
Fig. 1.
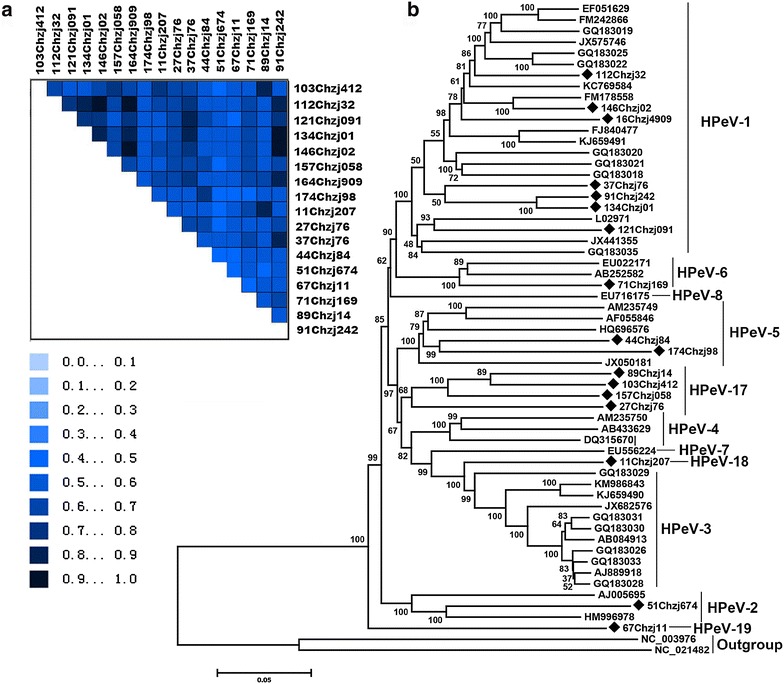
Sequence divergence and phylogenetic analysis based on the complete genome of HPeVs in the present study. a Nucleotide sequence divergence of the 17 HPeVs in this study was established using AliGROOVE software. The scores were ranging from 0 to +1 indicating the non-random similarity. The darker blue indicated the higher non-randomized accordancy between pairwise sequence comparisons. Strain names in this study were listed on top and the right hand side of the matrix; b neighbor joining phylogenetic tree was constructed with Mega5.0 from multiple alignments of the complete genome of the 17 HPeVs in the present study and other 47 HPeVs with complete genome sequence available in GenBank. Two Ljungan virus strains were included as outgroup. Bootstrap values are indicated at the nodes. The scale bar indicates the number of substitutions per position for a unit branch length. Viruses identified in the present study were displayed with diamonds