

Abstract
Harvest index (HI), the ratio of seed mass to total biomass of the aboveground plant parts, is an important trait for harvestable yield of crops. Unfortunately, HI of Brassica napus is lower than that of other economically important crops. To identify candidate genes associated with high HI, a genome-wide association study of HI and four HI-related traits was conducted with 520 B. napus accessions cultivated in both Yunnan and Chongqing. We detected 294 single nucleotide polymorphisms significantly associated with the abovementioned traits, including 79 SNPs that affected two or more traits. Differentially expressed genes between extremely high- and low-HI accessions were identified in 8 tissues at two cultivated regions. Combination of linkage disequilibrium and transcriptome analyses revealed 33 functional candidate genes located within the confidence intervals of significant SNPs associated with more than one trait, such as SHOOT GRAVITROPISM 5 (Bna.SGR5), ATP-CITRATE LYASE A-3 (Bna.ACLA-3) and CAROTENOID CLEAVAGE DIOXYGENASE 1 (Bna.CCD1), their orthologs in the Arabidopsis thaliana have been shown to play key roles in photosynthesis, inflorescence, and silique development. Our results provide insight into the molecular mechanisms underlying establishment of high-HI B. napus and lay a foundation for characterization of candidate genes aimed at developing high-HI B. napus varieties.
Harvest index (HI), also named the coefficient of economic yield, is the ratio of economic yield (seeds) to the total shoot dry matter, and represents a plant’s ability to convert photoassimilates into economic products. HI is closely associated with the transport and distribution of photosynthates, organ development, and plant architecture. Therefore, yield could be enhanced by increasing the HI of crops, or vise versa1.
Rapeseed (Brassica napus, AACC, 2n = 38) is one of the most widely cultivated oilseed crops, worldwide. The global average yield of rapeseed production improved significantly, from 0.52 t/hm2 in 1961 to over 1.64 t/hm2 in 20072, but the HI of rapeseed still remains much lower than that of other economically important crops, such as rice (Oryza sativa), wheat (Triticum aestivum), maize (Zea may), and soybean (Glycine max)3. HI is highly heritable, though it is still influenced by various environmental factors, crop photosynthetic characteristics, vascular properties, panicle traits, and nitrogen nutrition levels1. Therefore, it has been recognized as an important breeding target for yield improvement in B. napus.
Genome-wide association studies (GWASs) are a powerful approach for gaining insight into the genetic architecture of complex traits in many crops, and have been used to identify loci and candidate genes. In B. napus, GWASs have revealed genetic loci underlying traits of interest at relatively high resolution. Li et al.4 performed a GWAS of thousand seed weight (TSW) and related quality traits, and identified two single nucleotide polymorphisms (SNPs) significantly associated with TSW4. Eight quantitative trait loci (QTLs) associated with plant height and five associated with branch number were identified in a GWAS, based on the abovementioned genotype data5. Several candidate genes associated with seed germination and vigor were also identified in a GWAS analysis6. Recently, a GWAS of HI and HI-related traits of B. napus was carried out using a natural population of 155 B. napus cultivars7. The authors identified 9 significant SNPs associated with HI in the B. napus C subgenome, which explained 3.42% of the phenotypic variation7. Taken together, these findings illustrate the promise of GWAS for detecting QTLs that control important complex agronomic traits.
Identification of significant QTLs controlling HI and HI-related traits of B. napus, using GWAS, is a crucial step for further improvement of B. napus yield. To increase accuracy in QTL identification, and identify efficiently QTLs responsible for environmental variation, we cultivated 520 B. napus accessions at two extremely different environments in China, under the same cultivation regime. Linchang, Yunnan Province (YN) is a typical high-yield production region of B. napus. In this region, the average yield is over 5.3 tons per hectare8, whereas in Chongqing (CQ), the standard B. napus production region in the Yangtze River Basin, the yield is only about 2.7 tons per hectare9,10. Phenotypic data was collected and used to conduct GWAS for identification of significant SNPs controlling HI-related traits. Transcriptome analyses of 8 tissues from extremely high- and low-HI B. napus accessions were conducted to identify candidate genes within the confidence intervals of significant SNPs. These results not only provide insight into the genetic mechanisms underlying high-HI formation in B. napus, but lay a solid foundation for further functional validation of candidate genes and marker-based breeding of B. napus varieties for high HI.
Results
Variation in HI and four HI-related traits
HI and four HI-related traits (biomass yield (BY), canopy biomass yield (CBY), stem dry weight (ST), and seed yield (SY)) in the lines grown in three natural environments (CQ in 2013 (13CQ), CQ in 2014 (14CQ) and YN in 2014 (14YN)) and two virtual environments, (trait difference (14YN-CQ) and trait variation (14VTL) between YN and CQ in 2014) were analyzed using descriptive statistics and Analysis of Variance (ANOVA) (Table 1). Among the five investigated traits, the coefficient of variation (CV) of SY was higher than that of the other traits in most environments, reflecting large variation of SY within the entire GWAS panels. The average CV of HI was higher in the lines grown at YN than CQ. The CVs of different traits in 14VTL ranged from 18.98% to 48.31%, and those of SY and HI were highest, indicating that this trait was sensentive to environmental variation.
Table 1. Mean values and heritability of HI and four HI-related traits.
| Traita | Env.b | Number of accessions Tested | Mean ± SD | Range | CV (%) | h2 (%) |
|---|---|---|---|---|---|---|
| CBY | 13CQ | 476 | 66.17 ± 15.1 | 22.38~129.63 | 22.82 | 74.71 |
| 14CQ | 516 | 52.85 ± 14.04 | 16.32~94.27 | 26.57 | ||
| 14YN | 302 | 107.67 ± 21.08 | 49.00~177.5 | 19.57 | ||
| 14YN-CQ | 300 | 52.47 ± 23.44 | −31.37~112.3 | 29.21 | ||
| 14VTL | 300 | 0.64 ± 0.27 | −0.43~1.41 | |||
| ST | 13CQ | 476 | 26.79 ± 6.8 | 9.50~54.88 | 25.53 | 82.13 |
| 14CQ | 516 | 37.41 ± 9.56 | 13.99~69.63 | 25.55 | ||
| 14YN | 302 | 58.31 ± 15.51 | 27.67~128.67 | 26.61 | ||
| 14YN-CQ | 300 | 19.39 ± 15.18 | −20.62~74.37 | 31.72 | ||
| 14VTL | 300 | 0.38 ± 0.27 | −0.43~1.12 | |||
| BY | 13CQ | 476 | 92.97 ± 20.5 | 36.31~184.50 | 22.05 | 72.71 |
| 14CQ | 515 | 90.29 ± 21.65 | 37.33~159.00 | 23.98 | ||
| 14YN | 302 | 165.56 ± 31.51 | 76.67~265.5 | 19.03 | ||
| 14YN-CQ | 300 | 71.39 ± 34.49 | −37.82~167.95 | 26.96 | ||
| 14VTL | 300 | 0.54 ± 0.25 | −0.33~1.21 | |||
| SY | 13CQ | 496 | 22.92 ± 7.8 | 1.46~53.39 | 33.98 | 77.04 |
| 14CQ | 516 | 18.10 ± 6.18 | 2.82~40.85 | 34.14 | ||
| 14YN | 302 | 32.15 ± 10.74 | 6.34~61.07 | 33.41 | ||
| 14YN-CQ | 300 | 12.77 ± 12.14 | −34.51~49.56 | 48.31 | ||
| 14VTL | 300 | 0.46 ± 0.46 | −1.46~1.60 | |||
| HI | 13CQ | 476 | 0.23 ± 0.05 | 4.8%~42% | 22.00 | 75.20 |
| 14CQ | 514 | 0.20 ± 0.042 | 4.5%~33% | 21.00 | ||
| 14YN | 302 | 0.19 ± 0.054 | 5.2%~31% | 28.20 | ||
| 14YN-CQ | 300 | −0.01 ± 0.07 | −0.21~0.16 | |||
| 14VTL | 300 | −0.08 ± 0.37 | −1.33~0.90 |
aCBY, canopy biomass yield; ST, stem dry weight; BY, biomass yield; SY, seed yield per plant.
bEnvironments: 13CQ, CQ in 2013; 14CQ, CQ in 2014; 14YN, YN in 2014; 14YN−CQ: 14YN−14CQ 14VTL, 2 × (14YN−14CQ)/(14YN + 14CQ).
SD, standard deviation; CV(%), coefficient of variation. h2, broad-sense heritability.
The five evaluated traits exhibited continuous variation in the accessions, both in CQ and YN (Supplementary Fig. S1), indicating that they were quantitative traits controlled by multiple genes. The estimate of broad-sense heritabilities ranged from 72.71% to 82.13%, suggesting that the majority of phenotypic variation for these traits, in B. napus, is attributed to genetic effects. In general, plants grown in the high-yield region had a larger ST and lower HI (Table 1), implying that photosynthetic products might be restrained in the stems of the accessions that have a lower transport efficiency. Hence, it should be possible to increase the B. napus yield and HI by increasing phloem transport of photosynthetic products from the stems to siliques.
Population structure and model selection
Based on a previous study, the 520 B. napus accessions were divided into two subgroups (P1 and P2)11 (Supplementary Table S1). We estimated the relative kinships12, and found 68.87% of the kinship coefficients ranged from 0 to 0.05, with 55.32% of pairwise kinship coefficients being 0 (Fig. 1a), indicating that most accessions have no, or only a weak genetic relationship with the other accessions and have only a small effect on the results of GWAS. Principal component analysis (PCA) demonstrated that the top two eigenvectors clearly separated these accessions into two subpopulations, which is consistent with the pattern of the genetic structure of the GWAS population (Fig. 1b).
Figure 1. Distribution of pairwise relative kinship coefficients and PCA of 520 B. napus inbred lines.

(a) Relative kinship coefficients and (b) PCA results. Only kinship coefficients of 0 to 0.5 are shown. Blue triangles and red circles represent the B. napus accessions in group 1 and group 2, respectively. Subpopulation in PCA plot analysis was assigned based on STRUCTURE analysis results from our previous study.
Previous studies showed that kinship (K) and population structure (Q, PCA) can result in false associations in GWAS13. For all traits, we performed association analyses to evaluate the performance of six statistical models (i.e., the naive, Q, PCA, K, Q + K, and PCA + K models) (Supplementary Fig. S2 and Table 2). Generally, the observed -log10(P-value) values from the naive model greatly deviated from the expected distribution, followed by the K and Q models, whereas the PCA + K model showed a better effect than the other models for most traits, and consequently this model was used for association study in our experiment.
Table 2. Summary of significantly associated SNPs for HI and four HI-related traits.
| Trait | Env. | NSSc | Chromosome | R2 d |
|---|---|---|---|---|
| CBY | 13CQ | 22 | A03/A06/A07/A08/A09/C03/C05/C07/C08 | 4.37%~5.96% |
| 14CQ | 2 | A03/A06 | 3.71%~5.24% | |
| 14YN | 0 | |||
| 14YN-CQ | 1 | C01 | 8.24% | |
| 14VTL | 1 | A08 | 8.09% | |
| ST | 13CQ | 123 | A08/A03 | 4.54%~9.69% |
| 14CQ | 3 | A03 | 4.51%~6.43% | |
| 14YN | 2 | A03/C06 | 7.31%~8.51% | |
| 14YN-CQ | 0 | |||
| 14VTL | 3 | A08/A10 | 7.81%~8.13% | |
| BY | 13CQ | 83 | A03/A04/A05/A06/A07/A08/A09/C01/C02/C03/C04/C05/C07/C08/C09 | 4.56%~8.36% |
| 14CQ | 5 | A03/A09/C02/C05 | 3.47%~4.53% | |
| 14YN | 0 | |||
| 14YN-CQ | 0 | |||
| 14VTL | 0 | |||
| SY | 13CQ | 10 | A02/A03/A05/A09/C01/C05/C06/C08 | 5.38%~7.49% |
| 14CQ | 2 | A03/C04 | 4.24%~4.25% | |
| 14YN | 0 | |||
| 14YN-CQ | 1 | A04 | 8.12% | |
| 14VTL | 6 | A03/A04/A06 | 7.96%~11.01% | |
| HI | 13CQ | 12 | A01/A02/A07/A09/A10/C04/C05/C09 | 4.63–5.66% |
| 14CQ | 1 | A07 | 4.76% | |
| 14YN | 2 | A10/C06 | 7.86%~8.51% | |
| 14YN-CQ | 7 | A01/A06/A07/C03/C06 | 7.43%~9.61% | |
| 14VTL | 8 | A06/A07/C01/C06 | 5.51%~8.68% |
NSS, number of significantly associated SNPs; R2, phenotypic variance.
Association mapping
Based on the optimal models, we detected 294 SNPs (P < 3.14E-05) significantly associated with HI and four HI-related traits (Fig. 2 and Supplementary Table S2), including 30, 26, 131, 88, and 19 SNPs associated with HI and with CBY, ST, BY, and SY, respectively. Under environment 13CQ, a total 250 significant SNPs were identified, followed by environments 14VTL, 14CQ, 14YN-CQ and 14YN, in which 18, 13, 9 and 4 significant SNPs, respectively, for different traits were detected.
Figure 2. Manhattan plots of association analysis for HI under five environments.

Manhattan plot of GWAS results for (a) 13CQ-HI, (b) 14CQ-HI, (c) 14YN-HI, (d) 14YN-CQ-HI, and (e) 14VTL-HI. The dashed horizontal red line depicts the significance threshold (−log101/31,839 = 4.5).
Among these SNPs, 80 could be identified repeatedly, including 79 SNPs which associated with multiple traits (Fig. 2 and Supplementary Table S3). For example, 16 SNPs were simultaneously associated with BY, CBY, and ST. The marker Bn-A06-p23865356 was closely related to 14VTL-HI, 14VTL-SY, and 14YN-CQ-HI (Supplementary Fig. S3), whereas Bn-scaff_15763_1-p506136 on chromosome C06 was simultaneously associated with 14YN-HI, 14YN-ST, 14VTL-HI, and 14YN-CQ-HI (Supplementary Fig. S4). These significant SNPs associated with more than one trait were chosen for further candidate gene identification.
To identify the genotypes associated with relatively low biomass and high seed yield, genotypic classification analysis was performed by comparing the mean trait values between lines carrying different alleles of all significant SNPs. For most of the significant SNPs, higher seed yield of plants with favorable alleles possessed higher biomass yield (Supplementary Table S2). We also identified a few alleles of significant SNPs showing favorable effects on seed yield, but no effects on biomass yield at YN, such as Bn_scaff_19170_1_p1132242, Bn_scaff_19526_1_p67 and Bn_A09_p25738298 (Supplementary Fig. S5), which are interesting genotypes for further study.
Identification of differentially expressed genes
To identify differentially expressed genes (DEGs) between extremely high- and low-HI B. napus lines at CQ and YN, 8 tissues harvested from four extremely high- and low-HI lines were separatively pooled as 32 independent samples (2 environments × 2 kinds of HI lines × 8 tissues). As a result, a total of 64 libraries (2 biological replicates per sample) were constructed and used for sequencing. After removing low quality and contaminant reads, a total of 1,385,050,482 125-bp clean paired-end reads were acquired, with an average of 21.64 million reads per sample, containing ~330 Gb of sequence data. On average, 83.62% of the input reads mapped uniquely to the B. napus reference genome14 (Supplementary Table S4).
RNA sequencing (RNA-seq) results showed that the number of DEGs was greatest in seeds harvested 20 days after flowering on the main inflorescence at CQ (2129 up-regulated and 1889 down-regulated genes), and on the primary branch at YN (816 up-regulated and 901 down-regulated genes) (Fig. 3a). The average number of DEGs between the high- and low-HI B. napus accessions grown at CQ (899 DEGs) was much higher than that in plants grown at YN (467 DEGs) (Fig. 3b), indicating that transcriptomic variation might be negatively correlated with yield in B. napus. At CQ, seeds on the main inflorescence (SM) showed the highest transcriptomic variation, whereas genes in seeds on the primary branch (SP) displayed highest transcriptomic variation at YN. Thus, stems and seeds appear to be important vegetative and reproductive tissues underlying differences in HI, since transcriptomic variation in these tissues was higher than in the other tissue types analysed.
Figure 3. Comparison of DEGs between extremely high- and low-HI lines grown under two different environments.

(a) Number of up- and down-regulated DEGs in 8 different tissues of plants grown at YN and CQ. (b) Average number of up- and down-regulated DEGs in all tissues of plants grown at YN and CQ. The “up” and “down” indicate up- and down-regulated genes, respectively.
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis
GO enrichment analysis identified 3391 and 3704 significantly enriched GO terms from amongst the DEGs at CQ and YN, respectively (Supplementary Table S6). Among the significantly enriched level-2 GO terms (Fig. 4), nutrient reservoir activity (GO:0045735) was the GO term for Molecular Function with the highest overrepresentation (false discovery rate (FDR) = 6.31E-39). Extracellular region (GO:0005576) was the most overrepresented GO (FDR = 1.63E-65) among the GO terms for Cell Component. In addition, DEGs identified at YN showed higher enrichment for Biological Processes than those grown at CQ (Fig. 4). Cell wall organization or biogenesis (GO:0071554, FDR = 1.67E-48), photosynthetic membrane (GO:0034357, FDR = 2.57E-51), and enzyme regulator activity (GO:0030234, FDR = 1.21E-21) were the most enriched level-3 GO terms in Biological Process, Cell Component, and Molecular Function, respectively (Supplementary Figs S6, S7 and S8).
Figure 4. Significantly enriched level 2 GO terms of DEGs.

First letters (c and y) in the group names represent the cultivation region (CQ and YN, respectively). The “up” and “down” indicate that the GO enrichment results were derived from up- and down-regulated genes, respectively.
KEGG enrichment analysis identified 92 and 54 significantly enriched pathways associated with the up- and down-regulated genes, respectively (Supplementary Table S6). Among those significantly enriched pathways, flavonoid biosynthesis (ath00941), glucosinolate biosynthesis (ath00966), pentose and glucuronate interconversions (ath00040), phenylpropanoid biosynthesis (ath00940), starch and sucrose metabolism (ath00500), and cutin, suberine and wax biosynthesis (ath00073) were more abundant than other pathways, and significantly enriched in multiple samples. The ancient, conserved flavonoid biosynthesis and phenylpropanoid biosynthesis pathways might play critical roles in plant adaptation to environmental stresses, which seemed to be more important for B. napus cultivation at CQ. As the difference in HI among the B. napus accessions was mainly caused by differences in transport efficiency from source to sink organs, these DEGs over-represented in starch and sucrose metabolism might control the photoassimilate partitioning and allocation from the site of synthesis to regions of utilization and/or storage and underlie the variation in HI among the B. napus accessions.
Identification of candidate genes
To identify candidate genes, we combined the GWAS and RNA-seq results, and only selected genes differentially expressed (DE) between the extremely high- and low-HI accessions, or encoded transcription factors (TFs) associated with high HI within the linkage disequilibrium (LD) blocks. Calculation of the LD blocks of 21 SNPs associated with more than one trait showed that 14 SNPs were located in LD blocks ranging in size from 231~498 kb, containing 35~128 genes (Supplementary Table S3). The 500-kb flanking sequences surrounding the remaining 7 significant SNPs were regarded as LD intervals.
In the 21 LD blocks, 73 genes encoding TFs and 174 DEGs were identified. The biological functions of the candidates were analyzed by performing Protein Basic Local Alignment Search Tool (BLASTP) searches against all Arabidopsis proteins. Of the 10 TFs that were DE within the LD intervals, 4 were regarded as important candidates. For example, BnaA06g34390D was located within the LD block of Bn-A06-p23865356 and encoded orthologs of Arabidopsis SHOOT GRAVITROPISM 5 (AtSGR5) which is a regulator of auxin biosynthesis and transport and, thus, of the early events of gravitropism in inflorescence stems15 (Fig. 5). In addition, several DEGs were also chosen as their biological functions were critical for variation of HI, such as BnaA02g13010D and BnaA02g12860D, two genes located within the LD intervals of marker Bn-A02-p10067386, and encoded orthologs of TEOSINTE BRANCHED1, CYCLOIDEA, PCF1 (AtTCP1), CYTOCHROME P450, FAMILY 735, SUBFAMILY A, and POLYPEPTIDE 2 (AtCYP735A2) in Arabidopsis16; BnaA03g38660D was located 32 kb away from the marker Bn-A03-p20375514, and encoded an ortholog of Arabidopsis VACULOLAR SORTING RECEPTOR 3 (AtVSR3), which is essential for pollen tube growth17; BnaA08g06910D was 164 kb upstream of marker Bn-A08-p7814328, and encoded an ortholog of Arabidopsis HEAT SHOCK TRANSCRIPTION FACTOR A1D (AtHSFA1D), a TF known to confer thermotolerance in plants18. All candidate genes within the LD blocks associated with development and stress tolerance might contribute to the differences in HI and HI-related traits listed in Supplementary Table S3.
Figure 5. Candidate gene in the LD interval on chromosome A06 identified through GWAS.

Top, the candidate gene region associated with HI and SY on chromosomes A06, as determined by GWAS. Middle, pairwise LD estimates in the haplotype block of the SNP marker Bn-A06-p23865356 on chromosome A06. Bottom, heatmap of candidate genes based on fold change of all genes in the haplotype block in 8 tissues of plants grown at YN and CQ. The dashed line represents the most important candidate gene in the LD heatmap and expression level.
Validation of DEGs by quantitative real-time polymerase chain reaction (qRT-PCR)
To validate the RNA-seq results, we selected 10 DEGs and evaluated their expression levels by qRT-PCR. All amplified PCR products were recovered for sequencing. The Pearson correlation coefficients between RNA-Seq and qRT-PCR data were calculated based on the log2 fold change. As shown in Supplementary Figs S9 and S10, the expression patterns of DEGs detected by qRT-PCR showed significant similarity (r2 = 0.9322) with the RNA-Seq data, confirming the reliability and accuracy of the RNA-seq data.
Discussion
HI is a complex agronomic trait that is influenced by a variety of physiological and environmental factors, and varies widely among crops. For modern varieties of the most intensively-cultivated grain crops, including rice19, wheat20, barley (Hordeum vulgare)21, and maize22, HI values range from 0.4 to 0.63,23. Genetic and agronomic improvements have greatly increased the yield potential of Glycine max (soybean), which is associated with an increase in HI from around 0.35 to 0.533. According to previous reports, the average HI for B. napus was 0.28 (n = 117)24, only higher than that of oat (0.21), with values ranging from 0.04 to 0.4125. In our study, the HI ranged from 0.048 to 0.42 and 0.052 to 0.34 for plants grown at CQ and YN, respectively, a finding consistent with earlier studies. although the HI was relatively stable in B. napus, and the observed variation was less than that of most other field crops examined24, some B. napus accesscions with high HI ( > 0.4) were identified, suggesting that the potential for futher yield increase in B. napus.
Correlation analyses showed that SY was significantly and positively related to HI (Supplementary Table S6), which is in line with the findings of several previous studies7,26. In another important crop, soybean, yield increase was also positively associated with HI27. These results suggest that yield can be enhanced in B. napus by increasing HI at both high-yield and standard production regions in China.
In the present study, ST was not correlated with HI in plants grown at CQ in 2013 and 2014, and tended to be negatively associated with HI in those grown at YN in 2014. The stem is an important organ in B. napus, accumulating large amount of stored photosynthate and nitrogen during the vegetative stage, and reallocating these assimilates and most nitrogen to seeds during the filling stage28. The significant negative correlation observed between the ST and HI indicates that phloem translocation of photoassimilates and their partitioning to seeds is a key factor limiting yield improvement under high-yield growth conditions. A similar phenomenon was observed in hybrid rice, namely that poor translocation and partitioning of assimilates to the grain resulted in limited grain filling and a low HI29. In addition, there was a significant positive relationship between HI and CBY.
In the Brassicaceae, silique walls are the major photosynthetically active organ after flowering and contribute assimilates and nutrients to fill seed growth30. Plants with higher CBY may be expected to possess more siliques, and thus to produce more assimilates and have a higher SY and HI. Furthermore, we noticed that the relationship between BY and HI was different in plants grown at CQ and YN. A similar inconsistency was observed in soybean grown in different environments27, indicating that HI is more complex than other HI-related traits, and might be easily affected by differences in cultivation environments. In addition, CBY and BY were both significantly positively related to SY, which is consistent with previous evidence31, suggesting yield potential greatly depends on biomass accumulation in B. napus.
In GWASs, general linear models (GLMs) often lead to false positives (type I errors), whereas mixed linear models (MLMs) are more stringent and can result in false negatives (type II errors)32. The PCA model has been widely used to correct for population stratification in GWASs33 and after integrating the kinship, this mixed PCA + K model gave significant improvement in reducing spurious associations, and thus has been broadly used in GWASs of several crops4. In the current study, six models, involving different combinations of K, Q, and PCA were used to analyze all of the traits examined this our study. As with early studies, the mixed PCA + K model was best in removing false-positive association signals (Table 2), indicating that population structure correction in a PCA model was more efficient than the STRUCTURE algorithm at eliminating false positives34. Hence, to reduce the number of false positives and negatives, only optimal models for each trait were considered when identifying association signals.
Over the past several years, several QTLs for important traits in B. napus were identified in GWAS, providing relatively higher mapping resolution and more candidate loci than in studies based on biparental populations35. In our study, a large natural population composed of 520 B. napus accessions was phenotyped by the 60K Brassica SNP array. GWAS results showed that the most significant SNPs for HI in plants grown at CQ and YN, in 2014, were located on chromosomes C05 and A07, and explained 5.66% and 4.76% of the phenotypic variation, respectively. For BY, a previous GWAS only detected a single significant SNP, which explained 0.76% of phenotypic variation7, whereas the most significant SNP we identified on chromosome A03, in our GWAS results from 13CQ, explained 8.36% of phenotypic variation. Arguable, this identified higher contribution rate could well be related to the larger population size employed in our GWAS. Consistent with this notion, rice scientists have perfomed whole-genome resequencing on 3000 rice accessions to obtain a global representation of genetic and functional diversity36.
Recently, Körber et al.37 idenfitifed only two significantly associated SNPs for SY due to the fact that this trait was examined at only two locations37, and Zhao et al.31 detected 9 consensus QTLs for SY, mainly located on chromosomes A02, A06, C01, and C0631. In the present study, significantly associated SNPs Bn-A02-p10067386 and Bn-scaff_16547_1-p307962, located on chromosomes A02 and C06, respectively, were closely co-localized with previously identified QTLs for SY, indicating that these QTLs were stable and could be repeatedly detected in different environments, and thus suitable for marker-assisted selection in high-yield breeding of B. napus.
The average yield of B. napus cultivated at YN is much higher than that in standard production regions. To elucidate how the environment affects HI and HI-related traits, we performed GWAS in virtual environments 14VTL and 14YN-CQ, to identify the significantly associated signals for each trait responsible for environmental change. We detected 8 and 7 significant SNPs for HI in environments 14VTL and 14YN-CQ, respectively, and of these 3 could be found repeatedly in the two environments. In addition, Bn-scaff_15763_1-p506136 was significantly associated with HI in both 14YN and 14YN-CQ environments. These results indicate that these associated signals were stable, and this method would be a very useful strategy for identifying loci that control plant responses to environmental variation.
Combining GWAS and RNA-seq can provide both the power and resolution needed to identify DE candidate genes, and has proven to be more successful than either strategy alone38,39. Based on integrative analyses of RNA-seq and GWAS in B. napus, 8 and 24 candidate genes putatively associated with drought tolerance and stem resistance to Sclerotinia sclerotiorum, respectively, were identified38,40. More recently, QTL fine mapping, digital gene expression (DGE) profiling, and whole-genome re-sequencing (WGS) were combined to identify a NODULIN 26-LIKE INTRINSIC PROTEIN gene involved in regulating boron uptake efficiency in B. napus39. Applying a similar strategy, we identified 73 TFs and 174 genes that were DE in at least one tissue from a total of 1113 genes within the LD blocks of 21 significant SNPs that associated with more than one trait. Taken together, these findings demonstrate the efficacy of combining strategies for rapid identification of candidate genes associated with complex traits.
The SNP Bn-A02-p10067386, associated with both HI and SY, is of particular interest. Within the LD block, BnaA02g12860D encodes an ortholog of AtCYP735A2, which is a cytochrome P450 monooxygenase that catalyzes the trans-hydroxylation of cytokinins (CKs) to form trans-zeatin (tZ)-type CK41. In Arabidopsis this gene defines the size of rosette leaves, inflorescence stems, and the number of flowers and siliques. We note that expression of BnaA02g12860D was significantly up-regulated in the leaf (Le) and silique pericarps, located on the main inflorescence (SPM) of the high HI accessions grown at CQ, implying that this gene might encode a regulator of HI and SY.
The BnaA03g38390D gene, located within the LD block of Bn-A03-p20375514 associated with BY, CBY, and ST at 13CQ, encodes an ortholog of the nuclear transcription factor Y, subunit B 7 (AtNF-YB7)42. Overexpression of AtNF-YB7 resulted in earlier seedling establishment and higher biomass than controls through an increase in carbon assimilation and a reduction in transpiration. In this regard, BnaA03g38390D showed significantly higher expression in BM of the high HI accessions at CQ. Hence, the role of this gene in shoot development of the high HI accessions deserves further study.
We identified BnaA06g34390D within the LD block of the Bn-A06-p23865356, and established that it encoded an ortholog of AtSGR5 (Fig. 5). As a C2H2-type TF, AtSGR5 is involved in the early events of gravitropism in the inflorescence stem15. Up-regulation of BnaA06g34390D expression was detected in the SPM and silique pericarps on the primary branch (denoted as SPP) at CQ, implying that seed yield at CQ and HI might be affected by the angle of the SP, becasue silique wall photosynthesis is known to be important for seed filling. Further functional characterization of BnaA06g34390D is needed to reveal the physiological significance of the involvement of this gene in the variation of HI in B. napus. It is notable that in this LD bock, three significant SNPs were detected specifically in the virtual environments, which were regarded as candidate loci for HI and SY associated with environment variations, implying environment-specific effects on HI and SY.
Within the LD interval of the Bn-A08-p20354609, BnaA08g26390D was identified as the most plausible candidate gene, and it encodes an ortholog of ATP-CITRATE LYASE A-3 (ACLA-3) in Arabidopsis. Importantly, this gene was significantly up-regulated in all 8 tissues in plants grown at YN and CQ. A previous study showed that plants with reduced ACL activity have a complex bonsai phenotype, with features such as miniaturized organs, smaller cells, aberrant plastid morphology, and reduced cuticular wax deposition43. Hence, the significantly higher expression of BnaA08g26390D in vegetative tissues might be important for accumulating more photosynthates in the higher HI B. napus accessions.
The LD block of the Bn-scaff_15763_1-p506136 is another noteworthy region, as here, a gene encoding a leucine-rich repeat (LRR) family protein, BnaC06g17400D was significantly up-regulated in seeds of the high HI B. napus accessions grown at YN, but repressed in plants grown at CQ (Fig. 6). Functional analysis of BnaC06g17400D on STRING (Search Tool for the Retrieval of Interacting Genes/Proteins, http://string-db.org)44 revealed that the protein was the ortholog of AT3G59510 (Leucine-rich repeat (LRR) family protein), and interacted with AT4G24190 (HEAT SHOCK PROTEIN 90.7), which encodes an endoplasmic reticulum (ER)-resident HSP90-like protein and is required for shoot apical meristem, root apical meristem, and floral meristem formation and pollen tube elongation45. Hence, we propose that BnaC06g17400D is also a key regulator of high HI formation at both CQ and YN.
Figure 6. Candidate gene in the LD interval on chromosome C06 identified through GWAS.

Top, the candidate gene region associated with HI and ST on chromosome C06, as determined by GWAS. Middle, pairwise LD estimates in the haplotype block of the SNP marker Bn-scaff_15763_1-p506136 on chromosome C06. Bottom, heatmap of candidate genes based on fold change of all the genes in the haplotype block in 8 tissues of plants grown at both YN and CQ. The dashed line represents the candidate gene associated with high HI in the LD heatmap and expression level.
Within the confidence interval of Bn-scaff_16197_1-p1970599, BnaC08g33690D encodes CAROTENOID CLEAVAGE DIOXYGENASE 1 (CCD1), which plays essential roles in photosynthesis, plant development, and abiotic stress adaptation46. The expression levels of those candidate genes were significantly up-regulated in vegetative tissues in plants grown at CQ and YN. Thus, these genes should also be considered as important candidates for HI variation.
In this study, we identified several significant SNPs and candidate genes associated with HI and HI-related traits. However, we realized that the accuracy of our GWAS results is impacted, especially for such complex traits, because only representative environments were chosen for field trials. Hence, further studies are needed to validate repeatability of these significant SNPs, under multiple environments, and biological functions for these candidate genes will need to be experimentally verified.
In summary, our study demonstrated that HI is a complex agronomic trait that is influenced by both environmental and genotypic factors. The substantial phenotypic variation for HI and HI-related traits in B. napus, at both high-yield and standard production regions provides an excellent opportunity to dissect the genetic architecture of these complex traits, with a focus on the effect of environmental variation. Using a combination of GWAS and RNA-seq, we identified a large number of novel loci associated with HI, and predicted 33 candidate genes. These genes can be used in marker-based breeding programmes for increased HI.
Methods
Plant materials and field trials
The natural population of B. napus provided by the Chongqing Rapeseed Engineering Research Center, China, consisted of 520 genetically diverse inbred lines (Supplementary Table S1). Most of these accessions were derived from research institutions in China, but several were introduced from other countries, including Germany, Australia, and Canada.
All rapeseed inbred lines were planted, in triplicate, in a randomized block design. Each plot contained three rows; 10 plants per row, 20 cm between plants within each row, and 30 cm between rows. Plants were cultivated under natural environments, as follows: 520 accessions were cultivated in Beibei, Chonqging (29°45′ N, 106°22′ E, 238.57 m, CQ), for two consecutive years (2012–13 and 2013–14, denoted as 13CQ and 14CQ) and, based on the flowering time observed in 2013 at CQ, 320 accessions were selected for planting at Lincang, Yunnan Province (23°43′ N, 100°02′ E, 1819.50 m, YN) in 2014 (14YN). All accessions were sown at the end of September in both YN and CQ, and harvested at the beginning of May (CQ) or the end of April (YN) in the following year.
After harvesting, plants were divided into canopy and stem, and dried at 50 °C to a constant weight in a ventilated drying oven. Canopy biomass yield (CBY) and stem dry weight (ST) were defined as the dried weight of canopy and stems, respectively. Biomass yield (BY) was represented as the sum of CBY and ST. Seed yield,per plant (SY), was determined by measuring the dry weight of all seeds produced per plant. Harvest index (HI) was represented as the ratio of SY to BY.
To identify the significant associated signals for each trait responsible for environmental variation, two virtual environments were derived from the abovementioned five environments. The first virtual environment was the trait difference between YN and CQ in 2014 (denoted as 14YN-CQ) and the second virtual environment was the trait variation between two the locations in 2014 (denoted as 14VTL), which was calculated by the following formula: 2 × (YN − CQ)/(CQ + YN). The mean value of the ten plants with the same height in the same plot represents the trait value of a replication, and the mean value of the three replications was regarded as the phenotypic value of the target trait.
Statistical analysis
The ANOVA was conducted using GLM procedure in SAS version 8.0 (SAS Institute Inc.), with the following model: yger = μ + αg + βe + (αβ)ge + γr(e) + εger, where yger was the phenotypic trait value of the gth genotype in the eth environment for the rth replicate, μ the grand mean, αg the genotypic effect of the gth genotype, βe the effect of the eth environment, (αβ)ge the interaction effect of the gth genotype and the eth environment, γr(e) the block effect within the eth environment, and εger the residual. All effects were modeled as random. The CORR procedure in SAS version 8.0 (SAS Institute Inc.) was used to calculate the Pearson’s correlation coefficients between traits of interest. The broad-sense heritability was calculated as: 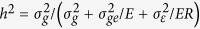 , where
, where 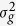 ,
, 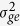 and
and 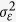 are estimates of the variances of genotype, genotype × environment interactions, and error, respectively. E is the number of environments, and R is the number of replications per environment.
are estimates of the variances of genotype, genotype × environment interactions, and error, respectively. E is the number of environments, and R is the number of replications per environment.
Genotyping analysis
Genotyping of the 520 B. napus accessions was carried out in Huazhong Agricultural University, Wuhan, China, using the Brassica 60 K Illumina Infinium SNP array. The SNP data were analyzed by Illumina BeadStudio genotyping software, as previously described11. SNPs with minor allele frequencies (MAFs) of <0.05, or lacking a call higher than 0.9 were excluded from analysis. After processing, 31,839 SNP markers were retained for further analysis. To determine the physical localization of SNPs, flanking sequences were subjected to BLASTN searches against the B. napus genome, with an E-value cut-off of 1E-514,47. SNPs with a maximum Bit-Score were retained as unique SNPs, and subjected to further analysis.
Population structure and genome-wide association analysis
The population structure (Q) was inferred using STRUCTURE 2.3.4, as we previously reported11. The relative kinship matrix (K) and principal component (PC) of the natural population were calculated using TASSEL 5.2.112. Linkage disequilibrium (LD) between all pairs of SNP markers was estimated as the correlation of allele frequencies (r2) using TASSEL 5.2.1.
The effects of population structure (Q, PCA) and kinship (K) were estimated by a GWAS using the following six models: (a) naive model, which does not control for Q and K; (b) Q model, which controls for the percentage of admixture (Q matrix); (c) PCA model, which uses the top 10 PCs as fixed effects; (d) K model, which controls for K; (e) Q + K model, which uses both K as a random effect and the Q matrix; and (f) P + K model, which controls for both PCA and K. The former three models were performed using a GLM, while the remaining models were performed using an MLM in TASSEL 5.2.112. The optimal model for each trait was selected based on a Quantile-quantile (Q-Q) plot in ggplot248, by plotting the −log10(p) values of each SNP against the expected value. Suggestive (1/n) P-value threshold was set to control the genome-wide type 1 error rate, and n represented the number of SNP markers. The Manhattan plot was produced by qqman software49.
RNA-seq and identification of differentially expressed genes
To identify differentially expressed genes (DEGs) between extremely high and low HI lines, total RNA was pooled at equal molar amounts from four extremely high and low HI B. napus lines (Supplementary Table S7). For each type of line, eight tissues, including mature leaves (Le) and stems (St), and buds on the primary branch (BP) and the main inflorescence (BM), were harvested at flowering stage 63–65, according to the BBCH (Biologische Bundesanstalt, Bundessortenamt and CHemical industry) scale50. Seeds and silique pericarps on the main inflorescence and the primary branch were harvested at 20 days after flowering. For each sample, two biological replicates, each harvested from three independent plants, were collected for RNA-seq and qRT-PCR analysis.
Sequencing library preparation and sequencing reactions were conducted at the Biomarker Technologies Corporation (Beijing, China). RNA-seq data were deposited in the NCBI under the accession number SRP072900. All sequencing reads were subjected to quality filtering using Trimmomatic-0.3351, and mapped to the B. napus reference genome using STAR 2.4.2a with default parameters. Gene expression levels were quantified by the cuffquant program in terms of fragments per kilobase of exon per million mapped fragments (FPKM)52. The cuffdiff program was used to test for statistically significant differences in gene expression between two samples. DEGs were identified based on the following criteria: false discovery rate (FDR) <0.05 and absolute fold change >253. To identify and classify transcription factors (TFs) in B. napus, the putative amino acids were aligned with Arabidopsis TFs downloaded from the Plant Transcription Factor Database v3.0 (http://planttfdb.cbi.pku.edu.cn) and The Arabidopsis Information Resource (http://www.arabidopsis.org)54.
GO and KEGG enrichment analyses of DEGs
To determine the biological significance of DEGs, all B. napus genes were annotated by BLASTP analysis against the Arabidopsis proteome (TAIR10) with an E-value cut-off of 1E-546. GO enrichment analysis was implemented using BinGO v2.4.4, and significantly enriched GO terms (FDR <0.05) were displayed using ggplot248,55,56. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was conducted on the KOBAS 2.057. All GO and KEGG statistical tests were corrected for multiple comparisons (Benjamini Hochberg method).
Identification of candidate genes
To define regions of interest for identification of candidate genes, the haplotype blocks were investigated with Haploview 4.258, and the LD blocks were defined by the Four Gamete Rule with a fourth haplotype frequency cutoff of 0.1. All genes within the LD block were considered for identification of candidates. For significantly associated markers outside of the LD blocks, the 250-kb flanking regions on either side of the markers were used to identify candidates. Differentially expressed (DE) candidate genes were identified by screening the DEGs within the LD blocks or flanking regions of significantally associated markers.
Validation of DEGs by qRT-PCR
Ten DE candidate genes identified in the abovementioned analyses were assayed by qRT-PCR. Gene-specific primers were designed using Geneious Pro 8.1.559 (Supplementary Table S8). The total RNA was the same as used for RNA sequencing. cDNA synthesis, qRT-PCR cycling conditions, amplification efficiency, and specificity assessment were as previously described60. Two independent biological replicates, each with three technical replicates were analyzed per sample. Relative expression levels were calculated using the 2−ΔΔCt method, with Bna.ACT7 and Bna.UBC21 as internal controls61.
Additional Information
How to cite this article: Lu, K. et al. A combination of genome-wide association and transcriptome analysis reveals candidate genes controlling harvest index-related traits in Brassica napus. Sci. Rep. 6, 36452; doi: 10.1038/srep36452 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
We are grateful to Professor William Lucas from University of California, Davis for critical reading of the manuscript. This work was supported by grants from the National Natural Science Foundation of China (U1302266, 31271756, and 31571701), the 973 Project (2015CB150201), the National Key Research and Development Plan (2016YFD0101007), the 863 Project (2013AA102602), the National Key Technology Support Program (2013BAD01B03-12) and the 111 Project (B12006).
Footnotes
Author Contributions J.L. and Y.L. conceived and designed the experiments; K.L., Z.X., H.J., L.P., C.Q., M.F., B.H. and L.T. performed the experiments, Y.L. and X.X. analyzed the data, and K.L., Z.X., H.J. and J.L. wrote the paper. All authors reviewed the manuscript.
References
- Zhang J. & Yang J. Improving harvest index is an effective way to increase crop water use efficiency. In Proceedings of the 4th International Crop Science 286 (2004). [Google Scholar]
- Turchini G. & Mailer R. J. Rapeseed (canola) oil and other monounsaturated fatty acid-rich vegetable oils. Fish oil Replace. Altern. lipid sources Aquac. Feed. 161–208 (2011). [Google Scholar]
- Hay R. K. M. Harvest index: a review of its use in plant breeding and crop physiology. Ann. Appl. Biol. 126, 197–216 (1995). [Google Scholar]
- Li F. et al. Genome-wide association study dissects the genetic architecture of seed weight and seed auality in rapeseed (Brassica napus L.). DNA Res. 1–13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F. et al. A genome-wide association study of plant height and primary branch number in rapeseed (Brassica napus). Plant Sci. 242, 169–177 (2016). [DOI] [PubMed] [Google Scholar]
- Hatzig S. V. et al. Genome-wide association mapping unravels the genetic control of seed germination and vigor in Brassica napus. Front. Plant Sci. 6, 221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X. et al. Unravelling the complex trait of harvest index in rapeseed (Brassica napus L.) with association mapping. BMC Genomics 16, 379 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai H. Rapeseed yield per hectare at Lincang city hit a record in China. http://www.lincang.gov.cn/Jrlc/Bmdt/201602/55471.html (2014).
- Zhang F. et al. Yield and agronomic traits of winter rapeseed cultivars registered in China from 2001 to 2010. Chin. J. Oil. Crop Sci. 34, 239–244 (2012). [Google Scholar]
- Cai G. et al. Genetic dissection of plant architecture and yield-related traits in Brassica napus. Sci. Rep. 6, 21625 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu C.-M. et al. Identification of candidate genes for seed glucosinolate content using association mapping in Brassica napus L. Genes (Basel). 6, 1215–1229 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury P. J. et al. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635 (2007). [DOI] [PubMed] [Google Scholar]
- Flint-Garcia S. A. et al. Maize association population: A high-resolution platform for quantitative trait locus dissection. Plant J. 44, 1054–1064 (2005). [DOI] [PubMed] [Google Scholar]
- Chalhoub B. et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science. 345, 950–953 (2014). [DOI] [PubMed] [Google Scholar]
- Morita M. T. et al. A C2H2-type zinc finger protein, SGR5, is involved in early events of gravitropism in Arabidopsis inflorescence stems. Plant J. 47, 619–628 (2006). [DOI] [PubMed] [Google Scholar]
- Takei K., Yamaya T. & Sakakibara H. Arabidopsis CYP735A1 and CYP735A2 encode cytokinin hydroxylases that catalyze the biosynthesis of trans-Zeatin. J. Biol. Chem. 279, 41866–41872 (2004). [DOI] [PubMed] [Google Scholar]
- Avila E. L. et al. Expression analysis of Arabidopsis vacuolar sorting receptor 3 reveals a putative function in guard cells. J. Exp. Bot. 59, 1149–1161 (2008). [DOI] [PubMed] [Google Scholar]
- Liu H. C., Liao H. T. & Charng Y. Y. The role of class A1 heat shock factors (HSFA1s) in response to heat and other stresses in Arabidopsis. Plant, Cell Environ. 34, 738–751 (2011). [DOI] [PubMed] [Google Scholar]
- Bueno C. S. & Lafarge T. Higher crop performance of rice hybrids than of elite inbreds in the tropics: 1. Hybrids accumulate more biomass during each phenological phase. F. Crop. Res. 112, 229–237 (2009). [Google Scholar]
- Zhang X., Chen S., Sun H., Pei D. & Wang Y. Dry matter, harvest index, grain yield and water use efficiency as affected by water supply in winter wheat. Irrig. Sci. 27, 1–10 (2008). [Google Scholar]
- Kemanian A. R., Stöckle C. O., Huggins D. R. & Viega L. M. A simple method to estimate harvest index in grain crops. F. Crop. Res. 103, 208–216 (2007). [Google Scholar]
- D’Andrea K. E., Otegui M. E. & De La Vega A. J. Multi-attribute responses of maize inbred lines across managed environments. Euphytica 162, 381–394 (2008). [Google Scholar]
- Yang J. & Zhang J. Crop management techniques to enhance harvest index in rice. J. Exp. Bot. 61, 3177–3189 (2010). [DOI] [PubMed] [Google Scholar]
- Unkovich M., Baldock J. & Forbes M. Variability in harvest index of grain crops and potential significance for carbon accounting: Examples from Australian agriculture. Adv. Agron. 105, 173–219 (2010). [Google Scholar]
- Hirth J. R., Haines P. J., Ridley A. M. & Wilson K. F. Lucerne in crop rotations on the Riverine Plains 2. Biomass and grain yields, water use efficiency, soil nitrogen, and profitability. Aust. J. Agric. Res. 52, 279–293 (2001). [Google Scholar]
- Shi J., Li R., Zou J., Long Y. & Meng J. A dynamic and complex network regulates the heterosis of yield-correlated traits in rapeseed (Brassica napus L.). PLoS One 6, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bruin J. L. & Pedersen P. Growth, yield, and yield component changes among old and new soybean cultivars. Agron. J. 101, 124–130 (2009). [Google Scholar]
- Schjoerring J. K., Bock J. G. H., Gammelvind L., Jensen C. R. & Mogensen V. O. Nitrogen incorporation and remobilization in different shoot components of field-grown winter oilseed rape (Brassica napus L.) as affected by rate of nitrogen application and irrigation. Plant Soil 177, 255–264 (1995). [Google Scholar]
- Yang J. et al. Grain and dry matter yields and partitioning of assimilates in japonica/indica hybrid rice. Crop Sci. 42, 766–772 (2002). [Google Scholar]
- Bennett E. J., Roberts J. A. & Wagstaff C. The role of the pod in seed development: strategies for manipulating yield. New Phytol. 190, 838–853 (2011). [DOI] [PubMed] [Google Scholar]
- Zhao W. et al. Genome-Wide Identification of QTL for seed yield and yield-related traits and construction of a high-density consensus map for QTL comparison in Brassica napus. Front. Plant Sci. 7, 17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace J., Gardner C., Romay C., Ganapathysubramanian B. & Lübberstedt T. Genome-wide association analysis of seedling root development in maize (Zea mays L.). BMC Genomics 16, 47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X. et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 42, 961–967 (2010). [DOI] [PubMed] [Google Scholar]
- Montilla-Bascon G. et al. Genome-wide association study for crown rust (Puccinia coronata f. sp. avenae) and powdery mildew (Blumeria graminis f. sp. avenae) resistance in an oat (Avena sativa) collection of commercial varieties and landraces. Front. Plant Sci. 6, 103 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q. et al. Genetic variation and association mapping for 12 agronomic traits in indica rice. BMC Genomics 16, 1067 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.-Y., Wang J. & Zeigler R. S. The 3,000 rice genomes project: new opportunities and challenges for future rice research. Gigascience 3, 8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Körber N. et al. Agronomic and seed quality traits dissected by genome-wide association mapping in Brassica napus. Front. Plant Sci. 7, 386 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. et al. Identification of putative candidate genes for water stress tolerance in canola (Brassica napus). Front. Plant Sci. 6, 1058 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y. et al. Transcriptomics-assisted QTL Fine Mapping for the Rapid Identification of a NODULIN 26-LIKE INTRINSIC PROTEIN gene regulating boron efficiency in allotetraploid rapeseed. Plant. Cell Environ (2016). [DOI] [PubMed] [Google Scholar]
- Wei L. et al. Genome-wide association analysis and differential expression analysis of resistance to Sclerotinia stem rot in Brassica napus. Plant Biotechnol. J. 14, 1368–1380 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiba T., Takei K., Kojima M. & Sakakibara H. Side-Chain Modification of Cytokinins Controls Shoot Growth in Arabidopsis. Dev. Cell 27, 452–461 (2013). [DOI] [PubMed] [Google Scholar]
- Han X. et al. Overexpression of the poplar NF-YB7 transcription factor confers drought tolerance and improves water-use efficiency in Arabidopsis. J. Exp. Bot. 64, 4589–4601 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatland B. L., Nikolau B. J. & Wurtele E. S. Reverse genetic characterization of cytosolic acetyl-CoA generation by ATP-citrate lyase in Arabidopsis. Plant Cell 17, 182–203 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D. et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein E. M. et al. Plant endoplasmin supports the protein secretory pathway and has a role in proliferating tissues. Plant J. 48, 657–673 (2006). [DOI] [PubMed] [Google Scholar]
- Zhao S. et al. Molecular characterization of carotenoid biosynthetic genes and carotenoid accumulation in Lycium chinense. Molecules 19, 11250–11262 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginestet C. ggplot2: elegant graphics for data analysis. J Roy Stat Soc A 174, 245–246 (2011). [Google Scholar]
- Turner S. D. qqman: an R package for visualizing GWAS results using Q-Q and manhattan plots. bioRxiv (2014). [Google Scholar]
- Lancashire P. D. et al. A uniform decimal code for growth stages of crops and weeds. Ann. Appl. Biol. 119, 561–601 (1991). [Google Scholar]
- Dobin A. et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei B. et al. RNA sequencing analysis reveals transcriptomic variations in tobacco (Nicotiana tabacum) leaves affected by climate, soil, and tillage factors. Int. J. Mol. Sci. 15, 6137–6160 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J., Zhang H., Kong L., Gao G. & Luo J. PlantTFDB 3.0: A portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 42 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoot M. E., Ono K., Ruscheinski J., Wang P. L. & Ideker T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 27, 431–432 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S., Heymans K. & Kuiper M. BiNGO: A Cytoscape plugin to assess overrepresentation of Gene Ontology categories in biological networks. Bioinformatics 21, 3448–3449 (2005). [DOI] [PubMed] [Google Scholar]
- Xie C. et al. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett J. C., Fry B., Maller J. & Daly M. J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005). [DOI] [PubMed] [Google Scholar]
- Kearse M. et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K. et al. Genome-wide survey and expression profile analysis of the mitogen-activated protein kinase (MAPK) gene family in brassica rapa. PLoS One 10, 1–26 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu C. et al. Comparative transcriptome analysis of recessive male sterility (RGMS) in sterile and fertile Brassica napus lines. PLoS One 10, 1–18 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.