

Abstract
Purpose
We have incorporated a positron emission tomography (PET) functionality in T cells expressing a CD19-specific chimeric antigen receptor (CAR) to non-invasively monitor the adoptively transferred cells.
Procedures
We engineered T cells to express CD19-specific CAR, firefly luciferase (ffLuc), and herpes simplex virus type-1 thymidine kinase (TK) using the non-viral-based Sleeping Beauty (SB) transposon/transposase system adapted for human application. Electroporated primary T cells were propagated on CD19+ artificial antigen-presenting cells.
Results
After 4 weeks, 90 % of cultured cells exhibited specific killing of CD19+ targets in vitro, could be ablated by ganciclovir, and were detected in vivo by bioluminescent imaging and PET following injection of 2′-deoxy-2′-[18F]fluoro-5-ethyl-1-β-D-arabinofuranosyl-uracil ([18F]FEAU).
Conclusion
This is the first report demonstrating the use of SB transposition to generate T cells which may be detected using PET laying the foundation for imaging the distribution and trafficking of T cells in patients treated for B cell malignancies.
Keywords: Chimeric antigen receptor, Immunotherapy, B cell malignancy
Introduction
Genetic modification of T cells to redirect their specificity to cell-surface tumor-associated antigens (TAAs) via stable expression of chimeric antigen receptors (CARs) is a successful strategy for adoptive immunotherapy. Multiple early-phase clinical trials evaluating the safety and efficacy of adoptive transfer of CAR+ T cells into patients with advanced B cell malignancies are under way or have been completed [1, 2]. We have previously generated a CAR that recognizes CD19, a B cell TAA. This CAR consists of a human CD19-specific scFv fused to modified IgG4 Fc and hinge regions, the CD28 intracellular domain, and the CD3-ξ intracellular domain. Gene transfer of this CD19-specific CAR into T cells redirects specificity towards CD19+ tumors [3]. To help resolve safety and cost-related issues associated with retroviral- or lentiviral-mediated transduction of T cells with CARs, we recently evaluated the Sleeping Beauty (SB) transposon/transposase system and electro-transfer of associated DNA plasmids to achieve non-viral genomic integration. CD19-specific CAR+ T cells were numerically expanded on K562-derived artificial presenting cells (aAPC) co-expressing human CD19 and an array of co-stimulatory molecules including CD86, CD137L, as well as a membrane-bound mutein of interleukin (IL)-15. CD19 served to specifically propagate the genetically modified T cells, leaving those cells that did not integrate the transposon to die from neglect. This method of expansion resulted in the outgrowth of sufficient numbers of CD19-specifc CAR+ T cells for clinical applications [4, 5]. Thus, we have initiated three US Food and Drug Administration-sanctioned clinical trials to adoptively transfer autologous [6] and allogeneic clinical-grade CAR+ T cells into patients with B cell malignancies after hematopoietic stem-cell transplantation (HSCT).
Accurate assessment of infused T cell persistence and targeting to tumors in a clinical setting is an essential component of managing effective adoptive immunotherapy. Current methods of assessing infused T cell persistence and function employ polymerase chain reaction to amplify specific gene markers (e.g., CAR (persistence), interferon-γ (IFN-γ; function)) from infused T cells serially sampled from peripheral blood and tumor tissue [7]. Other techniques include the assessment of biomarkers (e.g., CAR-specific antibodies) via immunohistochemical analysis of tissue sections, flow cytometry analysis of peripheral blood or tumor biopsies, and assessment of TAA-mediated stimulation of IFN-γ production [8, 9].
These methods of assessing T cell function and frequency, however, carry inherent limitations. They present intermittent data obtained from longitudinal sampling events and do not provide a comprehensive whole-body view of the temporal and spatial distribution of infused T cells. Genetic modification of T cells with CD19-specific CAR and a co-reporter suitable for whole-body non-invasive imaging provides a method for repetitive monitoring that can inform on the therapeutic potential of adoptive T cell therapy for CD19+ B cell malignancies. Positron emission tomography (PET) is an established imaging technology currently employed in the clinic that relies on the detection of radioactive tracers which incorporate within the cells of interest. Herpes simplex virus (HSV)-1 thymidine kinase (TK) and its variants, such as sr39 [10], can be employed as PET reporter genes because they phosphorylate radiolabeled purine or pyrimidine nucleoside analogs such as 9-(4-[18F]fluoro-3-hydroxymethylbutyl)guanine ([18F]FHBG; approved for clinical use [11]) and 2′-deoxy-2′-[18F]fluoro-5-ethyl-1-β-D-arabinofuranosyl-uracil ([18F]FEAU), the latter of which is a more sensitive radiotracer [12]. Intracellular TK-mediated phosphorylation entraps these radiotracers and the foci of the radiotracer accumulation can be spatially delineated using positron emission tomography (PET) [13]. PET has been successfully used to detect TK+ tumor-specific T cells in mouse tumor models [13–15], TK+ polyclonal T cells intravenously injected into non-human primates [16], and TK+ T cells intracranially infused into the site of tumor resection in a patient with glioblastoma multiforme [11].
In the present study, we demonstrate that human T cells can be genetically modified by single transposition or triple transposition achieved by electroporation of one or three SB transposons coding for CAR or CAR, TK, and ffLuc transgenes to generate T cells that are (a) CD19 specific and (b) detectable using bioluminescent imaging (BLI) and PET following adoptive transfer into immuno-deficient mice and administration of the radiotracer [18F]FEAU.
Materials and Methods
Plasmids
One SB transposon plasmid CD19RCD28/pSBSO expresses the human codon-optimized (CoOp), second-generation CoOpCD19RCD28 CAR in the SB transposon DNA plasmid pSBSO under the control of the human elongation factor-1α hybrid promoter and flanked by inverted/direct repeats [5]. Another SB transposon plasmid ffluc-neo/pSBSO expresses myc-tagged firefly luciferase (ffLuc) fused to CoOp neomycin phosphotransferase (Neo) for bioluminescent imaging (BLI). A cytoplasm-retargeted mutant of wild-type HSV1-derived TK was designed by inserting the leucine-rich nuclear export signal (NES) of mitogen-activated protein kinase cDNA of Xenopus (aa 32–52) between His9 and Ala10 at the N-terminus [13]. Additionally, arginines 25, 26, 32, and 33 of the nuclear localization sequence (NLSm) of TK were mutated to glycine. The CoOpNES-NLSm-TK fragment was synthesized by GeneArt (Germany) and cloned into the plasmid Flag-TK-Hygro/pΔSBSO at the NruI site, replacing the wild-type HSV1-derived TK, to produce an N-terminal FLAG-tagged NES-NLSm-TK fragment with a C-terminal-fused and hygromycin phosphotransferase (Hygro) gene (3[FLAG]-NES-NLSm-TKCoOpHygro). The fusion transgene was cloned into the SB vector pSBSO using AgeI and KpnI sites to generate the SB transposon plasmid 3[FLAG]-NES-NLSm-TKCoOpHygro/pSBSO (Fig. 1a).
Fig. 1.

Schematic of 3[FLAG]-NES-NLSm-TKCoOpHygro imaging construct, transposon and transposase vectors, and CAR+TK+ffLuc+ T cell preparation schematic. a The codon-optimized (CoOp) herpes simplex virus (HSV)-1 thymidine kinase (TK) transgene was modified by mutating its nuclear localization signal (NLSm), inserting nuclear export signal (NES) between His9 and Ala10, and inserting a FLAG sequence at the N-terminus. This coding sequencing is expressed in frame with a hygromycin phosphotransferase (Hygro) gene in a pSBSO DNA plasmid backbone. b DNA plasmids for three SB transposon coding for CD19-specific CAR (CD19RCD28), 3[FLAG]-NES-NLSm-TKCoOpHygro, and myc-ffLuc-Neo(CoOp). Also shown is the DNA plasmid coding for the SB transposase SB11 (pKan-CMV-SB11). c Schematic of triple transposition depicting electroporation and numeric expansion of CAR+ or CAR+TK+ffLuc+ T cells on aAPC (clone no. 4) in the presence of hygromycin B (0.2 mg/ml), neomycin (0.8 mg/ml), IL-2 (50 U/ml), and IL-21 (30 ng/ml). Abbreviations: hEF1-alpha/p constitutive human elongation factor-1α hybrid promoter, 3[FLAG] three repeats of the FLAG epitope (amino acids DYKDDDDK), CD19RCD28(CoOp) codon-optimized CD19-specific CAR, 3[FLAG]NES-NLMm-TK-Hygro codon-optimized TK mutein containing N-terminal 3[FLAG] sequence, NES, and mutated NLS fused with hygromycin resistance gene, myc-ffLuc-Neo(CoOp) codon-optimized myc epitope-firefly luciferase-neomycin phosphotransferase fusion, IR/DR SB-inverted/direct repeats, BGH polyadenylation signal from bovine growth hormone, ColE1 replication origin, Kan/R kanamycin-resistance gene, EM7 EM7 promoter derived from T7 promoter, Hygro hygromycin phosphotransferase, SV40 polyA SV40 polyadenylation site.
Cell Lines and Their Propagation
Daudi cells co-expressing β2-microglobulin (Daudiβ2m cells), NALM-6 pre-B cells (American Type Culture Collection, Manassas, VA, USA) were cultured as described previously [17]. CD19-negative NALM-6 cells were generated in our laboratory by CRISPR-Cas9 knockout of the CD19 chromosomal loci. Complete knockout of CD19 was confirmed by flow cytometry analysis of cell surface antigen. Selective in vitro expansion of genetically modified T cells was carried out using a K562-derived aAPC line (clone no. 4) co-expressing CD19, CD64, CD86, CD137L, and a membrane-bound IL-15 (mIL-15; co-expressed with enhanced green fluorescent protein) [5]. TK+ U87 cells were cultured in Dulbecco’s modified Eagle’s medium/nutrient mixture F12 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10 % fetal bovine serum (FBS) [18]. All cell lines were tested for Mycoplasma infection and stored in a research cell bank upon receipt.
Generation of CAR+ T Cells Using the SB System
CD19-specific CAR+ and CAR+TK+ffLuc+ T cells were generated from peripheral blood mononuclear cells (PBMC) using SB transposition as described previously [5]. Briefly, 1 to 2 × 107 mononuclear cells, isolated from blood via Ficoll-Paque density gradient centrifugation (GE Healthcare, Piscataway, NJ, USA), were resuspended in 100 μl of Nucleofector solution (Human T Cell Nucleofector Kit, Lonza, Basel, Switzerland) along with CAR SB transposon (CD19RCD28/pSBSO; 15 μg) and SB11 transposase (pKan-CMV-SB11; 5 μg) supercoiled DNA plasmids, transferred to a single cuvette, and electroporated using a Nucleofector II (Lonza; program U-14) on day 0 of the culture cycle. The cells were allowed to rest for 2 to 3 h at 37 °C in serum-free, phenol red-free RPMI medium (HyClone, South Logan, UT, USA), cultured overnight in phenol-free RPMI medium containing 10 % FBS, and stimulated the next day (day 1) with γ-irradiated (100 Gy) aAPC (clone no. 4) at a T cell/aAPC ratio of 1:2. Additional γ-irradiated aAPC were added every 7 days at a 1:2 ratio. Soluble recombinant human IL-21 (eBioscience, San Diego, CA, USA) was added at a concentration of 30 ng/ml the day after electroporation, and soluble recombinant human IL-2 (Chiron, Emeryville, CA, USA) was added to the cultures at 50 U/μλ beginning 7 days after electroporation. Cytokines were re-added three times a week over the course of the culture. CAR+TK+ffLuc+ T cells were generated independently from three different donors.
Generation of CAR+TK+ffLuc+ T Cells Using the SB System
Triple transposition was achieved by mixing CD19RCD28/pSBSO (7.5 μg), ffluc-neo/pSBSO (7.5 μg), 3[FLAG]-NES-NLSm-TKCoOpHy/pSBSO (7.5 μg), and SB transposase pKan-CMV-SB11 (5 g) supercoiled DNA plasmids in a single cuvette, as shown in Fig. 1b and electroporated and expanded as described above. Hygromycin B (0.2 mg/ml; Invitrogen) and G418 (0.8 mg/ml; Invitrogen) were added to the electroporated cells beginning on day 5 and additional γ-irradiated aAPC were added every 7 days at a 1:2 ratio. Soluble recombinant human IL-21 was added at a concentration of 30 ng/ml the day after electroporation, and soluble recombinant human IL-2 was added to the cultures at 50 U/ml beginning 7 days after electroporation. Hygromycin, G418, and cytokines were re-added three times a week over the course of the culture.
Chromium Release Assay
The cytolytic activity of T cells was examined using a 4-h chromium release assay (CRA) as previously described [19]. Briefly, CD19-specific T cells were incubated with 5 × 103 Cr-51-labeled target cells in a V-bottomed 96-well plate (Corning Life Sciences, Tewksbury, MA, USA). To determine specific cytolysis, intracellular Cr-51 release was measured using a TopCount NXT (Perkin-Elmer, Boston, MA, USA) and the resulting data were reported as the mean ± standard deviation (SD) of three experiments performed each performed in triplicate from different donor T cells.
Ganciclovir-Mediated Ablation of CAR+ TK+ffLuc+ T Cells
T cells expressing TK were evaluated for conditional ablation using ganciclovir (GCV; Sigma-Aldrich, St. Louis, MO, USA). 4 × 105 T cells were co-cultured in triplicate with 8 × 105 γ-irradiated aAPC in a 24-well plate in the absence or presence of graded doses of GCV (0.01–10.00 μM) in a volume of 2 ml/well of complete RPMI medium (HyClone) containing 50 U/ml soluble recombinant human IL-2 and 30 ng/ml soluble recombinant human IL-21. On day 7, viable T cells were counted based on Trypan blue exclusion using Cellometer (Nexcelom Bioscience, Lawrence, MA, USA). The results are expressed as mean percentages of the final cell numbers relative to starting counts derived from triplicate wells.
Flow Cytometry
Approximately 106 T cells in a volume of 100 μl of complete RPMI medium were stained with PE conjugated to F(ab′)2 fragment of goat antibody specific for human Fcγ (Catalog number H10104; Invitrogen) to detect cell surface expression of the CD19-specific CAR. Alternatively, an APC-conjugated anti-idiotype antibody developed in our laboratory (anti-CD19-CAR-APC) targeting CD19-CAR was used to measure CAR expression. Non-specific antibody binding was blocked using FACS wash buffer (2 % FBS in phosphate-buffered saline (PBS)). Surface staining reactions using antibodies against CD3-PerCP-Cy5.5, CD4-PE, CD8-FITC (Catalog numbers 340949, 347327, and 347313; Biosciences, San Jose, CA, USA) and CD19-CAR were performed for 30 min at 4 °C. To measure TK expression, cells were washed, fixed, permeabilized (for 20 min at 4 °C with 100 μl of Cytofix/Cytoperm buffer), and stained with a monoclonal APC-conjugated antibody specific against FLAG (catalog number PJ315; Prozyme, Hayward, CA, USA). Non-specific antibody intracellular binding was blocked by inclusion of 10 % heat-inactivated human serum AB (Gemini Bio-Products) in the FACS wash buffer. Data acquisition was performed with a FACSCalibur (BD Biosciences) using CellQuest (version 3.3; BD Biosciences). Data analysis was performed using the FlowJo software program (version 7.2.2; TreeStar).
Confocal Microscopy
CAR+ and CAR+ TK + ffLuc + T cells were fixed and permeabilized in 100 μl of Cytofix/Cytoperm buffer for 20 min at 4 °C (BD Biosciences) followed by washing in Perm/wash buffer (1×; BD Biosciences, CA) containing 10 % heat-inactivated human serum AB (Gemini Bio-Products, West Sacramento, CA, USA). Cells were then stained with the anti-FLAG APC mAb (PhycoLink) at a 1:200 dilution for 20 min at 4 °C, washed twice in Perm/wash buffer (1×; BD Biosciences, CA) containing 10 % heat-inactivated human serum AB. After washing, the cells were transferred onto slides in 20 μl of Prolong Gold anti-fade agent containing DAPI (Invitrogen) and cover slips were mounted and sealed along the edge. After drying at room temperature, cells were examined under a Leica TCS SP2-SE confocal microscope using oil immersion lens (×63 objective). Single-scan images were acquired in a 1024 × 1024 format with a line averaging of 8 and overlaid.
[18F]FEAU Synthesis
The radiotracer [18F]FEAU was synthesized using previously described methods [12]. The decay-corrected radiochemical yield for [18F]FEAU was 18 % measured at the end of bombardment with a radiochemical purity greater than 99 % and specific activity greater than 2 Ci/μmol (>74 GBq/μmol) assessed at the end of synthesis.
In Vitro Uptake of [18F]FEAU
Radiotracer uptake studies were performed as described previously [18]. Briefly, 107 T cells were centrifuged and resuspended in 6 ml of RPMI medium containing 10 % FBS, [14C]thymidine (0.01 μCi/ml; Moravek, Brea, CA, USA), [3H]FEAU (0.1 μCi/ml; Moravek), and [18F]FEAU (2.5 μCi/ml at time 0). After incubation for 120 min, the cells were pelleted by centrifugation (3000 rpm for 30 s) in a conical 15 ml tube, and a 50-μl aliquot of the supernatant was transferred to a pre-weighed scintillation tube. The remainder of the media was removed, and the pellets were frozen on dry ice transferred into pre-weighed scintillation tubes, and thoroughly resuspended in 0.1 ml of Soluene-350 (Perkin-Elmer, Boston, MA, USA) before adding 1 ml of Insta-Fluor Plus scintillation fluid (Perkin-Elmer). To quantitate [18F]FEAU uptake by T cells, the radioactive gamma emissions of cell pellets and medium samples were measured using a Packard Cobra Quantum gamma counter. Following the decay of F-18 activity, the radioactive beta emissions of the media and cell pellets were measured within precalibrated energy ranges using a Packard Tri-Carb 3100TR scintillation counter to quantitate the accumulation of [3H]FEAU and [14C]thymidine (Thd). The ratios of radioactivity accumulation of the cells to the media were determined as ((disintegrations per minute/g cells)/(disintegrations per minute/g media)) and plotted over time. Linear regression analysis was used to determine the accumulation rate expressed as grams media per gram cells per minute or minutes.
Bioluminescent Imaging of T Cells
All animal studies described were approved by the Institutional Animal Care and Use Committee (IACUC) under internal Animal Care and Use Form (ACUF) 00001131-RN01. All applicable institutional and/or national guidelines for the care and use of animals were followed. BLI was performed using approximately 8-week-old NOD.Cg-PrkdcscidIl2rgtm1wjl/SzJ mice under protocol. T cells (1.5 × 107 cells/100 μl of PBS) were intravenously injected into the mice via the tail vein. Graded doses of T cells (3.75 × 106, 7.50 × 106, and 1.50 × 107 cells/100 μl of PBS) were subcutaneously injected into flanks. BLI was performed using a Xenogen IVIS 200 system 5 min after intraperitoneal injection of D-luciferin (2 mg/kg in PBS; Caliper Life Sciences, Hopkinton, MA, USA). The mice were anesthetized using isoflurane (2 %) in 98 % oxygen and placed in prone positions in the imaging chamber. Images were acquired for 1 and 5 min exposure periods.
PET Imaging of T Cells
Mice were anesthetized as before in a secure prone position. A bolus injection of [18F]FEAU (3.7 MBq in 100 μl of saline) was administered via the tail vein. Following a 2-h wait period, static PET images were acquired on an Inveon PET/computed tomography (CT) system (Siemens, Germany). PET images were reconstructed using ordered subset expectation maximization algorithm. Regional radioactivity concentrations of [18F]FEAU were determined in ROI drawn on the reconstructed images. The radiotracer uptake levels were determined and expressed as mean percent injected dose per gram (%ID/g) ± SD.
Results
SB-Mediated Gene Transfer and Propagation of CAR+TK+ffLuc+ T Cells
We previously used the SB system and aAPC to generate CD19-specific CAR+ T cells and have adapted this approach to gene transfer for human application [6]. To render these T cells detectable by PET and BLI, we developed a new technique dubbed “triple transposition” based on the synchronous electro-transfer of three SB transposons and SB transposase to generate CAR+ T cells that co-expressed a mutein of TK and ffLuc. The TK transgene was modified to enhance direct phosphorylation of [18F]-FEAU and trapping within the cytosol by mutating the NLS and adding a NES to the N-terminus, as described previously [13]. An N-terminal FLAG epitope tag, for detection with a FLAG-specific antibody, and a C-terminal hygromycin phosphotransferase sequence, for in vitro selection, were fused in frame with the modified TK to create the complete transgene referred to as 3[FLAG]-NES-NLSm-TKCoOpHygro. After electroporation, genetically modified T cells were propagated for 4 weeks by co-culture with aAPC (clone no. 4) [4, 20] in the presence of IL-2/IL-21 and cytocidal concentration of hygromycin B as illustrated in Fig. 1c. The electroporated/propagated T cells were assessed for expression of the two transgenes (anti-CD19-CAR (anti-idiotype) and Fc-specific antibody to reveal cell-surface CAR expression and FLAG-specific mAb to reveal intracellular TK expression) and approximately 95 % of the T cells expressed both CAR and TK (Fig. 2a). The majority the gated lymphocyte population (99 %) expressed CD3 of which over 90 % co-expressed CD4 at Day 28. Conversely, a higher proportion of untransduced T cells were CD8+ (68.7 %) versus CD4+ (26.3 %). Confocal microscopy revealed that FLAG-tagged TK was predominantly expressed in the cytoplasm of genetically modified T cells and was not detectable in CAR+TKneg T cells (Fig. 2b).
Fig. 2.

Expression of CAR and TK transgenes. a Genetically modified T cells numerically expanded for 4 weeks on aAPC were analyzed using flow cytometry. Dot plots depict percent expression of CAR (anti-CD19-CAR-APC) versus CD3-PerCP-Cy5.5, CD4-PE versus CD8-FITC, and CAR (anti-Fcγ-PE) versus TK (anti-FLAG-APC) within lymphocyte-gated populations after electro-transfer of DNA plasmids expressing CAR or CAR and TK. Representative data from three different experiments are shown. b Confocal images of genetically modified T cells after staining with DAPI (nucleus, blue) and a monoclonal anti-FLAG mAb (TK, red) indicating the absence of TK in CAR+ T cells and nuclear localization of TK in CAR+TK+ffLuc+ T cells. Scale bars represent 1 μm.
Genetically Modified T Cells Exhibit Redirected Specificity for CD19
CAR+TK+ffLuc+ T cells were assessed by CRA for specific lysis of CD19+ targets. CAR+TK+ffLuc+ T cells demonstrated a significant increase of killing of CD19+ NALM-6 and Daudiβ2m targets. Lysis of CD19neg NALM-6 cells was not detected indicating antigen-specific targeting and lysis by the AR+TK+ffLuc+ T cells. This T cell effector function was not significantly different from that observed with CAR+TKneg T cells consistent with the conclusion that the expression of TK by triple transposition affected neither the specificity nor the cytotoxicity of the genetically modified T cells (Fig. 3a).
Fig. 3.

Redirected specificity of genetically modified T cells and sensitivity to GCV. a Specific lysis of CD19+ NALM-6, CD19neg NALM-6, and CD19+ Daudiβ2m, cells by CAR+TK+ and CAR+TKneg T cells in a 4 h CRA. Lysis of CD19neg NALM-6 cells demonstrates background killing. Results represent average values derived from three different donor T cells. Mean ± SD values for three experiments each performed in triplicate are shown. b Bar graph showing the in vitro sensitivity of T cells to GCV. CAR+TK+ffLuc+ (gray bars) and CAR+TKneg (white bars). Viable T cells were co-cultured on aAPC with graded doses of GCV and counted after 7 days based on Trypan blue exclusion. The results are expressed as mean percentages of the final cell numbers relative to the starting counts. Mean ± SD values for triplicate wells are shown.
Ganciclovir-Mediated Ablation of CAR+TK+ T Cells
Ectopic expression of TK renders the genetically modified T cells susceptible to conditional ablation by GCV. This is desirable to control potential adverse events attributed to the infused T cells. We observed total ablation of CAR+TK+ffLuc+ T cells in the presence of 1 μM GCV with negligible impact on the survival of CAR+TKneg T cells (Fig. 3b). These data reveal that the TK mutein as expressed after triple transposition retains its enzymatic activity and functions to suicide genetically modified upon addition of GCV pro-drug.
Accumulation of Radiotracers In Vitro by CAR+TK+ffLuc+ T Cells
The uptake of radiotracer reflects the catalytic rate of TK and intracellular accumulation in genetically modified T cells. We compared the uptake of [18F]FEAU that was synthesized by us with commercially available [3H]FEAU. CAR+TK+ffLuc+ T cells demonstrated similar rates of [3H]FEAU and [18F]FEAU accumulation, 0.147 and 0.163 min−1, respectively (Fig. 4). The final cells-to-medium accumulation ratios of [3H]FEAU and [18F]FEAU at 120 min were 19.0 and 16.8, respectively. Such ratios are consistent with those observed in HVS1tk-expressing U87 cells, used as positive control for PET in our previous studies [18]. The rate of [14C]thymidine accumulation was 0.22 min−1, yielding a cell:medium accumulation ratio of 26 at 120 min. These values indicate T cell viability (based on the rate of uptake) and proliferation (based on final cells-to-medium ratio) and are consistent with use of viable CAR+TK+ffLuc+ T cells harvested while undergoing expansion on aAPCs. In summary, these data demonstrate that CAR+TK+ffLuc+ T cells retain their proliferative activity and express high levels enzymatically active HSV1tk that is adequate for detection in vivo by PET imaging.
Fig. 4.
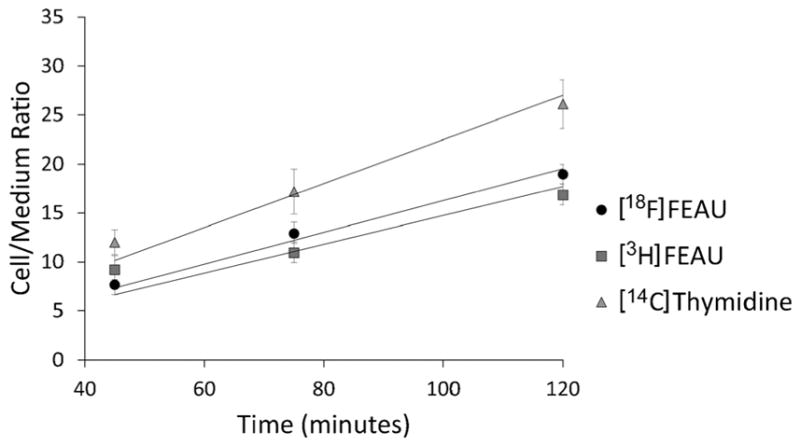
In vitro uptake of radiotracers by CAR+TK+ffLuc+ T cells. Genetically modified T cells exhibit similar uptake of commercial [3H]FEAU and [18F]FEAU. Uptake of thymidine reveals viability of T cells over the course of the assay. Mean ± SD values for triplicate wells are shown.
In Vivo Detection of CAR+TK+ffLuc+ T Cells Using [18F]FEAU PET and Bioluminescence Imaging
To demonstrate that the TK mutein could trap sufficient radiotracer for in vivo imaging, the CAR+TK+ffLuc+ T cells were pre-labeled ex vivo with [18F]FEAU and subsequently injected into mice via the tail vein to be visualized by PET. The cells were first imaged using BLI to confirm the presence of live T cells, followed by PET/CT imaging. The infused T cells were prominently detected in the lungs owing to direct trafficking through the right ventricle and subsequent entrapment in the alveolar microvasculature (Fig. 5a). To determine the sensitivity of PET, we injected subcutaneously graded numbers of genetically modified CAR+TK+ffLuc+ T cells that have been pre-labeled in vitro with [18F]FEAU. The observed PET signal increased proportionately with the number of T cells injected (Fig. 5b). TK+ U87 cells (106 cells/100 μl) served as the positive control for comparison. The radioactivity/cm3 in the region of injected T cells also increased in linear proportion to the number of injected T cells. BLI confirmed the presence of live T cells at the sites of subcutaneous injections. Next, we assessed the ability to detected infused unlabeled CAR+TK+ffLuc+ cells upon in vivo administration of [18F]FEAU. We subcutaneously injected graded numbers of CAR+TK+ffLuc+ T cells in 0.1 mm3 volumes followed by intravenous injection of 3.7 MBq [18F]FEAU 30 min later. The injection sites containing 1.5 × 107 and 7.5 × 106 CAR+TK+ffLuc+ T cells could be reliably detected by PET 2 h later (Fig. 5c). T cell population injection sites coincided with focal points of radiotracer uptake. Non-specific abdominal radiotracer accumulation was observed consistent with urinary clearance (kidneys and bladder) of the radiotracer. Skeletal focal points of radioisotope accumulation were also noted. Skeletal accumulation is plausibly attributed to uptake of free F-18 by bone. Again, BLI confirmed the presence of viable T cells at the injection sites.
Fig. 5.

In vivo PET imaging of CAR+TK+ffLuc+ T cells infused in mice. a T cells pre-labeled ex vivo with [18F]FEAU were intravenously injected and immediately imaged by PET/CT and BLI. The T cells were predominantly located in the lungs owing to direct trafficking via the tail vein to the right ventricle. b Increasing numbers of pre-labeled T cells subcutaneously injected and imaged using PET/CT and BLI. Injected TK+ U87 cells served as positive control for PET imaging. c Contralateral subcutaneous injections of unlabeled CAR+TK+ffLuc+ T cells (15 × 106 and 7.5 × 106 cells) were imaged 2 h after intravenous injection of [18F]FEAU (100 μCi/100 μl saline). T cell population injection sites coincide with focal points of radiotracer uptake. Non-specific abdominal radiotracer accumulation is consistent with urinary clearance (kidneys and bladder) of the radiotracer. Skeletal radioisotope accumulation was also noted and attributed to free fluorine-18 uptake by bone.
Discussion
PET provides a clinically applicable imaging approach using existing infrastructure to non-invasively monitor the efficacy and potency of T cell therapies, as defined by persistence, trafficking, and anti-tumor activity. Longitudinal imaging of infused T cells is rendered feasible by the genetically enforced expression of TK. Previously, we adapted the SB transposon for human application to generate CAR+ T cells for investigational therapy of CD19+ B cell malignancies (IND no. 14193, 14577, and 14739). In the present study, we advanced the SB system using triple transposition to co-express a CD19-specific CAR, mutein of TK derived from HSV-1 and ffLuc to redirect their specificity towards CD19+ tumor cells and enable in vivo imaging in mice using BLI and PET.
Triple transposition appears advantageous for the expression of three transgenes in one T cell since it overcomes limitations on the cargo-load of the SB transposon [21] and facilitates the mixing and matching of genes of interest from a catalog of DNA plasmids to add specific functionalities that enable tumor targeting, selective ablation, and PET imaging. GCV-mediated elimination of infused CAR+TK+ T cells is a practical means of preventing or controlling graft-versus-host disease (GvHD) in patients receiving adoptive T cell therapy and has been demonstrated in mouse models and clinical trial settings [22–24]. However, TK transgene expression is potentially immunogenic in the immunocompetent host rendering infused T cells susceptible to immune-mediated clearance by the recipient’s immune response leading to their premature elimination [24, 25]. Our system provides a means to overcome this limitation, where two CAR+ T cell products that do and do not express TK can be easily generated. Clinical-grade CAR+TK+ T cells can be initially administered to reveal biodistribution of infused cells and be conditionally ablated in the event of toxicity. This can then be followed by CAR+ T cells with the potential for long-term persistence and improved therapeutic potential. This is feasible since the cost to generate a clinical-grade DNA plasmid is approximately 1/10th the cost to manufacture and release recombinant retrovirus used to transduce T cells for clinical trials. Proof of principle was established in this paper showing that SB-generated CAR+TK+ffLuc+ T cells functioned to recognize and ablate CD19+ tumor cells (similar to CAR+TKneg T cells) and could be conditionally ablated with GCV.
Numerical expansion of T cells following electroporation using the Sleeping Beauty system has been well established for the production of clinical-grade CAR+ T cells. This method requires four weekly stimulation cycles with aAPC in the presence of IL-2 and IL-21 and yields stable transgene expression in over 90 % of the T cell population in 28 days [26, 27]. In contrast, virally transduced T cells are typically prepared for infusion in a process that takes approximately 2 weeks, where T cells from apheresis products are stimulated with CD3/CD28 beads, transduced with retro-or lentiviral particles in Retronectin-coated bags, and further expanded for a few days prior to infusion. The yield of CAR+ T cells by viral transduction is, however, lower (<60 %) than that produced by the Sleeping Beauty system (>90 %) [28, 29].
Retrovirus has previously been used to enforce expression of TK transgene and image macaque T cells with [18F]-FEAU [22, 30]. However, the sensitivity of detection in humans is not well established. To improve the ability of TK to enzymatically trap radiotracer within the T cell cytoplasm, we removed the NLS and introduced a NES. After confirming expression of the TK mutein in the cytoplasm we validated our ability to generate [18F]FEAU and the observed cells-to-medium radiotracer accumulation ratio of 16.8 (at 2 h) is comparable with previously reported values in other cell types that were successfully imaged by PET [16, 18, 31].
The sensitivity of PET to image T cells was assessed by two methods. Firstly, genetically modified T cells were preloaded ex vivo with [18F]FEAU to gauge the potential for imaging under circumstances that reveal the minimum number of detectable cells in a defined volume under conditions in which background radioactivity levels is negligible. As anticipated, these pre-labeled CAR+TK+ffLuc+ T cells injected via the tail vein were observed in the lungs. Secondly, we assessed the feasibility of PET to detect CAR+TK+ffLuc+ T cells after they had been administered in vivo. PET after intravenous administration of [18F]FEAU could reliably detect genetically modified T cells, albeit at a much lower limit. Tenfold more T cells were needed for in vivo labeling compared with in vitro-labeled T cells, due to (i) higher body background radioactivity after intravenous administration of [18F]FEAU and (ii) limited cellular access to circulating [18F]FEAU in subcutaneous tissue, which is underperfused compared with tissues such as lung and spleen, resulting in (iii) reduced accumulation of [18F] FEAU in CAR+TK+ffLuc+ T cells administered in vivo. Thus, we expect increased levels of radiotracer uptake by CAR+TK+ffLuc+ T cells in a therapeutic setting, where infused T cells would be expected to traffic to tissues with unhindered access to [18F]FEAU. The sensitivity will further increase if the infused T cells are within a focal point (tumor or draining lymph node) or undergo sustained proliferation.
The sensitivity of PET for detection of CAR+TK+ffLuc+ T cells is a function of expression level of the reporter enzyme, its catalytic activity, stability, and the administered dose of [18F]FEAU. Expression level relies on robust promoter activity to yield consistent levels of protein and multiple stable genomic integrations (transgene copy number) to maximize the reporter gene product levels. Stable expression of HSV1tk in T cells in all the studies described to date was mediated by retroviral transduction methods, which yield an average of three to four integrations per cell compared with an average of one integration per cell in SB-mediated gene transfer. Higher doses of administered [18F]FEAU can enhance the sensitivity of PET for detection of genetically modified T cells expressing lower levels of HSV1tk. However, the dose of [18F]FEAU that can be safely administered in patients is limited by radiation toxicity concerns. For reference, the clinically acceptable intravenous dose of [18F]FHBG was determined as 530 MBq [32] and 254.62 MBq was safely administered in a clinical study [11]. PET imaging of HSV1tk+ T cells has been demonstrated in a variety of mouse immunotherapy models using [18F]FEAU, [18F]FIAU, and [18F]FHBG as radiotracers [13–15]. Homing of these HSV1tk+ T cells was detected qualitatively with foci of radiotracer accumulation at the tumor site. A more quantitative study by Yaghoubi et al. demonstrated clinical PET detection of HSV1tk+ T cells in a patient with glioblastoma multiforme infused intracranially with an accumulative dose of 1 × 109 cytolytic HSV1tk+ T cells over a period of 5 weeks and detected at the tumor site with [18F]FHBG [11]. However, quantitative analysis describing cell number per unit volume of tumor tissue was not performed to reasonably allow for comparison of sensitivity with the SB system. Such a study is warranted to establish the feasibility of SB transposition for the detection of adoptive T cell therapy in a clinical setting.
Conclusions
We adapted the SB transposon/transposase system and harnessed our existing aAPC propagation platform to produce genetically modified T cells using an approach that is readily amenable to clinical translation. This process of triple transposition allows for stable expression of three functional transgenes to redirect T cell specificity, render them susceptible to conditional ablation, and facilitate their imaging by BLI and PET. Imaging with [18F]FEAU appears to be a candidate approach to examine the long-term biodistribution and persistence of infused genetically modified T cells. Such a non-invasive imaging approach is critical for assessing the efficacy of adoptive transfer of CAR+ T cells in humans.
Acknowledgments
The authors would like to thank Dr. Carl June (University of Pennsylvania) for help in generating and providing aAPC clone no. 4 and Dr. Perry Hackett (University of Minnesota) for help with the SB system. Grant support includes R01 (CA124782, CA120956, CA141303), R33 (CA116127), P01 (CA148600), Albert J Ward Foundation, Burroughs Wellcome Fund, Gillson Longenbaugh Foundation, Cancer Prevention and Research Institute of Texas, CLL Global Research Foundation, National Foundation for Cancer Research, and Pediatric Cancer Research Foundation.
Abbreviations
- [18F]-FEAU
2′-deoxy-2′-[18F]fluoro-5-ethyl-1-β-D-arabinofuranosyl-uracil
- [18F]-FHBG
9-(4-[18F]fluoro-3-hydroxymethylbutyl)guanine
- aAPC
Artificial presenting cells, K562-derived
- BLI
Bioluminescent imaging
- CAR
Chimeric antigen receptor
- CoOp
Codon optimized
- CRA
Chromium release assay
- CT
Computed tomography
- GCV
Ganciclovir
- HSCT
Hematopoietic stem-cell transplantation
- HSV-1
Herpes simplex virus-1
- Hygro
Hygromycin phosphotransferase
- mIL-15
Membrane-bound interleukin-15
- Neo
Neomycin phosphotransferase
- NES
Nuclear export signal
- NLS
Nuclear localization sequence
- NLSm
Nuclear localization sequence, mutated
- PET
Positron emission tomography
- PBMC
Peripheral blood mononuclear cells
- SB
Sleeping Beauty
- TAA
Tumor-associated antigen
- [14C]-Thd
[14C]thymidine
- TK
Thymidine kinase, herpes simplex virus type-1
Footnotes
Compliance with Ethical Standards
Conflict of Interest
The technology described in this publication was advanced through research conducted at the University of Texas MD Anderson Cancer Center (MD Anderson) by Laurence Cooper. In January 2015, the technology was licensed by MD Anderson for commercial application to ZIOPHARM Oncology, Inc., and Intrexon Corporation, in exchange for equity interests in each of these companies. Laurence Cooper, Amer M. Najjar, Pallavi R. Manuri, Simon Olivares, Tiejuan Mi, Helen Huls, Richard E. Champlin, Brian Rabinovich, and Dean A. Lee are eligible in accordance with the Rules of Board of Regents of The University of Texas System to share in the proceeds of the disposition of the equity received by MD Anderson as a result of the licensing of this technology. On May 7, 2015, Dr. Cooper was appointed as the Chief Executive Officer at ZIOPHARM. Dr. Cooper is now a Visiting Scientist at MD Anderson where he continues to help supervise the development of this technology. The research being reported in this publication is research in which The University of Texas MD Anderson Cancer Center has an institutional financial conflict of interest. Because MD Anderson is committed to the protection of human subjects and the effective management of its financial conflicts of interest in relation to its research activities, MD Anderson has implemented an Institutional Conflict of Interest Management and Monitoring Plan to manage and monitor the conflict of interest with respect to MD Anderson’ s conduct of this research.
References
- 1.Jena B, Dotti G, Cooper LJ. Redirecting T-cell specificity by introducing a tumor-specific chimeric antigen receptor. Blood. 2010;116:1035–1044. doi: 10.1182/blood-2010-01-043737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cooper LJ, Jena B, Bollard CM. Good T cells for bad B cells. Blood. 2012;119:2700–2702. doi: 10.1182/blood-2011-12-398719. [DOI] [PubMed] [Google Scholar]
- 3.Kowolik CM, Topp MS, Gonzalez S, et al. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006;66:10995–11004. doi: 10.1158/0008-5472.CAN-06-0160. [DOI] [PubMed] [Google Scholar]
- 4.Singh H, Manuri PR, Olivares S, et al. Redirecting specificity of T-cell populations for CD19 using the Sleeping Beauty system. Cancer Res. 2008;68:2961–2971. doi: 10.1158/0008-5472.CAN-07-5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh H, Figliola MJ, Dawson MJ, et al. Reprogramming CD19-specific T cells with IL-21 signaling can improve adoptive immunotherapy of B-lineage malignancies. Cancer Res. 2011;71:3516–3527. doi: 10.1158/0008-5472.CAN-10-3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kebriaei P, Huls H, Jena B, et al. Infusing CD19-directed T cells to augment disease control in patients undergoing autologous hematopoietic stem-cell transplantation for advanced B-lymphoid malignancies. Hum Gene Ther. 2012;23:444–450. doi: 10.1089/hum.2011.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bollard CM, Aguilar L, Straathof KC, et al. Cytotoxic T lymphocyte therapy for Epstein-Barr virus + Hodgkin’s disease. J Exp Med. 2004;200:1623–1633. doi: 10.1084/jem.20040890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Savoldo B, Goss JA, Hammer MM, et al. Treatment of solid organ transplant recipients with autologous Epstein Barr virus-specific cytotoxic T lymphocytes (CTLs) Blood. 2006;108:2942–2949. doi: 10.1182/blood-2006-05-021782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Science Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ray P, Tsien R, Gambhir SS. Construction and validation of improved triple fusion reporter gene vectors for molecular imaging of living subjects. Cancer Res. 2007;67:3085–3093. doi: 10.1158/0008-5472.CAN-06-2402. [DOI] [PubMed] [Google Scholar]
- 11.Yaghoubi SS, Jensen MC, Satyamurthy N, et al. Noninvasive detection of therapeutic cytolytic T cells with 18F-FHBG PET in a patient with glioma. Nature Clin Practice. 2009;6:53–58. doi: 10.1038/ncponc1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soghomonyan S, Hajitou A, Rangel R, et al. Molecular PET imaging of HSV1-tk reporter gene expression using [18F]FEAU. Nature Protocols. 2007;2:416–423. doi: 10.1038/nprot.2007.49. [DOI] [PubMed] [Google Scholar]
- 13.Ponomarev V, Doubrovin M, Serganova I, et al. Cytoplasmically retargeted HSV1-tk/GFP reporter gene mutants for optimization of noninvasive molecular-genetic imaging. Neoplasia. 2003;5:245–254. doi: 10.1016/S1476-5586(03)80056-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dubey P, Su H, Adonai N, et al. Quantitative imaging of the T cell antitumor response by positron-emission tomography. Proce Natl Acad Sci (USA) 2003;100:1232–1237. doi: 10.1073/pnas.0337418100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koehne G, Doubrovin M, Doubrovina E, et al. Serial in vivo imaging of the targeted migration of human HSV-TK-transduced antigen-specific lymphocytes. Nature Biotech. 2003;21:405–413. doi: 10.1038/nbt805. [DOI] [PubMed] [Google Scholar]
- 16.Dotti G, Tian M, Savoldo B, et al. Repetitive noninvasive monitoring of HSV1-tk-expressing T cells intravenously infused into nonhuman primates using positron emission tomography and computed tomography with 18F-FEAU. Mol Imaging. 2009;8:230–237. [PMC free article] [PubMed] [Google Scholar]
- 17.Serrano LM, Pfeiffer T, Olivares S, et al. Differentiation of naive cord-blood T cells into CD19-specific cytolytic effectors for posttrans-plantation adoptive immunotherapy. Blood. 2006;107:2643–2652. doi: 10.1182/blood-2005-09-3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Najjar AM, Nishii R, Maxwell DS, et al. Molecular-genetic PET imaging using an HSV1-tk mutant reporter gene with enhanced specificity to acycloguanosine nucleoside analogs. J Nucl Med. 2009;50:409–416. doi: 10.2967/jnumed.108.058735. [DOI] [PubMed] [Google Scholar]
- 19.Manuri PV, Wilson MH, Maiti SN, et al. piggyBac transposon/transposase system to generate CD19-specific T cells for the treatment of B-lineage malignancies. Hum Gene Ther. 2010;21:427–437. doi: 10.1089/hum.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davies JK, Singh H, Huls H, et al. Combining CD19 redirection and alloanergization to generate tumor-specific human T cells for allogeneic cell therapy of B-cell malignancies. Cancer research. 2010;70:3915–3924. doi: 10.1158/0008-5472.CAN-09-3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karsi A, Moav B, Hackett P, Liu Z. Effects of insert size on transposition efficiency of the sleeping beauty transposon in mouse cells. Marine Biotech. 2001;3:241–245. doi: 10.1007/s101260000072. [DOI] [PubMed] [Google Scholar]
- 22.Kornblau SM, Stiouf I, Snell V, et al. Preemptive control of graft-versus-host disease in a murine allogeneic transplant model using retrovirally transduced murine suicidal lymphocytes. Cancer Res. 2001;61:3355–3360. [PubMed] [Google Scholar]
- 23.Kornblau SM, Aycox PG, Stephens C, McCue LD, Champlin RE, Marini FC. Control of graft-versus-host disease with maintenance of the graft-versus-leukemia effect in a murine allogeneic transplant model using retrovirally transduced murine suicidal lymphocytes. Exp Hematol. 2007;35:842–853. doi: 10.1016/j.exphem.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Traversari C, Marktel S, Magnani Z, et al. The potential immunogenicity of the TK suicide gene does not prevent full clinical benefit associated with the use of TK-transduced donor lymphocytes in HSCT for hematologic malignancies. Blood. 2007;109:4708–4715. doi: 10.1182/blood-2006-04-015230. [DOI] [PubMed] [Google Scholar]
- 25.Berger C, Flowers ME, Warren EH, Riddell SR. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood. 2006;107:2294–2302. doi: 10.1182/blood-2005-08-3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huls MH, Figliola MJ, Dawson MJ, et al. Clinical application of Sleeping Beauty and artificial antigen presenting cells to genetically modify T cells from peripheral and umbilical cord blood. J Vis Exp: JoVE. 2013:e50070. doi: 10.3791/50070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh H, Huls H, Kebriaei P, Cooper LJ. A new approach to gene therapy using Sleeping Beauty to genetically modify clinical-grade T cells to target CD19. Immunol Rev. 2014;257:181–190. doi: 10.1111/imr.12137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hollyman D, Stefanski J, Przybylowski M, et al. Manufacturing validation of biologically functional T cells targeted to CD19 antigen for autologous adoptive cell therapy. J Immunother. 2009;32:169–180. doi: 10.1097/CJI.0b013e318194a6e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tumaini B, Lee DW, Lin T, et al. Simplified process for the production of anti-CD19-CAR-engineered T cells. Cytotherapy. 2013;15:1406–1415. doi: 10.1016/j.jcyt.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marini FC, 3rd, Kornblau SM. Production and culture of HSVtk transduced suicidal lymphocytes induces variable changes in the lymphocyte subset composition. Bone Marrow Transpl. 1999;23:907–915. doi: 10.1038/sj.bmt.1701738. [DOI] [PubMed] [Google Scholar]
- 31.Singh H, Najjar AM, Olivares S, et al. PET imaging of T cells derived from umbilical cord blood. Leukemia. 2009;23:620–622. doi: 10.1038/leu.2008.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yaghoubi S, Barrio JR, Dahlbom M, et al. Human pharmacokinetic and dosimetry studies of [(18)F]FHBG: a reporter probe for imaging herpes simplex virus type-1 thymidine kinase reporter gene expression. J Nucl Med. 2001;42:1225–1234. [PubMed] [Google Scholar]