
Figure 3. Transcriptome-wide RNA structure probing technologies.
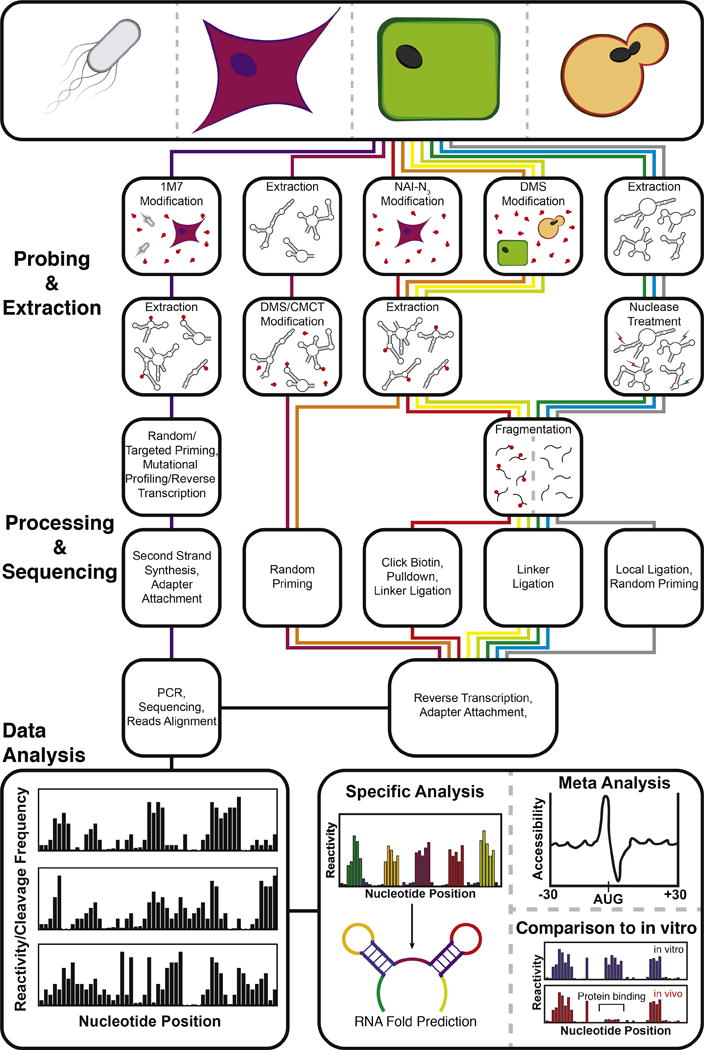
Transcriptome-wide RNA structure probing uses chemical probing or enzymatic cleavage to introduce covalent modifications or directly cleave RNA in a structure dependent fashion, respectively. Modification or cleavage positions are detected through processing steps followed by next-generation sequencing. Bioinformatic processing of the resulting sequencing reads yields a measure of chemical modification ‘reactivity’ or enzymatic cleavage frequency at each nucleotide. High reactivities correspond to flexible nucleotide positions that are not participating in RNA structures or bound by cellular factors. High enzymatic cleavage frequencies give information on structure depending on the characteristics of the nucleases used. These values can be used for several types of specific analyses, such as constrained RNA folding, averaging meta-analysis of reactivities across the entire transcriptome, and comparisons with in vitro probing data. An outline of the steps for SHAPE-Map [40] and in-cell SHAPE-Seq [47] (purple) CIRS-seq [38] (maroon), icSHAPE [39] (red), structure-seq [36] (orange), DMS-seq [35] (yellow), Mod-seq [37] (light green), PARS [30] (green), FragSeq [29] (blue), and RPL [41] (grey), is shown. Further technique details can be found in Table 1.