronger cytotoxic effect on MCS as compared to CQ and Lys-01 as shown by viability assay (Figure 3E). Interestingly, clonogenic cell survival assay showed that while 12 μM CQ or Lys-01 still allowed about 80% of treated cells to form colonies, the same concentration of SAL completely inhibited the capacity of cells to survive treatment (Figure 3F).
SAL blocks the autophagic flux in cancer stem cells
SAL was identified as a breast CSC-specific cytotoxic agent using a model of immortalised and transformed breast carcinoma cells (HMLER) carrying CSC properties [40]. Using the same model, Yue and colleagues reported that SAL selectively targets breast CSC through inhibition of autophagy [43]. HMLER cells are composed of two different cell populations differing in their CSC properties and in CD24 expression (Figure S2A) [51]. CD44+/CD24+ cells have an epithelial phenotype and no ability to make mammospheres while CD44+/CD24low cells have properties of CSC, including capacity to form mammospheres. First, we observed also in these cells that 2 μM SAL blocks the autophagic flux both at pH 7.4 and pH 6.8 as measured by LC3-II in the presence of BafA1 (Figure S2C).
In order to confirm the data on autophagic flux obtained by WB, we transfected HMLER CD24+ and CD24low cells with an LC3 tandem probe in which LC3 is tagged to GFP and RFP. Because the acidic pH of lysosomes quenches the green fluorescence of GFP, this probe helps to identify and count autophagosomes (GFP+RFP+ yellow dots) and autolysosomes (GFP-RFP+ red dots). Cells were exposed overnight to medium buffered at pH 7.4 or 6.8 and treated with SAL (2 μM) or starved with Earle's Balanced Salt Solution (EBSS) for 6 hours (Figure 4A). As expected, EBSS-treated cells showed an increased number of red dots in all conditions examined, indicating that starvation-induced autophagy occurs in all cells (Figure 4B). Treatment with SAL induced the accumulation of yellow dots in all conditions tested as compared to control and such effect was significantly greater in both cell lines cultured at acidic pH (Figure 4B).
Figure 4. Effects of SAL on autophagy in HMLER cells by fluorescence microscopy.
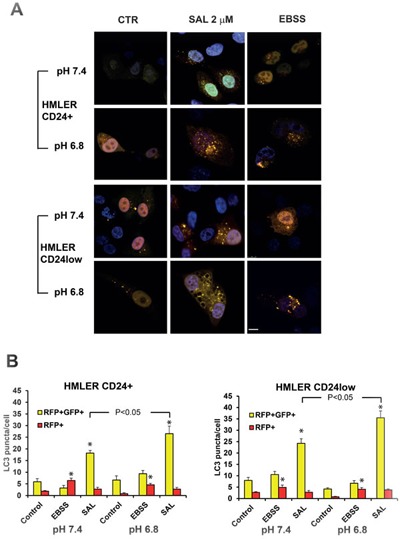
A. HMLER CD24+ and CD24low cells were transfected with RFP-GFP-LC3 plasmid and then cultured with medium buffered at pH 7.4 or pH 6.8 overnight. SAL (μM) was added at different concentrations for 6 hours and cells treated with EBSS were used as positive control. Cells were fixed and observed with confocal microscopy. B. The number of yellow and red dots per cell was analysed and data in the charts are expressed as mean ± SE from at least 15 cells/condition. Scale bar: 10 μm. The * indicates a P < 0.05 for differences between treatment groups and controls.
To further investigate the sensitivity of HMLER cells to autophagy inhibition we tested the activity of different SAL concentrations at pH 7.4 and pH 6.8. First, we observed that the basal autophagic turnover of LC3-II seems to be higher in CD24+ cells as compared to CD24low cells (Figure 5A). The analysis of the autophagic flux based on LC3-II turnover showed that SAL reduced the autophagic flux in a dose-dependent manner in both cell lines and at both pH 7.4 and pH 6.8 (Figure 5A-5C). Quantification of the autophagic flux calculated based on BafA1-induced accumulation of SQSTM1 supported the data obtained with LC3-II (Figure 5B, 5C).
Figure 5. Effects of SAL on autophagy in HMLER cells by western blot.
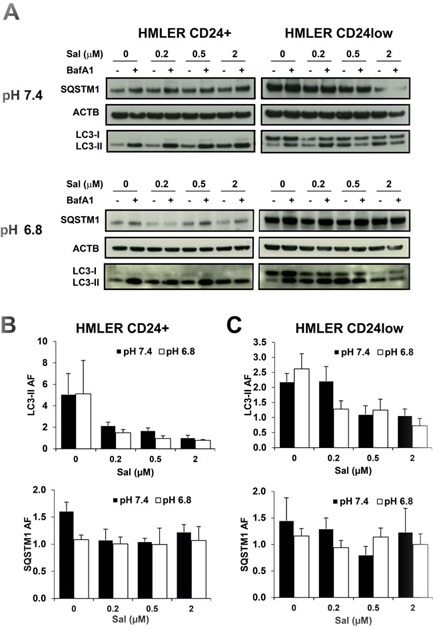
A. HMLER CD24+ and CD24low cells were plated and cultured with medium buffered at pH 7.4 or pH 6.8 overnight. SAL (μM) was added at different concentrations for 6 hours, with or without BafA1 (100 nM) during the last 2 hours incubation. The picture shows one representative WB analysis of four different experiments. Quantification of LC3-II and SQSTM1 is shown in panel B. for CD24+ cells and panel C. for CD24low cells and data are expressed as mean and SE from four independent experiments.
In conclusion, these data suggest that very low SAL concentrations are able to inhibit the autophagic flux in HMLER cells cultured at acidic pH.
Acidosis enhances the cytotoxicity of salinomycin against cancer cells
The inability of CQ to block autophagy in acidic conditions is associated with a complete lack of cytotoxicity in cancer cells under acidic conditions. Therefore, we performed cytotoxic assays to test whether low pH affected the cytotoxic effects of SAL. We used parental HCT116 and Me30966 cells cultured at pH 7.4 and their low pH-adapted sublines cultured at pH 6.8 (HCT116pH7.4 and HCT116pH6.8) and exposed to increasing SAL concentrations for 48 hours. Microscopic observation of cells indicated a massive vacuolization at pH 6.8 already 6 hours after treatment with SAL, as shown for Me30966 cells in Figure 6A. SAL showed a dramatic and significantly increased cytotoxicity in low pH-adapted cells (Figure 6B-6C), with a 10-fold difference in IC50 values (Table 1). These effects were not dependent on the phenotype of acid-adapted cells since parental HCT116 and Me30966 cells exposed to acidic medium only for the duration of the treatment were equally sensitive as their acid-adapted sublines (Figure 6D-6E), suggesting that the increased SAL cytotoxicity in acidic conditions are likely dependent only on the pH of the culture medium. Conversely, the cytotoxic effects of SAL in acid-adapted cells cultured at pH 7.4 were comparable to those observed for the parental cell lines (Figure 6D-6E).
Figure 6. Cytotoxic activity of SAL on parental and acid-adapted HCT116 and Me30966 cells.
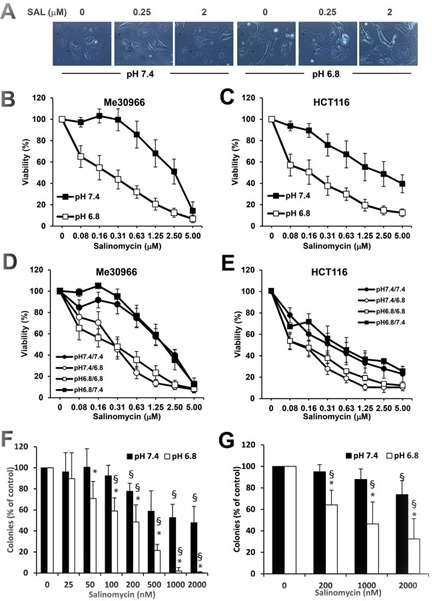
A. Phase contrast microscopy pictures of Me30966 cells treated with SAL for 5 hours. B.-C. Viability assay with acid phosphatase on Me30966, HCT116 and their pH 6.8 adapted sublines after 48 hours treatment with different concentrations of SAL. D.-E. Parental and low pH-adapted Me30966 (D) and HCT116 (E) cells were plated in their respective medium at pH 7.4 and pH 6.8. The next day the medium was replaced with either medium with the same pH or medium with a different pH. Viability was assessed after 48 hours treatment with different concentrations of SAL. The experiment was repeated five times in triplicate wells and data show means and SD. F.-G. Clonogenic assay on parental and pH 6.8-adapted HCT116 cells treated with SAL for 8 hours (F) or 48 hours (G) and then left in drug-free medium for 6 days. Data in B-E are obtained from at least three different experiments and expressed as mean ± SD. Differences between groups in F-G were analysed with paired T-test. * indicates a P < 0.05 for differences between pH 7.4 and pH 6.8 while § indicates a P < 0.05 for differences between SAL-treatment and controls. Non significant differences are not shown.
Table 1. The IC50 (μM) of SAL in HCT116 and Me30966 cells is reported.
| HCT116 | Me30966 | |
|---|---|---|
| pH 7.4 | 1.63 ± 0.77 | 2.46 ± 0.62 |
| pH 6.8 adapted | 0.12 ± 0.06 | 0.22 ± 0.15 |
| pH 6.8 transient | 0.13 ± 0.07 | 0.54 ± 0.19 |
To analyse the effects of SAL on clonogenic cell survival we used HCT116pH7.4 and HCT116pH6.8 cells and exposed them to different SAL concentrations for 48 hours, followed by culturing cells in drug-free medium for 6 days. Again, the inhibiting effect of SAL on clonogenic growth was dramatically increased in cells cultured at acidic pH (Figure 6F). In addition, we performed clonogenic survival assay on parental HCT116 cells cultured at pH 7.4 and pH 6.8 in the presence of SAL for 8 hours and then kept in drug-free medium for 6 days. The data indicate that even short exposure to SAL in acidic conditions is effective in reducing clonogenic cell survival while very mild effects were observed in cells treated at pH 7.4 (Figure 6G).
Acidic conditions improve the ability of SAL to kill cancer stem cells
SAL was shown to be more selective towards breast CSC [40] and it is currently investigated as a promising compound to develop in anti-CSC therapy [52]. We showed that autophagy inhibition by SAL is increased at acidic conditions. We were therefore interested in investigating the effects of pH on cytotoxic activity of SAL in HMLER CD24+ and CD24low cells at pH 7.4 and 6.8. First, we confirmed also in these cells that the cytotoxic activity of CQ is totally inhibited by even mild acidosis (Figure S2B). Thus, we tested the sensitivity of these cells to the dimeric CQ derivative Lys-01 and we observed that CD24low cells are more sensitive to Lys-01 with respect to CD24+ cells (IC50 13.5 ± 1.7 vs 30.3 ± 9 μM, P = 0.07), indicating that breast CSC are more sensitive to autophagy inhibition (Figure 7A). As expected, the CD24low cells are more sensitive to SAL as compared to CD24+ cells in standard culture conditions at pH 7.4 (P < 0.05), in line with their increased susceptibility to autophagy inhibition (Figure 7B and Table 2). Interestingly, the cytotoxic effects of SAL on both cell lines were dramatically amplified in acidic culture conditions (Figure 7B and Table 2), as already observed for HCT116 and Me30966 cells. Importantly, the IC50 of SAL for CD24low cells was 10-fold lower than that for CD24+ cells, suggesting an even more selective effect of SAL on these cells at acidic conditions (P < 0.05, Table 2). For example, massive vacuolization was observed in CD24low cells exposed to SAL for only 5 hours and cultured at acidic pH while this phenomenon was not seen at pH 7.4 (Figure S2D).
Figure 7. Cytotoxic activity of SAL on breast cancer stem cells.
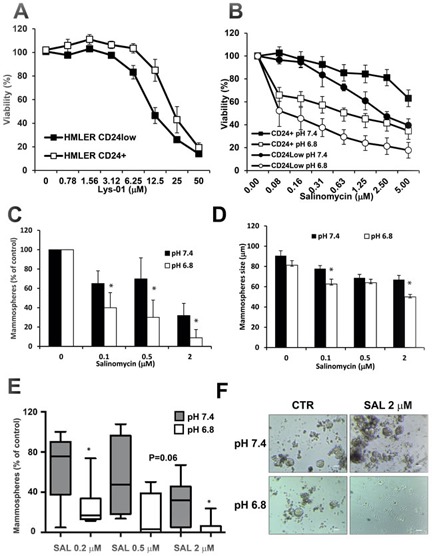
A. Cell viability of HMLER CD24+ and CD24low cells after treatment with the autophagy inhibitor Lys-01 for 48 hours. B. Cell viability of HMLER CD24+ and CD24low cells cultured with medium buffered at different pH and treated with SAL for 48 hours. C. CD24low cells were treated with SAL for 48 hours at two pH conditions and viable cells were allowed to form mammospheres for 6 days. Salinomycin reduces the sphere-forming capacity of CD24low cells and the effects are increased when treatment is done in acidic conditions. D. Mammospheres size from experiments in panel D is shown. Data in panels C-D are obtained from four different experiments and expressed as mean ± SD. E. CSC were isolated from six patients with breast carcinoma. Single cell suspensions obtained from patients-derived mammospheres were cultured in medium buffered at pH 7.4 or 6.8 for 10 days and the number of mammospheres was evaluated by phase contrast microscopy. The box plots indicate medians and 25-75 percentiles and 5-95 percentiles. Differences between groups were analyzed by Wilcoxon signed-rank test. F. Phase contrast microscopy pictures of CSC-derived mammospheres from patient 4 with and without treatment with SAL 2 μM at pH 7.4 and pH 6.8. Scale bar: 100 μm. In all charts the * indicates P < 0.05.
Table 2. The ICC50 (μM) of SAL in HMLER CD24+ and CD24low cells is reported.
| CD24+ | CD24low | |
|---|---|---|
| pH 7.4 | 7.6 ± 1.2 | 3.1 ± 0.3 |
| pH 6.8 | 1.8 ± 0.3 | 0.14 ± 0.06 |
SAL has pH-dependent effects on inhibition of mammosphere formation
We used HMLER CD24low cells to investigate whether the pH-dependent activity of SAL also affects the CSC property of mammospheres formation. First, we cultured adherent CD24low cells at pH 7.4 and pH 6.8 in presence of increasing concentrations of SAL for 48 hours and investigated the sphere-forming ability of viable single cells after treatment. As shown in Figure 7C, at physiological pH 7.4 culture conditions cells treated with SAL formed significantly less mammospheres as compared to untreated cells (P < 0.05). However, this effect was dramatically increased in cells that were treated with SAL under acidic conditions, with < 10% mammospheres formed with SAL 2 μM at pH 6.8 (P < 0.05, Figure 7C). In line with this observation, we found that the effects of SAL on reducing the size of mammospheres were also significantly enhanced in acidic conditions (Figure 7D). Similar results were obtained when single cell suspension of CD24low cells were allowed to form mammospheres in the presence or absence of SAL for 7 days (Figure S3A-B).
Acidic conditions improve the ability of SAL to inhibit mammosphere formation from breast cancer tissue derived stem cells
In order to test the potential relevance of these findings in clinical settings we have analysed the activity of SAL on patient derived breast CSC. Clinical characteristics of tumor patients are provided in Table 3. Briefly, tumor cells were isolated from patient-derived breast cancer tissues and cultured in selective medium as described in Materials and Methods in order to obtain mammospheres (see Figure S3C as example). Mammospheres were then trypsinized and single cells suspensions were cultured at pH 7.4 and pH 6.8 in absence or presence of three SAL concentrations. Newly formed mammospheres were counted after 10 days in culture. First, we could observe that different pH culture conditions did not significantly affect the absolute number of mammospheres formed in untreated cultures (23 ± 8 at pH 7.4 vs 17 ± 5 at pH 6.8, P = 0.13). As observed for mammospheres from HMLER CD24low cells, SAL inhibited the formation of mammospheres in standard culture conditions in a dose-dependent manner (Figure 7E). Notably, the ability of SAL to inhibit mammosphere formation from patients-derived CSC was dramatically increased in acidic conditions, indicating that SAL has a strong pH-dependent activity also on freshly derived patients CSC (Figure 7F).
Table 3. Clinical features of breast carcinoma patients.
| Patient ID | Tumor type | ER+ (%) | PR+ (%) | Her2+ (%) | KI67+ (%) | NHG |
|---|---|---|---|---|---|---|
| 1 | IDC | 100 | 0 | 0 | 21 | II |
| 2 | IDC | 95 | 100 | 0-1 | 35 | III |
| 3 | DCIS | NA | NA | NEG | NA | II-III |
| 4 | IDC | 95 | 0 | 2 | 34 | III |
| 5 | IDC | 95 | 45 | 0 | 37 | III |
| 6 | IDC | 0 | 0 | 3 | 28 | III |
Salinomycin accumulates at higher concentrations in cells under acidic conditions
A major reason for the lack of anti-autophagic activity of CQ in cancer cells cultured in acidic conditions is the dramatically reduced entry of the drug into cells because of its chemical properties as a weak base with a pKa around 8.1 [36, 53]. SAL contains a carboxylic acid and is a weak acid with a reported experimental pKa of 6.4, which may favor its plasma membrane permeability in acidic conditions. Since SAL showed an improved ability to target cancer cells in acidic conditions we applied UPLC-MS/MS to investigate pH-dependent changes in intracellular SAL accumulation. These experiments were run with two concentrations of SAL (2-4 μM) and in three cell lines (HCT116, HMLER CD24+ and HMLER CD24low cells). The analysis in all cell lines indicated that SAL accumulates in a dose-dependent manner and that the intracellular amount of SAL is significantly higher (up to 50%) in cells cultured in acidic conditions (Figure 8). These results suggest that the intracellular accumulation of SAL may be favored by acidic conditions, which may contribute to the increased activity of SAL in acidic conditions.
Figure 8. Mass spectrometry data of pH-dependent intracellular SAL accumulation.
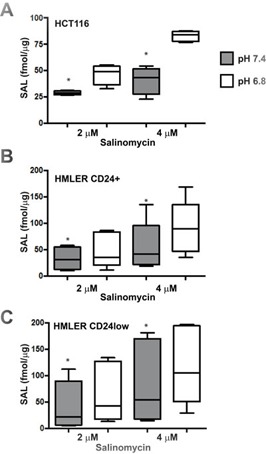
HCT116 A., HMLER CD24+ B. and CD24low C. cells were plated and the next day exposed to medium buffered at pH 7.4 or pH 6.8 in presence of 2 and 4 μM SAL for 3 hours. The amount of intracellular SAL was quantified by UPLC-MS/MS and expressed as fmol/ μg proteins. Data for HMLER cells represent mean ± SEM of five experimental replicates while data for HCT116 cells are representative of one of two experiments with similar results. Differences between groups were analysed by Wilcoxon signed rank test and the * indicates P < 0.05.
DISCUSSION
Tumor acidosis represents an important pathogenic factor in the progression towards malignant disease and it has been known for many decades to negatively impact on therapeutic efficacy [20, 54]. Autophagy is also known as an important protective mechanism against different forms of anticancer therapies and autophagy inhibition is recognized as a potential tool to improve efficacy of chemotherapy [25]. We and others have reported that melanoma and breast carcinoma cell lines adapt to chronic acidosis by upregulating autophagy, thus providing a rationale for targeting autophagy in acidic tumor regions to prevent malignant progression and improve therapeutic efficacy [34, 35]. The results of phase I/II clinical trials using HCQ as autophagy inhibitor in combination anticancer therapies have indicated potential clinical benefits for a subset of the treated patients but also poor efficacy [33]. Confounding factors for the explanation of these results are the autophagy-independent additional off-target effects of CQ reported recently [55, 56]. We showed that tumor acidosis is one of the factors that may negatively affect the efficacy of CQ in vivo [36]. In fact, CQ shows no cytotoxicity and no autophagy inhibition on cancer cells cultured in acidic conditions and in hypoxic tumor regions in a human colon carcinoma xenograft. Such limitations may possibly be overcome by the development of new CQ-derivatives optimized for clinical applications [25, 37]. This scenario prompted us to search for more effective autophagy inhibitors able to target cancer cells during tumor acidosis. The ionophore salinomycin was reported as a compound selectively targeting breast cancer stem cell [40]. The effects of SAL on cancer stem cells have been also reported for other cancer types including glioma and many carcinomas [52, 57–59]. It has been shown that SAL interferes with the maintenance of breast CSC by acting as inhibitor of autophagy and of lysosomal proteolytic function [43]. Here we confirm that SAL at doses 0.2-2 μM potently inhibits the autophagic flux in different tumor cells lines, including osteosarcoma, melanoma, colon carcinoma and breast carcinoma. Importantly, unlike CQ and its derivatives whose activity is to different degrees counteracted by acidosis, the ability of SAL to inhibit autophagic flux is potently increased in conditions of both transient and chronic acidosis. In line with the increased activity as autophagy inhibitor at acidic pH, we found that the cytotoxic activity of SAL was augmented by 5-10 fold in conditions of acidosis when tested on melanoma, colon and breast carcinoma cell lines, with SAL IC50 being in the range 100-500 nM in cells cultured at pH 6.8. We observed that the cytotoxic activity of SAL in terms of IC50 was similar in cancer cell lines transiently exposed to acidosis and in the same cell lines chronically adapted to acidosis, suggesting that SAL sensitivity may be dictated by medium pH and not by intrinsic properties of the cell lines. Such activity may be related to the pharmacological properties of SAL, which is a weakly acidic, lipophilic compound with a reported pKa of 6.4 due to its carboxylic group [60]. In fact, mass spectrometry analysis of SAL showed an increased intracellular accumulation in cells cultured at pH 6.8 as compared to cells cultured in medium at the physiological pH. However, whether such increased accumulation is alone responsible to explain the stronger activity of SAL in acidic conditions remains to be further investigated. In fact, SAL has an inhibitory effect on oxidative phosphorylation and it is generally considered an inhibitor of mitochondrial function [61, 62], causing a very fast acidification of the mitochondrial matrix. Cell survival under acidosis was reported in post-mitotic cells to be mediated by a homeostatic adaptive response leading to increased mitochondrial function, including maximal respiratory capacity [63]. Interestingly, we observed that acid-adapted HCT116 cells show a higher maximal respiratory capacity as compared to cells cultured at physiological pH (Pellegrini et al, unpublished data) and it is conceivable to hypothesize that SAL-mediated inhibition of mitochondrial function may contribute to increase the sensitivity of cancer cells under acidosis. Moreover, SAL is a potassium ionophore and very likely alters potassium gradients. Interestingly, other potassium ionophores like nigerycin inhibit lysosomal degradation and autophagic flux [64] and there is growing evidence that ions modulate autophagy in cancer [65].
Preclinical pharmacokinetic studies on SAL metabolism performed in mice treated with 5 mg/kg indicated that the Cmax of plasma SAL concentration was 1.7 μM with a very fast systemic elimination within 5 hours [66]. Interestingly, SAL 5 mg/kg had low systemic toxicity but induced neurotoxic effects which could be reduced using mitochondrial Na+/Ca2+ exchanger inhibitors without affecting the antitumor effects. In line with this, studies in mice indicated that SAL has reversible dose-dependent adverse effects on the male reproductive system [67]. Therefore, the poor water solubility and its toxicity still represent important limitations for clinical use of SAL, pointing out that SAL derivatives and/or nanoparticles are to be developed to reach higher local concentrations at tumor site [68]. In this context, there is only one report on the treatment of two cancer patients with low dose SAL (250 μg/kg) which showed mild but not long-term side effects [69].
Since SAL has been reported to inhibit CSC in several cancer models we investigated its activity in a model of breast CSC. Two sublines derived from the cell line HMLER and differing in their stem cell properties have been recently described [51]. We first observed that the subline HMLER CD24low, carrying CSC properties has an increased sensitivity to autophagy inhibition mediated by Lys-01 and SAL with respect to the non-CSC subline HMLER CD24+, confirming that autophagy plays a crucial role in maintaining survival and proliferation of CSC [41, 70, 71]. Interestingly, exposure to acidic medium increased the sensitivity of CD24+ cells by 5-fold whereas a 15-fold increase in sensitivity was observed in CD24low cells. In line with this, SAL was more effective at inhibiting autophagic flux in CD24low cells and as low as 200 nM SAL was sufficient to block autophagic flux in CD24low cells cultured in acidic conditions. CD24low cells were described to have CSC features among which the ability to form mammospheres in vitro [51]. We found that SAL inhibits mammosphere formation from CD24low cells in standard culture conditions and such inhibition is greatly amplified during acidosis. This indicates that cells with stem-like properties are more susceptible to autophagy inhibition. Moreover it also supports new findings suggesting that inhibition of lysosomal function may specifically target CSC in different cancer types [72, 73]. To further investigate the potential clinical relevance of our observation, we analysed the pH-dependent effects of SAL on CSC derived from patients with breast carcinoma. Breast CSC obtained from cancer patients showed a much greater sensitivity to SAL when cultured in acidic medium.
In conclusion, we identified SAL as a potent inhibitor of the autophagic flux active under tumor acidosis on both cancer cells and CSC. The development of SAL derivatives with lower toxicity for humans will be fundamental to test its efficacy in clinical settings.
MATERIALS AND METHODS
Chemical and antibodies
RPMI-1640 (SH30255.01), trypsin (SH40003.12), phosphate-buffered saline (PBS, SH40003.12) and Fetal Bovine Serum (FBS) (SV30160.03) were from HyClone. Sodium Bicarbonate (25080) and RPMI-1640 without NaHCO3 (51800) were from Gibco. Bafilomycin A1 (BafA1, B1793), Chloroquine di-phosphate salt (CQ, C6628), salinomycin (SAL, S4526), protease cocktail tablets EDTA-free, phosphatase inhibitors (P5726, P0044), insulin (I9278) and bovine serum albumin (BSA, A7906) were from Sigma. Recombinant human epidermal growth factor (EGF) was purchased from Invitrogen (PHG6045). The Lys-01 was synthetized by OncoTargeting AB (Sweden) and dissolved in DMSO. Protein assay dye reagent concentrate (500-0006), protein assay standard I (5000-0007) and dry milk (170-6404) were from Bio-Rad. The following antibodies were used: LC3B (Cell Signaling Technology, 2775) and β-actin (Sigma, A5441). HRP-conjugated anti-rabbit (NA934V) and anti-mouse (NXA931) antibodies, ECL system (RPN2106) and PVDF membranes (RPN303F) were from GE Healthcare.
Cell culture
Me30966, HCT116 and HOS-GFP-LC3 cell lines were cultured in RPMI-1640 medium supplemented with 10% FBS and antibiotics. The HOS cell line stably expressing the GFP-LC3 plasmid (HOS-GFP-LC3) was a kind gift from Gerry McInerney (MTC, Karolinska Institute, Stockholm). The cell lines HMLER CD24+ and HMLER CD24low were kindly provided by Dr. Anne-Pierre Morel (Centre Leon Berard, Lyon, France). These cells were grown in DMEM-F12 medium supplemented with 10% FBS, antibiotics, insulin (10 μg/ml), hydrocortisone (0.5 μg/ml), EGF (10 ng/ml) and puromycin (0.5 μg/ml). All cells lines were grown at 37°C in presence of 5% CO2. The low pH adapted HCT-116 (HCT116pH6.8) and Me30966 (Me30966pH6.8) cell lines were obtained by growing the parental cells in RPMI-1640 medium buffered at pH 6.8 for three months as previously described [36]. The different pH in media was buffered by adding different concentration of NaHCO3 and letting the media equilibrate overnight in the incubator at 5% CO2. Actual pH in media was measured before, during and after each experiment. Cell lines were tested using the LGC Standards Cell Line Authentication service.
High-content fluorescence microscopy
HOS-GFP-LC3 cells (7000 cells/well) were plated into a 96 well plate (Cell carrier, 6005550) and incubated at 37°C and 5% CO2 overnight. The medium was removed at the following day and replaced with medium at pH 7.4 or pH 6.8. SAL was added for 6 h and BafA1 for the last 2 h of the incubation. The medium was removed and the cells were incubated with 4% paraformaldehyde and 5μg/ml Hoechst 33342 (Thermo Fischer, H3570) in the dark at RT for 20 minutes. The paraformaldehyde was removed and the cells were washed three times using PBS and kept in PBS during acquisition. Images were captured using an ImageXpress scanner. The software MetaXpress was used to analyse the pictures defining GFP-positive dots of 0.5-8 μm as GFP-LC3-positive vesicles. ImageJ was used to compose the pictures of the different wavelenghts.
Multicellular spheroids
HCT116 cells (10000 cells in 370 μL) were added to each well of ultra low 96-well plates and the plates were inverted to allow the cells to sediment for 24 hr. After flipping the plates, newly formed MCS were allowed to sediment and were incubated for 5 days before treatment with compounds. At this stage MCS had a diameter of about 600 μm.
Immunohistochemistry
For experiments in which MCS were stained for LC3 expression by immunoistochemistry, MCS were treated with BafA1 (50 nM), CQ (50 μM), Lys-01 (50 μM) and SAL (2 μM) for 24 hours before processing samples. MCS from each treatment group (n = 24) were fixed in 2% buffered formalin, dehydrated, embedded in paraffin and 10 μm sections were obtained. The sections were deparaffinized with xylene, rehydrated and microwaved. Sections were stained with rabbit anti-human LC3B (Cell Signaling Technology, 2775) and visualized by avidin-biotin-peroxidase complex technique (Vector Laboratories, Burlingame, CA, USA). Counterstaining was done using Mayer's haematoxylin.
The analysis of LC3 expression was performed by two independent operators using the software Aperio ImageScope and the algorithm positive pixel count v9, as described previously [36]. Since the basal level of LC3 expression in untreated MCS is weak, treatment with any of the autophagy inhibitors tested will block turnover of LC3-II and induce increased LC3 expression. LC3 expression was calculated as number of positive pixels (including positive and strong positive pixels). The peripheral area (roughly 10 cellular layers) and the central area of each MCS, respectively characterising proliferating/normoxic cells and quiescent/hypoxic cells [74] were analysed and LC3 expression was normalised to total area and expressed as percentage.
Mammosphere formation
After treatment with SAL for 48 hours, HMLER CD24low cells were collected and resuspended in DMEM/F12 medium supplemented with B27 (1:50, Invitrogen, 17504044), 20 ng/ml EGF, 0.4% BSA, 0.5 μg/ml hydrocortisone, 4 μg/ml insulin, as previously described [43]. For each treatment group, cells were plated at 1 cell/well/150 μl in one ultralow attachment 96-wells plate (Costar, 7007), one full plate per condition. After 5-7 days, the number and size of mammospheres formed in each plate were counted and analysed. In some experiments, HMLER CD24low cells were directly cultured as single cell suspension in the presence of SAL for 7 days and the number of mammospheres formed was evaluated.
Clinical samples
Fresh biopsies and scrapings from patients with primary breast cancer (Table 3) were obtained from the Karolinska Hospital and immediately processed. Research on the tumor samples was approved by the local biobank at Karolinska University Hospital and the Regional Ethics Board in Stockholm, Sweden. Fresh biopsies and/or tumor cells were directly generated from surgically resected breast cancers at Karolinska University Hospital as previously described [75]. Tumor cells were immediately transferred into the selective medium DMEM/F12-GlutaMAX supplied with 10 ng/ml b-FGF, 20 ng/ml EGF, 5 μg/ml insulin and 0.4% BSA for the initiation of mammospheres. In general, scrapings were washed twice in PBS, the final cell pellets were re-suspended in the selective medium and stable mammospheres can be observed after 3 days (Figure S3C). Sensitivity to SAL was tested on single cell suspensions obtained from dissociated mammospheres and cultured in DMEM-F12 buffered at pH 7.4 or 6.8 at 1000 viable cells per well in 48-well plates (Corning Costar, MA) to form mammospheres. Plates were incubated for 10 days and the number of newly formed mammospheres was documented.
Studies on autophagic flux
Mild saponin extraction of cytosolic GFP-LC3-I allows quantification of autophagosome-associated GFP-LC3-II by flow cytometry in HOS-GFP-LC3 cells [48]. HOS-GFP-LC3 cells (100,000 cells) were plated into 20 cm2 dishes. The next day medium was replaced with medium at pH 7.4 or 6.8 for 24 hours. SAL was added for 6 hours and BafA1 (50 nM) was added during the last 2 hours incubation. Cells were collected by trypsinization and treated with 0.05% saponin (Biochemika, 47036) in PBS for 10 minutes at RT. After wash in PBS the cells were collected and immediately analysed by a FACSCalibur instrument using Cellquest software (Becton Dickinson). GFP fluorescence was collected from 10,000 cells/sample.
Analysis of the autophagic flux by Western blot was done on different cell lines. Experiments with parental cell lines were performed by plating cells in standard RPMI buffered at pH 7.4. The next day the medium was replaced with media buffered at the desired pH (7.4, 6.8 and 6.5). Low pH-adapted cell lines were plated in medium buffered at pH 6.8 and let adhere overnight. SAL was added at specified concentrations for 6 hours and 100 nM BafA1 was added during the last 2 hours incubation. Cells were then collected for further analysis by flow cytometry or Western blotting.
Analysis of autophagy in HMLER CD24+ and CD24low cells was performed using an mRFP-GFP-LC3 reporter. Adherent cells were transfected with the mRFP-GFP-LC3 plasmid by using lipofectamine 3000. The next day, medium was replaced with fresh medium buffered at the desired pH. After overnight incubation, SAL was added for 6 hours and EBSS-treated cells were used as controls. Cells were fixed using 2% paraformaldehyde and observed by laser scanning fluorescence microscopy. Autophagic flux was determined by quantification of the number of autophagosomes (yellow dots) and autolysosomes (red dots) per cell, counting at least 15 cells per condition.
Western blotting
Cells were washed with PBS on ice and collected by scraping in cold PBS. The cell pellet was lysed in RIPA buffer (150 mM NaCl, 50 mM Tris pH 7.4, 1% Nonidet P-40, 0.1% SDS and 0.5% sodium deoxycholate) in presence of protease and phosphatase inhibitors. The protein concentration was determined by Biorad Protein Assay (Biorad Laboratories) and equal amount of proteins (20 μg) was loaded on pre-casted acrylamide gels (4-12% SDS-PAGE, NuPage). The proteins were transferred from the gel to PVDF membrane for 2 hours at 4°C. Red Ponceau staining of the membranes verified the proper loading and transfer. Membranes were blocked in 5% blotting grade dry milk in TBS with 0.1% Tween (TBS-T) for 1 hour at room temperature and then incubated with primary antibodies diluted in 5% BSA in TBS-T overnight at 4°C. The next day membranes were washed and incubated for 1 hour at room temperature with the appropriate HRP-conjugated secondary antibody and the binding revealed by the ECL system.
Cell viability assay
Cells were plated into 96-wells plates and the next day treated with different concentrations of the specific compounds (SAL, CQ and Lys-01). Forty-eight hours after treatment cell viability was evaluated by using the acid phosphatase assay [76].
Clonogenic assay
HCT116 cell lines were plated as 300 cells/well in 6-well plates and treated with SAL for 8 or 48 hours, following culture for 6 days in drug-free media. Colonies were stained with Giemsa.
Clonogenic cell survival was also assessed on HCT116 MCS treated for three days with different compounds. After treatment, the MCS were collected, washed in PBS and single cell suspension was obtained after incubation with Accutase (Biolegend, 423201). Equal volumes of cells for each treatment condition were plated in triplicate in 6-well plates for 6 days, followed by staining the colonies with Giemsa. The software ImageJ was used to perform colony counting.
UPLC-MS/MS analysis of salinomycin intracellular content
The intracellular content of SAL was examined using a method commonly used for assessing drug uptake/accumulation [77]. In brief, HCT116, HMLER CD24+ and CD24low cells were plated in 24-wells plates overnight and the next day medium was replaced with fresh medium at pH 7.4 and pH 6.8. Immediately after, SAL (2-4 μM) was added to the cells for 3 hours. After the 3h incubation cells were washed once with PBS at room temperature and then acetonitrile was added and let evaporate. The intracellular accumulation of SAL was quantified by ultra-high performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS). The compounds accumulated were extracted with 200 μl acetonitrile/water (60/40) spiked with 50 nM warfarin as internal standard. The samples were centrifuged at 2465g and 4°C for 20 minutes. Accumulation of SAL was determined using a Waters Xevo triple quadrupole mass spectrometer with electrospray ionization coupled to an Acquity UPLC system (Waters, Milford, MA, USA). Compounds were separated with a 2 min elution at a flow rate of 0.5 ml/min in a Waters BEH C18 column, 2.1 × 50 mm (1.7 μm) at 60°C. Mobile phase consisted of 5% acetonitrile and 0.1% formic acid in water (solvent A), and 0.1% formic acid in acetonitrile (solvent B). The chromatographic run started at 20% solvent B and increased linearly to 99% from 0.5 to 1.2 min, followed by a hold until 1.6 min, and a return to initial conditions at 1.7 min. SAL was detected by monitoring the mass transition 749>241 at a cone voltage of 48V and a collision energy of 33V. SAL eluted at 1.75 min. For all cell lines the total protein content was measured in representative wells using the BCA Protein Assay Reagent Kit (Pierce Biotechnology, Rockford, IL, USA) according to manufacturer's instructions. The obtained protein contents were used for normalization between experiments and cell lines.
Statistical analysis
All data were obtained from at least three experimental replicates and presented as mean ± SEM, if not otherwise indicated. Data analysis was performed using Graphpad Prism 6.0 (GraphPad Software, CA). Differences between groups were analysed with parametric or non-parametric tests according to the distribution of the values. The specific test used is indicated in the legend to figures. The significance level was set as P < 0.05.
SUPPLEMENTARY MATERIAL FIGURES
Acknowledgments
We are grateful to Patrice Codogno and Maryam Mehrpur for helpful discussions and Ann-Charlotte Björklund and Sara Corvigno for technical help and sharing of reagents.
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
GRANT SUPPORT
This work was supported by grants from the Swedish Cancer Society (grant # CAN 2012/415) and the Association for International Cancer Research (grant #11-0522). Francesca Vittoria Sbrana was supported by a scholarship from the Fondazione C.M. Lerici (Italian Culture Institute, Stockholm). Pedram Kharaziha is supported by BRECT. Matheus Dyczynski is supported by a KI doctoral grant.
REFERENCES
- 1.Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nature reviews Cancer. 2012;12:487–93. doi: 10.1038/nrc3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012;491:364–73. doi: 10.1038/nature11706. [DOI] [PubMed] [Google Scholar]
- 3.Spugnini EP, Sonveaux P, Stock C, Perez-Sayans M, De Milito A, Avnet S, Garcia AG, Harguindey S, Fais S. Proton channels and exchangers in cancer. Biochimica et biophysica acta. 2014 doi: 10.1016/j.bbamem.2014.10.015. [DOI] [PubMed] [Google Scholar]
- 4.Dvorak HF. Rous-Whipple Award Lecture. How tumors make bad blood vessels and stroma. Am J Pathol. 2003;162:1747–57. doi: 10.1016/s0002-9440(10)64309-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McDonald DM, Baluk P. Significance of blood vessel leakiness in cancer. Cancer Res. 2002;62:5381–5. [PubMed] [Google Scholar]
- 6.Lamonte G, Tang X, Chen JL, Wu J, Ding CK, Keenan MM, Sangokoya C, Kung HN, Ilkayeva O, Boros LG, Newgard CB, Chi JT. Acidosis induces reprogramming of cellular metabolism to mitigate oxidative stress. Cancer Metab. 2013;1:23. doi: 10.1186/2049-3002-1-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xie J, Wu H, Dai C, Pan Q, Ding Z, Hu D, Ji B, Luo Y, Hu X. Beyond Warburg effect--dual metabolic nature of cancer cells. Sci Rep. 2014;4:4927. doi: 10.1038/srep04927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hashim AI, Zhang X, Wojtkowiak JW, Martinez GV, Gillies RJ. Imaging pH and metastasis. NMR in biomedicine. 2011;24:582–91. doi: 10.1002/nbm.1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindner D, Raghavan D. Intra-tumoural extra-cellular pH: a useful parameter of response to chemotherapy in syngeneic tumour lines. British journal of cancer. 2009;100:1287–91. doi: 10.1038/sj.bjc.6605022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schornack PA, Gillies RJ. Contributions of cell metabolism and H+ diffusion to the acidic pH of tumors. Neoplasia. 2003;5:135–45. doi: 10.1016/s1476-5586(03)80005-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gallagher FA, Kettunen MI, Day SE, Hu DE, Ardenkjaer-Larsen JH, Zandt R, Jensen PR, Karlsson M, Golman K, Lerche MH, Brindle KM. Magnetic resonance imaging of pH in vivo using hyperpolarized 13C-labelled bicarbonate. Nature. 2008;453:940–3. doi: 10.1038/nature07017. [DOI] [PubMed] [Google Scholar]
- 12.De Milito A, Canese R, Marino ML, Borghi M, Iero M, Villa A, Venturi G, Lozupone F, Iessi E, Logozzi M, Mina PD, Santinami M, Rodolfo M, et al. pH-dependent antitumor activity of proton pump inhibitors against human melanoma is mediated by inhibition of tumor acidity. Int J Cancer. 2009;127:207–19. doi: 10.1002/ijc.25009. [DOI] [PubMed] [Google Scholar]
- 13.Robey IF, Baggett BK, Kirkpatrick ND, Roe DJ, Dosescu J, Sloane BF, Hashim AI, Morse DL, Raghunand N, Gatenby RA, Gillies RJ. Bicarbonate Increases Tumor pH and Inhibits Spontaneous Metastases. Cancer Res. 2009 doi: 10.1158/0008-5472.CAN-07-5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerweck LE. Tumor pH: implications for treatment and novel drug design. Semin Radiat Oncol. 1998;8:176–82. doi: 10.1016/s1053-4296(98)80043-x. [DOI] [PubMed] [Google Scholar]
- 15.Gillies RJ, Robey I, Gatenby RA. Causes and consequences of increased glucose metabolism of cancers. J Nucl Med. 2008;49(Suppl 2):24S–42S. doi: 10.2967/jnumed.107.047258. [DOI] [PubMed] [Google Scholar]
- 16.Parks SK, Chiche J, Pouyssegur J. Disrupting proton dynamics and energy metabolism for cancer therapy. Nature reviews Cancer. 2013;13:611–23. doi: 10.1038/nrc3579. [DOI] [PubMed] [Google Scholar]
- 17.Raghunand N, He X, van Sluis R, Mahoney B, Baggett B, Taylor CW, Paine-Murrieta G, Roe D, Bhujwalla ZM, Gillies RJ. Enhancement of chemotherapy by manipulation of tumour pH. Br J Cancer. 1999;80:1005–11. doi: 10.1038/sj.bjc.6690455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tannock IF, Rotin D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989;49:4373–84. [PubMed] [Google Scholar]
- 19.De Milito A, Marino ML, Fais S. A rationale for the use of proton pump inhibitors as antineoplastic agents. Curr Pharm Des. 2012;18:1395–406. doi: 10.2174/138161212799504911. [DOI] [PubMed] [Google Scholar]
- 20.Tredan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99:1441–54. doi: 10.1093/jnci/djm135. [DOI] [PubMed] [Google Scholar]
- 21.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther. 2011;10:1533–41. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheong H, Lu C, Lindsten T, Thompson CB. Therapeutic targets in cancer cell metabolism and autophagy. Nat Biotechnol. 2012;30:671–8. doi: 10.1038/nbt.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J, Gewirtz DA, Karantza V, Kimmelman A, Kumar S, Levine B, et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34:856–80. doi: 10.15252/embj.201490784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lorin S, Hamai A, Mehrpour M, Codogno P. Autophagy regulation and its role in cancer. Seminars in cancer biology. 2013;23:361–79. doi: 10.1016/j.semcancer.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 25.Rebecca VW, Amaravadi RK. Emerging strategies to effectively target autophagy in cancer. Oncogene. 2015 doi: 10.1038/onc.2015.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, Han W, Lou F, Yang J, Zhang Q, Wang X, He C, Pan H. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4:e838. doi: 10.1038/cddis.2013.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S, Mikkelson T, Wang D, Chang YC, Hu J, McAfee Q, Fisher J, Troxel AB, et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy. 2014;10:1359–68. doi: 10.4161/auto.28984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rangwala R, Leone R, Chang YC, Fecher LA, Schuchter LM, Kramer A, Tan KS, Heitjan DF, Rodgers G, Gallagher M, Piao S, Troxel AB, Evans TL, et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy. 2014;10:1369–79. doi: 10.4161/auto.29118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rangwala R, Chang YC, Hu J, Algazy KM, Evans TL, Fecher LA, Schuchter LM, Torigian DA, Panosian JT, Troxel AB, Tan KS, Heitjan DF, DeMichele AM, et al. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy. 2014;10:1391–402. doi: 10.4161/auto.29119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barnard RA, Wittenburg LA, Amaravadi RK, Gustafson DL, Thorburn A, Thamm DH. Phase I clinical trial and pharmacodynamic evaluation of combination hydroxychloroquine and doxorubicin treatment in pet dogs treated for spontaneously occurring lymphoma. Autophagy. 2014;10:1415–25. doi: 10.4161/auto.29165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mahalingam D, Mita M, Sarantopoulos J, Wood L, Amaravadi RK, Davis LE, Mita AC, Curiel TJ, Espitia CM, Nawrocki ST, Giles FJ, Carew JS. Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy. 2014;10:1403–14. doi: 10.4161/auto.29231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vogl DT, Stadtmauer EA, Tan KS, Heitjan DF, Davis LE, Pontiggia L, Rangwala R, Piao S, Chang YC, Scott EC, Paul TM, Nichols CW, Porter DL, et al. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy. 2014;10:1380–90. doi: 10.4161/auto.29264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poklepovic A, Gewirtz DA. Outcome of early clinical trials of the combination of hydroxychloroquine with chemotherapy in cancer. Autophagy. 2014;10:1478–80. doi: 10.4161/auto.29428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marino ML, Pellegrini P, Di Lernia G, Djavaheri-Mergny M, Brnjic S, Zhang X, Hagg M, Linder S, Fais S, Codogno P, De Milito A. Autophagy is a protective mechanism for human melanoma cells under acidic stress. J Biol Chem. 2012;287:30664–76. doi: 10.1074/jbc.M112.339127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wojtkowiak JW, Rothberg JM, Kumar V, Schramm KJ, Haller E, Proemsey JB, Lloyd MC, Sloane BF, Gillies RJ. Chronic autophagy is a cellular adaptation to tumor acidic pH microenvironments. Cancer Res. 2012;72:3938–47. doi: 10.1158/0008-5472.CAN-11-3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pellegrini P, Strambi A, Zipoli C, Hagg-Olofsson M, Buoncervello M, Linder S, De Milito A. Acidic extracellular pH neutralizes the autophagy-inhibiting activity of chloroquine: implications for cancer therapies. Autophagy. 2014;10:562–71. doi: 10.4161/auto.27901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McAfee Q, Zhang Z, Samanta A, Levi SM, Ma XH, Piao S, Lynch JP, Uehara T, Sepulveda AR, Davis LE, Winkler JD, Amaravadi RK. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S A. 2012;109:8253–8. doi: 10.1073/pnas.1118193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vinogradov S, Wei X. Cancer stem cells and drug resistance: the potential of nanomedicine. Nanomedicine (Lond) 2012;7:597–615. doi: 10.2217/nnm.12.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klevebring D, Rosin G, Ma R, Lindberg J, Czene K, Kere J, Fredriksson I, Bergh J, Hartman J. Sequencing of breast cancer stem cell populations indicates a dynamic conversion between differentiation states in vivo. Breast cancer research. 2014;16:R72. doi: 10.1186/bcr3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–59. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gong C, Bauvy C, Tonelli G, Yue W, Delomenie C, Nicolas V, Zhu Y, Domergue V, Marin-Esteban V, Tharinger H, Delbos L, Gary-Gouy H, Morel AP, et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene. 2013;32:2261–72. doi: 10.1038/onc.2012.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolf J, Dewi DL, Fredebohm J, Muller-Decker K, Flechtenmacher C, Hoheisel JD, Boettcher M. A mammosphere formation RNAi screen reveals that ATG4A promotes a breast cancer stem-like phenotype. Breast cancer research. 2013;15:R109. doi: 10.1186/bcr3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yue W, Hamai A, Tonelli G, Bauvy C, Nicolas V, Tharinger H, Codogno P, Mehrpour M. Inhibition of the autophagic flux by salinomycin in breast cancer stem-like/progenitor cells interferes with their maintenance. Autophagy. 2013;9:714–29. doi: 10.4161/auto.23997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu D, Choi MY, Yu J, Castro JE, Kipps TJ, Carson DA. Salinomycin inhibits Wnt signaling and selectively induces apoptosis in chronic lymphocytic leukemia cells. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:13253–7. doi: 10.1073/pnas.1110431108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jangamreddy JR, Ghavami S, Grabarek J, Kratz G, Wiechec E, Fredriksson BA, Rao Pariti RK, Cieslar-Pobuda A, Panigrahi S, Los MJ. Salinomycin induces activation of autophagy, mitophagy and affects mitochondrial polarity: differences between primary and cancer cells. Biochimica et biophysica acta. 2013;1833:2057–69. doi: 10.1016/j.bbamcr.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 46.Li T, Su L, Zhong N, Hao X, Zhong D, Singhal S, Liu X. Salinomycin induces cell death with autophagy through activation of endoplasmic reticulum stress in human cancer cells. Autophagy. 2013;9:1057–68. doi: 10.4161/auto.24632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klose J, Stankov MV, Kleine M, Ramackers W, Panayotova-Dimitrova D, Jager MD, Klempnauer J, Winkler M, Bektas H, Behrens GM, Vondran FW. Inhibition of autophagic flux by salinomycin results in anti-cancer effect in hepatocellular carcinoma cells. PloS one. 2014;9:e95970. doi: 10.1371/journal.pone.0095970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eng KE, Panas MD, Hedestam GB, McInerney GM. A novel quantitative flow cytometry-based assay for autophagy. Autophagy. 2010:6. doi: 10.4161/auto.6.5.12112. [DOI] [PubMed] [Google Scholar]
- 49.Gillies RJ, Raghunand N, Karczmar GS, Bhujwalla ZM. MRI of the tumor microenvironment. J Magn Reson Imaging. 2002;16:430–50. doi: 10.1002/jmri.10181. [DOI] [PubMed] [Google Scholar]
- 50.Swietach P, Patiar S, Supuran CT, Harris AL, Vaughan-Jones RD. The role of carbonic anhydrase 9 in regulating extracellular and intracellular ph in three-dimensional tumor cell growths. J Biol Chem. 2009;284:20299–310. doi: 10.1074/jbc.M109.006478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PloS one. 2008;3:e2888. doi: 10.1371/journal.pone.0002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Prud'homme GJ. Cancer stem cells and novel targets for antitumor strategies. Current pharmaceutical design. 2012;18:2838–49. doi: 10.2174/138161212800626120. [DOI] [PubMed] [Google Scholar]
- 53.Jensen PB, Sorensen BS, Sehested M, Grue P, Demant EJ, Hansen HH. Targeting the cytotoxicity of topoisomerase II-directed epipodophyllotoxins to tumor cells in acidic environments. Cancer Res. 1994;54:2959–63. [PubMed] [Google Scholar]
- 54.Strambi A, De Milito A. Contribution of pH Alterations to the Tumor Microenvironment. In: Mazurek S, Shoshan M, editors. Tumor cell metabolism. Springer; 2015. pp. 173–96. [Google Scholar]
- 55.Maes H, Kuchnio A, Peric A, Moens S, Nys K, De Bock K, Quaegebeur A, Schoors S, Georgiadou M, Wouters J, Vinckier S, Vankelecom H, Garmyn M, et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer cell. 2014;26:190–206. doi: 10.1016/j.ccr.2014.06.025. [DOI] [PubMed] [Google Scholar]
- 56.Maycotte P, Aryal S, Cummings CT, Thorburn J, Morgan MJ, Thorburn A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy. 2012;8:200–12. doi: 10.4161/auto.8.2.18554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang C, Tian Y, Song F, Fu C, Han B, Wang Y. Salinomycin inhibits the growth of colorectal carcinoma by targeting tumor stem cells. Oncology reports. 2015;34:2469–76. doi: 10.3892/or.2015.4253. [DOI] [PubMed] [Google Scholar]
- 58.Xiao Z, Sperl B, Ullrich A, Knyazev P. Metformin and salinomycin as the best combination for the eradication of NSCLC monolayer cells and their alveospheres (cancer stem cells) irrespective of EGFR, KRAS, EML4/ALK and LKB1 status. Oncotarget. 2014;5:12877–90. doi: 10.18632/oncotarget.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mao J, Fan S, Ma W, Fan P, Wang B, Zhang J, Wang H, Tang B, Zhang Q, Yu X, Wang L, Song B, Li L. Roles of Wnt/beta-catenin signaling in the gastric cancer stem cells proliferation and salinomycin treatment. Cell death & disease. 2014;5:e1039. doi: 10.1038/cddis.2013.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lagas JS, Sparidans RW, van Waterschoot RA, Wagenaar E, Beijnen JH, Schinkel AH. P-glycoprotein limits oral availability, brain penetration, and toxicity of an anionic drug, the antibiotic salinomycin. Antimicrobial agents and chemotherapy. 2008;52:1034–9. doi: 10.1128/AAC.01041-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Manago A, Leanza L, Carraretto L, Sassi N, Grancara S, Quintana-Cabrera R, Trimarco V, Toninello A, Scorrano L, Trentin L, Semenzato G, Gulbins E, Zoratti M, et al. Early effects of the antineoplastic agent salinomycin on mitochondrial function. Cell death & disease. 2015;6:e1930. doi: 10.1038/cddis.2015.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Senkowski W, Zhang X, Olofsson MH, Isacson R, Hoglund U, Gustafsson M, Nygren P, Linder S, Larsson R, Fryknas M. Three-Dimensional Cell Culture-Based Screening Identifies the Anthelmintic Drug Nitazoxanide as a Candidate for Treatment of Colorectal Cancer. Molecular cancer therapeutics. 2015;14:1504–16. doi: 10.1158/1535-7163.MCT-14-0792. [DOI] [PubMed] [Google Scholar]
- 63.Khacho M, Tarabay M, Patten D, Khacho P, MacLaurin JG, Guadagno J, Bergeron R, Cregan SP, Harper ME, Park DS, Slack RS. Acidosis overrides oxygen deprivation to maintain mitochondrial function and cell survival. Nature communications. 2014;5:3550. doi: 10.1038/ncomms4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grinde B. Effect of carboxylic ionophores on lysosomal protein degradation in rat hepatocytes. Experimental cell research. 1983;149:27–35. doi: 10.1016/0014-4827(83)90377-4. [DOI] [PubMed] [Google Scholar]
- 65.Cecconi F, Jaattela M. Targeting ions-induced autophagy in cancer. Cancer cell. 2014;26:599–600. doi: 10.1016/j.ccell.2014.10.014. [DOI] [PubMed] [Google Scholar]
- 66.Boehmerle W, Muenzfeld H, Springer A, Huehnchen P, Endres M. Specific targeting of neurotoxic side effects and pharmacological profile of the novel cancer stem cell drug salinomycin in mice. Journal of molecular medicine. 2014;92:889–900. doi: 10.1007/s00109-014-1155-0. [DOI] [PubMed] [Google Scholar]
- 67.Ojo OO, Bhadauria S, Rath SK. Dose-dependent adverse effects of salinomycin on male reproductive organs and fertility in mice. PloS one. 2013;8:e69086. doi: 10.1371/journal.pone.0069086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mao X, Liu J, Gong Z, Zhang H, Lu Y, Zou H, Yu Y, Chen Y, Sun Z, Li W, Li B, Gao J, Zhong Y. iRGD-conjugated DSPE-PEG2000 nanomicelles for targeted delivery of salinomycin for treatment of both liver cancer cells and cancer stem cells. Nanomedicine (Lond) 2015;10:2677–95. doi: 10.2217/nnm.15.106. [DOI] [PubMed] [Google Scholar]
- 69.Naujokat C, Steinhart R. Salinomycin as a drug for targeting human cancer stem cells. Journal of biomedicine & biotechnology. 2012;2012:950658. doi: 10.1155/2012/950658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cufi S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Vellon L, Menendez JA. Autophagy positively regulates the CD44(+) CD24(−/low) breast cancer stem-like phenotype. Cell cycle. 2011;10:3871–85. doi: 10.4161/cc.10.22.17976. [DOI] [PubMed] [Google Scholar]
- 71.Espina V, Mariani BD, Gallagher RI, Tran K, Banks S, Wiedemann J, Huryk H, Mueller C, Adamo L, Deng J, Petricoin EF, Pastore L, Zaman S, et al. Malignant precursor cells pre-exist in human breast DCIS and require autophagy for survival. PloS one. 2010;5:e10240. doi: 10.1371/journal.pone.0010240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Salerno M, Avnet S, Bonuccelli G, Hosogi S, Granchi D, Baldini N. Impairment of lysosomal activity as a therapeutic modality targeting cancer stem cells of embryonal rhabdomyosarcoma cell line RD. PloS one. 2014;9:e110340. doi: 10.1371/journal.pone.0110340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sukhai MA, Prabha S, Hurren R, Rutledge AC, Lee AY, Sriskanthadevan S, Sun H, Wang X, Skrtic M, Seneviratne A, Cusimano M, Jhas B, Gronda M, et al. Lysosomal disruption preferentially targets acute myeloid leukemia cells and progenitors. The Journal of clinical investigation. 2013;123:315–28. doi: 10.1172/JCI64180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang X, Fryknas M, Hernlund E, Fayad W, De Milito A, Olofsson MH, Gogvadze V, Dang L, Pahlman S, Schughart LA, Rickardson L, D'Arcy P, Gullbo J, et al. Induction of mitochondrial dysfunction as a strategy for targeting tumour cells in metabolically compromised microenvironments. Nature communications. 2014;5:3295. doi: 10.1038/ncomms4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ma R, Fredriksson I, Karthik GM, Winn G, Darai-Ramqvist E, Bergh J, Hartman J. Superficial scrapings from breast tumors is a source for biobanking and research purposes. Laboratory investigation. 2014;94:796–805. doi: 10.1038/labinvest.2014.65. [DOI] [PubMed] [Google Scholar]
- 76.Friedrich J, Eder W, Castaneda J, Doss M, Huber E, Ebner R, Kunz-Schughart LA. A reliable tool to determine cell viability in complex 3-d culture: the acid phosphatase assay. J Biomol Screen. 2007;12:925–37. doi: 10.1177/1087057107306839. [DOI] [PubMed] [Google Scholar]
- 77.Vildhede A, Karlgren M, Svedberg EK, Wisniewski JR, Lai Y, Noren A, Artursson P. Hepatic uptake of atorvastatin: influence of variability in transporter expression on uptake clearance and drug-drug interactions. Drug metabolism and disposition. 2014;42:1210–8. doi: 10.1124/dmd.113.056309. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.