

Abstract
Type I interferons (IFN) are crucial mediators of human innate and adaptive immunity and are massively produced from plasmacytoid dendritic cells (pDC). IRF7 is a critical regulator of type I IFN production when pathogens are detected by Toll-like receptor (TLR) 7/9 in pDC. However, hyperactivation of pDC can cause life-threatening autoimmune diseases. To avoid the deleterious effects of aberrant pDC activation, tight regulation of IRF7 is required. Nonetheless, the detailed mechanisms of how IRF7 transcription is regulated in pDC are still elusive. MYC is a well-known highly pleiotropic transcription factor however the role of MYC in pDC function is not well defined yet. To identify the role of transcription factor MYC in human pDC, we employed a knockdown technique using human pDC cell line, GEN2.2. When we knocked down MYC in the pDC cell line, production of IFN-stimulated genes was dramatically increased and was further enhanced by the TLR9 agonist CpGB. Interestingly, MYC is shown to be recruited to the IRF7 promoter region through interaction with NCOR2/HDAC3 for its repression. In addition, activation of TLR9-mediated NF-kB and MAPK and nuclear translocation of IRF7 were greatly enhanced by MYC depletion. Pharmaceutical inhibition of MYC recovered IRF7 expression, further confirming the negative role of MYC in the antiviral response by pDC. Therefore, our results identify the novel immunomodulatory role of MYC in human pDC and may add to our understanding of aberrant pDC function in cancer and autoimmune disease.
Keywords: MYC (c-Myc), interferon, interferon regulatory factor (IRF), plasmacytoid dendritic cell, Toll-like receptor (TLR)
INTRODUCTION
pDC is a specialized type I IFN-producing immune cell that senses viral RNA and DNA through endosomal TLR7 and TLR9, respectively (1–3). Type I IFNs, mainly IFNα and IFNβ are pleiotropic cytokines that have the most potent antiviral and anti-tumoral activity in humans (4–6). IFNs are released by host cells for communication between immune cells to trigger the protective defenses of the host and help eradicate pathogens. However, uncontrolled type I IFN production has been implicated in several human autoimmune diseases, such as systemic lupus erythematosus (SLE) (7, 8), rheumatoid arthritis (RA) (9, 10), inflammatory bowel disease (IBD) (11) or psoriasis (12). To avoid the deleterious effects of aberrant pDC activation, tight regulation of type I IFN production in pDC is essential. Importantly, production of these type I IFNs in pDCs are mainly controlled by IRF7 (13). Regulation of IRF7 is complex and possesses many positive and negative feedback mechanisms. Transcriptional, posttranscriptional, translational, posttranslational and epigenetic (acetylation and methylation) regulation of IRF7 have been extensively studied (14–21). Nonetheless, detailed mechanisms of how IRF7 transcription and activation are directly regulated in pDC are still unclear.
MYC is a well-known highly pleiotropic transcription factor. MYC is a basic helix-loop-helix leucine zipper (bHLH) transcription factor (22) and specifically binds to the DNA sequence 5′-CACGTG-3′ or 5′-CAGCTG-3′ known as an E-box motif to activate its targets (23). The N-terminal transactivation domain of MYC is essential for its biological function and directly interacts with components of basal transcription machinery or chromatin remodeling (the SWI/SNF chromatin-remodeling complex and histone acetyltransferase (HAT)) (24–26). Additionally, several recent findings suggested that MYC represses its target genes by interfering with transcriptional activators (27) or by recruiting the corepressor complexes including histone deacetylases (HDAC) (28, 29) or DNA methyltransferase (30). Many studies have also revealed that MYC induces its oncogenic activity through transcriptional repression (22, 31). Interestingly, MYC activation is reported shown to downregulate many genes involved in the NF-κB response and STAT1, the central player in type I and II IFN signaling via direct inhibition of transcription in Burkitt lymphoma cells (32–34). Less, however, is known about how MYC regulates pDC function, and the direct target genes of MYC in pDC have not been identified.
Here, we describe a previously unrecognized function of MYC in the TLR-mediated negative regulation of antiviral immune response of pDCs. We demonstrate that transcriptional repression of IRF7 by MYC provides an additional layer of tight regulation of IRF7 that ensures the appropriate level of type I IFN production and prevents the development of autoimmune disease. Combination studies have identified the IRF7 gene as a critical target of MYC, although not its only target. Our results characterize the novel immunomodulatory role of transcription factor MYC in human pDCs and may add to our understanding of aberrant pDC function in cancer and autoimmune disease.
MATERIALS AND METHODS
Reagents and antibodies
CpGA (ODN2116), CpGB (ODN2006) and R848 were purchased from Invivogen. The following antibodies were used for immunoblotting: anti-MYC, anti-IRF7, and anti-IRAK1 (Santa Cruz Biotech); anti-TLR7 (Abcam); anti-MyD88 and anti-TLR9 (eBioscience); anti-STAT1, anti-NCOR2, anti-HDAC1, anti-HDAC2, anti-HDAC3, anti-HDAC4, anti-HDAC6, anti-HDAC7, anti-pIKKα/β, anti-pIκBα, anti-IκBα, anti-pJNK, anti-pp38, anti-TBK1, anti-pTBK1 and anti-pERK (Cell Signaling Technologies); and anti-GAPDH (Sigma).
NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce) was used to investigate nuclear translocation of IRF7 as per the manufacturer’s instructions. MYC inhibitor (c-Myc Inhibitor II - CAS 413611-93-5) was purchased from CalBiochem. HDAC inhibitors, valproic acid (VPA) was purchased from Invivogen.
Online data access
The microarray dataset described in this manuscript is deposited in the NCBI Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo, GEO Series accession number GSE70276). The ChIP-Seq dataset described in this manuscript is deposited in GEO (GSE70275).
Statistical analysis
Data are presented as mean values ± one standard deviation. p-values were calculated using an unpaired two-tailed Student’s t-test. P values <0.05 were considered statistically significant.
Cell lines and tissue cultures
pDCs were isolated from healthy donors by sorting as lineage-negative (CD3, CD14, CD15, CD16, CD19, CD20, CD25, CD56, and CD11c) cells using a pDC isolation kit (Miltenyi Biotec) to more than 98% purity. Purified pDC were cultured in medium (RPMI 1640, Invitrogen) supplemented with 10% fetal calf serum (FCS) and IL-3 (10ng/ml, R&D systems). GEN2.2 cells were kindly provided by Dr. Joël Plumas (Université Joseph Fourier, Grenoble, France) and cultured as described (35). Briefly, GEN2.2 cells were cultured in GlutaMax-RPMI (GIBCO) supplemented with 10% FBS (Atlanta), MEM-nonessential amino acid solution (GIBCO), sodium pyruvate and gentamycin. HEK293T cells were purchased from American Type Culture Collection (ATCC) and cultured in DMEM (GIBCO) supplemented with 10% FBS (HyClone), L-glutamine, penicillin/streptomycin, and sodium pyruvate (GIBCO).
RNA extraction, processing and statistical analysis for microarray
RNA isolation was performed using Qiagen RNeasy mini kit according to the manufacturer’s instructions. Amplified RNA was hybridized to Illumina HT-12 V4 beadchips (47,231 probes) and scanned on an Illumina Beadstation 500. Illumina’s GenomeStudio version 2011.1 with the Gene Expression Module v1.9.0 (Illumina, San Diego, CA) was used to generate signal-intensity values. For the analysis of NC vs MYC in GEN2.2 cells, ANOVA was used for differentially expressed genes (DEGs) analysis for CpGB post-treatment and DEGs were identified by Welch T-test p<0.05 with Benjamini-Hochberg correction and two-fold normalized difference to untreated controls (509 DEGs).
Knockdown in GEN2.2 cells
Transfection of GEN2.2 cells was performed as described (36). Briefly, GEN2.2 cells were nucleofected with nucleofector kit V (Amaxa) with 0.4 nmol of siRNA for each reaction of 3–5 × 106 cells. siMYC, siHDAC3 and siTBK1 were purchased from Dharmacon. The program number for electroporation was A033. The non-targeting control (NC) is a non-targeting pool from Dharmacon.
Quantitative PCR analysis
For qRT-PCR experiments, total RNA was isolated from cells with Qiagen RNeasy mini kit and reverse transcribed into cDNA using iScript cDNA synthesis kit (Bio-Rad). qRT-PCR for IL6, IL23A, TNFα, IL8, IFNα4, IFNβ, CXCL10, IRF7, MYC, PLSCR1, IFIT1, TLR7, TLR9, IFNa14, CCND1, p21, TLR7, TBK1 and TLR9 was performed with specific primers using the instructions from the manufacturer (SyBr Green mastermix, Bio-Rad). Primer sequences are available upon request.
Measurement of cytokine production from GEN2.2 cells using ELISA
GEN2.2 cells were stimulated with the indicated concentrations of CpGA, CpGB or R848 for 4 h (to detect TNFα, IL-6 and IL-8) or for 12 h (to detect IFNα and IFNβ). Concentrations of IFNα, INF-β, TNFα, IL-6 and IL-8 in the culture supernatants were measured by ELISA (PBL Biomedical Laboratories and R&D Systems).
ChIP-seq and ChIP-PCR
ChIP assays were performed using Chromatin Immunoprecipitation (ChIP) Assay Kit according to the manufacturer’s instructions (Upstate Biotechnologies). Briefly, unstimulated GEN2.2 cells were cross linked with 1% of formaldehyde for 10 min at 37°C, sonicated and subjected to immunoprecipitation with 2 μg of anti-MYC antibody (Santacruz Biotechnology, N262X), or rabbit IgG control. After reverse cross-linking and recovering the chromatin, the isolated chromatin was subjected to library preparation using NEXTflex Rapid DNA-Seq Kit (BIOO Scientific) according to manufacturer’s instruction with the following exceptions: 1) a bead size selection was performed before the PCR amplification and 2) the libraries were size-selected between 200–300 bp by 8% PAGE before sequencing. ChIP-seq libraries were sequenced 1×50 on a HiSeq 2500 (Illumina). The resulted sequences were aligned to the hg19 reference genome using Bowtie2. Downstream analysis was performed by the HOMER next-gen sequencing analytical suite (http://homer.salk.edu/homer/index.html) (37). Peak finding was performed with the “findPeaks” command line with the parameters of “-style factor” and “-i input”. Input library was generated from the sonicated chromatin of GEN2.2 cells before the chromatin immunoprecipitation of MYC. The command line, “annotatePeaks.pl”, was used for peak annotation and finding the MYC motif occurrences in peaks.
Immunoprecipitated DNA was quantified using RT-PCR and qRT-PCR by comparison to input sample signals. The signals were normalized to 1% total chromatin input of the anti-MYC or the control IgG sample. The results were expressed as fold difference between the two samples. Fold enrichment was determined from triplicate PCR reactions at promoter regions of IRF7.
ChIP assays for human primary pDCs were performed as described as above. 10×106 human primary pDCs were pooled from 3 different healthy donors for one ChIP experiments. Immunoprecipitated DNA was quantified using qRT-PCR by comparing to input sample signals. The signals were normalized to 1% total chromatin input of the anti-MYC, anti-NCOR2, anti-HDAC3 or the control IgG sample. The results were expressed as fold difference between the two samples. Fold enrichment was determined from triplicate PCR reactions at promoter regions of IRF7.
Luciferase assay
Cells transfected with the indicated plasmid were harvested and subjected to luciferase reporter assay using a dual luciferase assay according to the manufacturer’s instructions (Promega).
IP and IP-MASS using nuclear extracts
MYC-binding protein complexes purified from GEN2.2 cells were lysed in lysis buffer (50 mM Tris-HCl pH 7.4, 250 mM NaCl, 1 mM EDTA, 1% TRITON-100, 10% glycerol supplemented with a complete protease inhibitor and phosphatase inhibitor cocktail (Sigma)) and subjected to ultracentrifugation. Cleared lysates were incubated overnight with protein G agarose beads (Pierce) with the indicated antibodies. Beads were washed extensively with lysis buffer, separated on a 4–20% gradient polyacrylamide gel, and stained with Coomassie blue (Sigma). Unstimulated GEN2.2 cells were lysed. Nuclear extracts were fractionated as shown in Figure 4A and then immunoprecipitated with MYC specific antibody and rabbit IgG control. Then, immune complexes from MYC-IP were separated on a 4–20% Gradient SDS-PAGE gel (Thermo Scientific) and stained with Coomassie Blue for visualization. All bands were in–gel digested with trypsin, and the proteins were identified by nanoflow LC-MS/MS analysis with a nano-LC1000 (Thermo Scientific) coupled to a Thermo ORbitrap Velos™ (Thermo Scientific) mass spectrometer. Acquired MS spectra were processed by BioWorks software to convert data into peptide and protein composition information. All bands were analyzed by liquid chromatography-MS at the Proteomics Center, Baylor College of Medicine, Houston, TX.
Figure 4. Pharmaceutical inhibition of MYC enhanced IRF7 expression and type I IFN production in pDC.
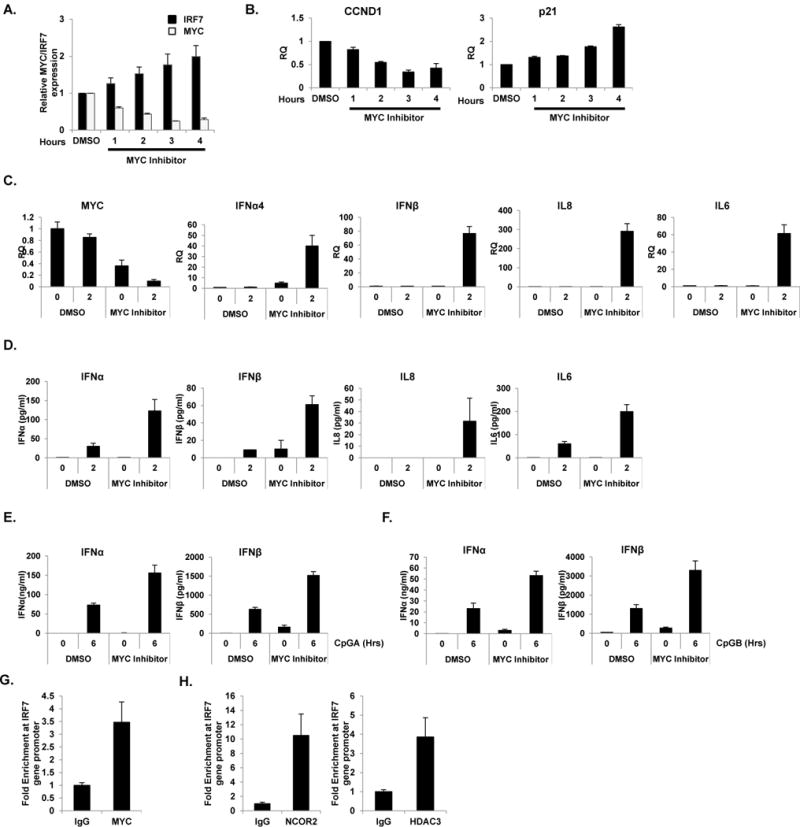
Pharmaceutical inhibition of MYC (MYC inhibitor) represses MYC and enhances IRF7 expression. (A) GEN2.2 cells were treated with 100 μM of MYC inhibitor (c-Myc Inhibitor II, CAS 413611-93-5, CalBiochem) and their mRNA was measured for MYC and IRF7. Data are representative of three independent experiments (average of triplicates ± standard deviation). (B) Effect of pharmaceutical inhibition of MYC was tested for CCND1. p21 was used as a control. Data are representative of three independent experiments (average of triplicates ± standard deviation). (C–D) Pharmaceutical inhibition of MYC enhanced CpGB-mediated type I IFN production as shown in the level of IFNα4, IFNβ, IL8 and IL6 in mRNA (C) and protein levels (D). Data are representative of three independent experiments (average of triplicates ± standard deviation). (E–F) Human primary pDC from six different healthy donors were pre-treated with 1μM MYC inhibitor for 0.5 h followed by stimulation with CpGA (1 μM, E) and CpGB (0.5 μM, F) for 6 h. IFNα and IFNβ production was measured by ELISA. Data are representative of six independent experiments (average of triplicates ± standard deviation). (G–H) Pools of 10×106 human primary pDC from ten different healthy donors stimulated with CpGB (0.5 μM) for 12 h were cross-linked and immunoprecipitated with anti-MYC (G) antibody, anti-NCOR2 and anti-HDAC3 (H) antibodies or rabbit IgG control. Amplification of the IRF7 promoter was visualized using qRT-PCR by comparing to input sample signals (1% of total crude). The levels of IRF7 promoter in MYC-bound or control IgG-bound DNA were normalized to input and expressed as fold difference between the two samples. Data are representative of three independent experiments (average of triplicates ± standard deviation)
RESULTS
MYC ablation in pDC cell lines revealed an inhibitory effect of MYC in antiviral immune response
To study the effects of MYC in the transcriptional signature of human pDC, MYC was ablated in the pDC cell line GEN2.2. GEN2.2 displays a similar phenotype and function to human primary pDCs (35). Knockdown experiments were performed in GEN2.2 cells to deplete Myc mRNA by Nucleofection and their mRNA levels were compared to that of non-specific siRNA control (NC). The knockdown efficiency of Myc mRNA and MYC protein was evaluated by qRT-PCR and by western blot analysis, respectively (Figure 1A and B). Then, MYC-ablated GEN2.2 cells were treated with CpGB (0, 2 and 4 h) for pDC maturation and activation. Cell lysates were hybridized on Illumina human beadchips, and analysis (Benjamini-Hochberg correction with p-value: 0.01) revealed a total of 509 DEG in Myc siRNA-transfected cells after CpGB treatment compared to untreated cells (Figure 1C and Table S1). We observed significantly increased transcription of ISGs (IFIT1, IFIT2, IFIT3, OAS1, OASL, ISG20, etc.) in MYC-knockdown pDCs compared to NC, indicating that MYC functions as a negative regulator of the IFN signaling pathway. Moreover, stimulation of MYC-depleted pDCs with CpGB significantly increased the transcription of pre-existing subset of ISGs and additional ISGs in a time-dependent manner (Figure 1C, marked with a dashed line). Moreover, the genes that were significantly upregulated at every time point in MYC-ablated cells (False Discovery Rate (FDR) 0.01, fold change >= 2) are represented as a gene symbol/time point connectivity network using Cytoscape 3.1.1 (38) (Figure 1D). It is noteworthy that most of the upregulated genes are ISGs, including genes that were upregulated at 0 and 2 h (IFI27, IFIT1, MX2, OAS1, OAS2, and STAT1) and ones that were upregulated at 0, 2 and 4 h (IRF7, IFI6, and IFI35). In addition, type I IFNs such as IFNA2, IFNA4, IFNA10, IFNA13, and IFNA14 were stimulated at 2 to 4 h post-treatment. The induced expression of these ISGs was validated by qRT–PCR and western blot analysis (Figure 1E and F). Subsequently, type I IFN protein and pro-inflammatory cytokine productions were measured using IFNα, IFNβ, IL6 and TNFα ELISA assay (Figure 1G) and greatly enhanced type I IFN and cytokine productions were observed when MYC was ablated in the pDC cell line.
Figure 1. MYC-depletion potentiates type I IFN production and signaling in pDC cell line.
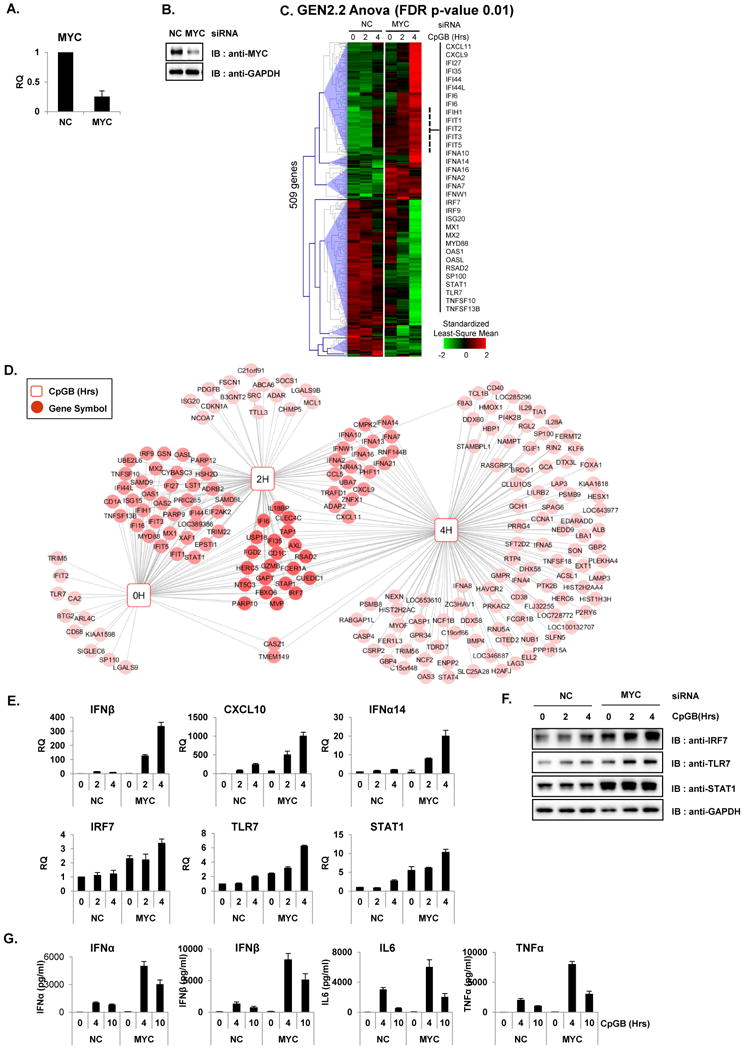
(A–B) GEN2.2 cells were nucleofected with siRNA against non-specific control (NC) or MYC and then analyzed for knockdown efficiency by qRT-PCR (A) and by western blot analysis (B). (C–E) GEN2.2 cell with siRNA against NC or MYC were treated with CpGB prior to hybridizing on Illumina HT-12 V4 beadchips. (C) Hierarchical clustering (Euclidian) of 509 DEGs that changed in MYC-ablated GEN2.2 cells vs. control at 0, 2 or 4 h after CpGB treatment (Benjamini-Hochberg correction p-value 0.01). The IFN-stimulated genes are highlighted in the dashed line, and representative genes are listed. Data are representative of at least three independent experiments. (D) Cytoscape network displaying the differentially upregulate genes (FDR 0.01 and FC greater than 2) in MYC-depleted pDC cells connected to the time point after CpGB treatment. Genes that were up-regulated at more than one time point were connected to a multiple time point hub and are represented as a darker color. (E–F) Microarray results shown in C were confirmed with qRT-PCR (E) and western blot analysis (F). mRNA levels of IFNβ, CXCL10, IFNα14, IRF7, TLR7 and STAT1 in MYC- and NC-siRNA nucleofected GEN2.2 cells are represented at the time points after stimulation with CpGB . Antibodies against IRF7, STAT1, TLR7 and GAPDH were used for western blot analysis. Data are representative of two independent experiments (average of triplicates ± standard deviation). (G) Enhanced proinflammatory cytokine production in MYC-depleted GEN2.2 cells. GEN2.2 cells treated with siRNA against MYC or NC for 48 h were further stimulated with CpGB or a control for the indicated time. IFNβ production was measured by ELISA using the supernatant from each experiment. All PCR results were normalized to the GAPDH and are presented as relative-fold changes with respect to NC siRNA-nucleofected GEN2.2 cells. Data are representative of three independent experiments (average of triplicates ± standard deviation).
MYC directly occupies IRF7 promoter to repress its expression
MYC is a well-known transcription factor that regulates the expression of many genes either as an enhancer or repressor by binding to consensus sequences (E-boxes). To identify direct targets of MYC, we performed chromatin immunoprecipitation/DNA sequencing (ChIP-Seq) in the GEN2.2 cell line. The ChIP-Seq result revealed that 46.2% of MYC-bound motif have enriched in E-box motif (5′-CACGTG-3′, p-value; 1e-847) as shown in Homer de novo motif analysis (Figure 2A). Intersection of the gene list from microarray (Figure 1A), ChIP-Seq results, and a promoter analysis using SABiosciences’ proprietary database DECODE (DECipherment Of DNA Elements) revealed 123 potential transcriptional targets of MYC, including IFITM3, IRF7, IRF9, ISG15, ISG20, SOCS1 and STAT1 (Figure 2B and Table S2). Interestingly, IRF7 was one of the most significantly enriched compared to non-specific input in ChIP-Seq. IRF7 is the key regulator of type I IFN production in pDC and contains E-box motif in the promoter region (5′-CACGTG-3′) (39). The occupancy of MYC in the IRF7 promoter region was confirmed by qRT-PCR using specific primers against the IRF7 promoter (Figure 2C and D). Occupancy of MYC on IRF7 promoter region was further confirmed with ChIP after knockdown of MYC (Figure 2E). As expected, in the absence of MYC (MYC KD), MYC occupancy on IRF7 promoter was dramatically reduced (Figure 2 F). Next, to validate these results, we conducted a luciferase reporter assay using the IRF7 promoter conjugated with luciferase. 293T cells stably expressing human TLR9 (Invivogen) were transiently transfected with an IRF7 promoter construct and siRNA against MYC. Interestingly, IRF7 promoter-Luc activity was significantly induced when MYC was ablated, indicating that MYC occupies the IRF7 promoter and represses its transcription (Figure 2G). The increase of IRF7 promoter activity in the absence of MYC was further augmented when stimulated with CpGB. These results demonstrate direct interactions between MYC and the IRF7 promoter region. Therefore, we hypothesized that MYC regulates pDC antiviral immune response through its transcriptional repression of IRF7.
Figure 2. IRF7 is a direct transcriptional target of MYC in pDC.
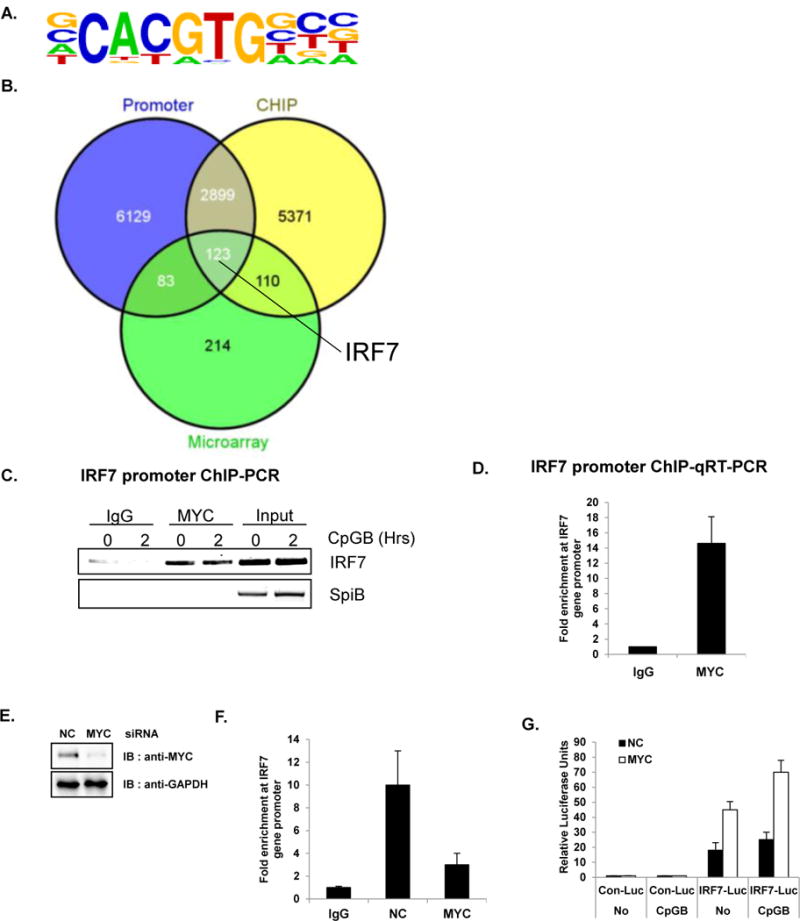
Direct target genes of MYC were identified using ChIP-Seq. (A–D) GEN2.2 (unstimulated or CpGB-treated) cells were cross-linked and immunoprecipitated with anti-MYC antibody or rabbit IgG control. In parallel, cross-linked input (without antibody) was put aside for the comparison. The bound DNAs were sequenced using HiSeq 2000 (Illumina) (A) The most highly enriched motif (46.2% of Targets, with p-value 1 e-847) were analyzed and presented using Homer de novo motif analysis (B) The Venn diagram shows the intersections of ChIP-Seq, promoter analysis, and microarray. Putative direct target genes of MYC (123 genes listed in Table S2) include IRF7. Venn diagrams were constructed using Venny. (C) The immunoprecipitated chromatin was isolated and used for semiquantitative RT-PCR with IRF7 promoter primers. MYC directly binds to the promoter region of IRF7. SpiB, another important transcription factor for pDC development was used as a negative control. Data are representative of three independent experiments. (D) Amplification of the IRF7 promoter was visualized using qRT-PCR by comparing to input sample signals (1% of total crude). The levels of IRF7 promoter in MYC-bound or control IgG-bound DNA were normalized to input and expressed as fold difference between the two samples. Data are representative of three independent experiments (average of triplicates ± standard deviation) (E) GEN2.2 cells were nucleofected with siRNA against NC or MYC and then analyzed for knockdown efficiency by western blot analysis. (F) GEN2.2 cells nucleofected with siRNA against NC or MYC were cross-linked and immunoprecipitated with anti-MYC antibody or rabbit IgG control. Amplification of the IRF7 promoter was visualized using qRT-PCR by comparing to input sample signals (1% of total crude). The levels of IRF7 promoter in MYC-bound or control IgG-bound DNA were normalized to input and expressed as fold difference between the two samples. Data are representative of three independent experiments (average of triplicates ± standard deviation) (G) Depletion of MYC showed an enhanced basal promoter activity of IRF7. HEK 293T cells stably expressing TLR9 were transiently co-transfected with luciferase reporter plasmid conjugated with control or human IRF7 promoter and transfected with MYC or NC siRNA. Twenty-four hours after transfection, cells were further stimulated with or without CpGB. Dual luciferase assays were performed 8 h after CpGB stimulation. All luciferase activity was normalized to the expression of the Renilla luciferase activity. The results were visualized as a fold of induction over the unstimulated- NC siRNA transfected GEN2.2 cells. Data are representative of three independent experiments (average of triplicates ± standard deviation)
MYC, NCOR2 and HDAC3 form a repressor complex to downregulate IRF7 expression through histone deacetylation
It is well established that MYC can inhibit the expression of its target genes through the recruitment of corepressors such as HDACs or histone methyl transferases (30, 40). In order to understand the molecular mechanisms by which MYC represses IRF7, we performed mass spectrometry-based immunoprecipitation analysis (IP-MASS) using a specific antibody against MYC. As seen in Figure 3A, MYC predominantly exists in the nuclear fraction of GEN2.2 where a nuclear fraction marker HDAC1 exists. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a control to show the purity of cytoplasmic and nuclear fractions. To purify the MYC-bound protein complexes, nuclear extracts from GEN2.2 were prepared, and the polypeptides bound to the MYC antibody or control IgG were separated by gradient polyacrylamide gel electrophoresis and visualized by Coomassie staining (Figure 3B). Several polypeptide bands were observed distinctly in the nuclear extracts pulled down with MYC antibody but not with control IgG. The polypeptide bands specific for anti-MYC pulldown were excised from the gel and analyzed by liquid chromatography mass spectrometry (LC-MS). Among many interesting candidates that were identified by IP-MASS to interact with MYC, those proteins with chromosome remodeling activities are listed in Table 1. Nuclear receptor co-repressor 2 (NCOR2; shown at 274 kDa in MYC pulldowns) was especially interesting because this transcriptional corepressor is known to recruit histone deacetylase to specific DNA promoter regions (41, 42). The interaction between NCOR2 and MYC was further confirmed by independent co-IP experiments using nuclear extracts from GEN2.2 (Figure 3C). There have been several reports suggesting possible interactions of MYC with HDAC3 or with NCOR2/HDAC3 (42–45). Thus, we hypothesized that MYC and/or NCOR2 recruit (or form a multi-protein complex with) specific HDAC(s) to the promoter region of their target genes and repress their expression. Therefore, another co-IP experiment was performed to see if specific HDAC(s) could interact with MYC. As shown in Figure 3D, HDAC3 could be specifically recruited to the MYC immune complexes, revealing that MYC and HDAC3 physically and endogenously assemble. Additionally, physical interaction of three protein MYC/NCOR2/HDAC3 was confirmed by co-IP for NCOR2 (Figure 3E). These findings further emphasize the epigenetic regulation of IRF7, especially supporting chromatin deacetylation by a MYC/NCOR2/HDAC3 complex to regulate the expression of IRF7 protein. To determine if chromatin structure plays a critical role in the regulation of IRF7, GEN2.2 cells were treated with HDAC inhibitor, valproic acid (VPA). The expression of IRF7 was significantly increased after the time course of VPA treatment compared to that of DMSO-treated GEN2.2 (Figure 3F), suggesting an essential role of epigenetic regulation when MYC/NCOR2/HDAC3 represses IRF7 expression in pDC. Importantly, consistent with our hypothesis that the MYC/NCOR2/HDAC3 ternary complex inhibits IRF7 transcription in pDC, the knockdown of HDAC3 in GEN2.2 cells showed a significantly higher level of IRF7 protein expression (Figure 3G) and type I IFN (IFNα and β) production (Figure 3H and I, measurements of mRNA and protein, respectively). Next, the effect of TLR9 ligand stimulation on the MYC/NCOR2/HDAC3 ternary complex formation was tested using co-IP experiments with MYC specific antibody followed by CpGB stimulation on GEN2.2 cells (Figure 3 J). CpGB stimulation did not affect and/or alter the interaction of ternary complex. These data directly suggest that HDAC3 contributes to MYC-dependent repression of IRF7 transcription and demonstrate that epigenetic changes on the IRF7 promoter by HDAC3 might serve as a mechanism to fine tune the hyper-activation of pDC.
Figure 3. MYC/NCOR2/HDAC3 complex repress type I IFN production and signaling in pDC.
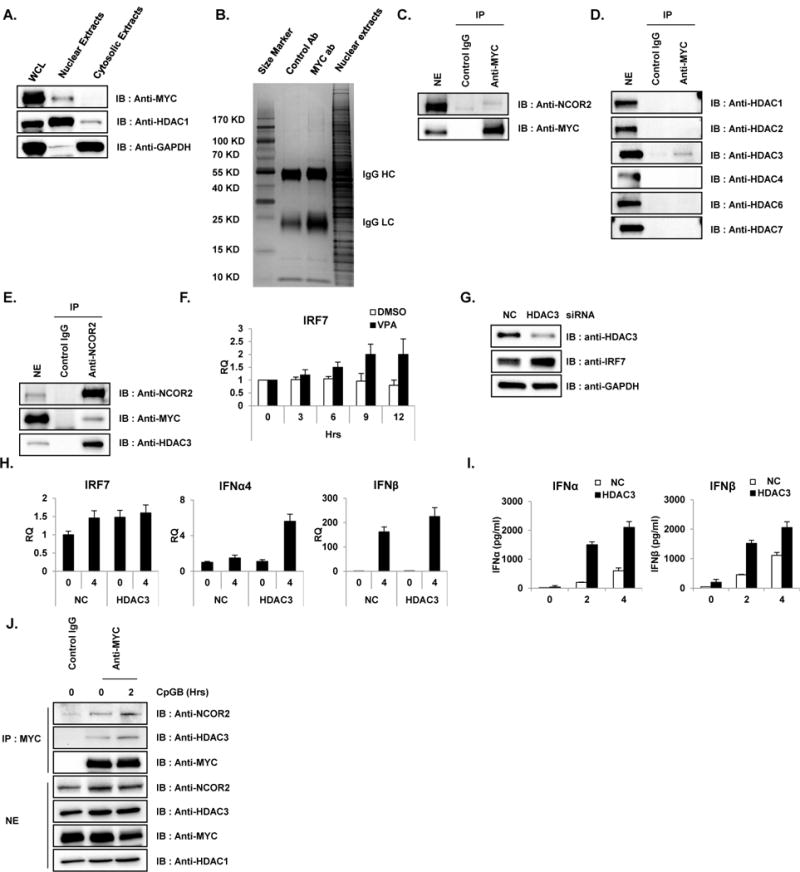
(A) Nuclear fraction of pDC cell line revealed the nuclear localization of MYC. Cytosolic and nuclear fractions from unstimulated GEN2.2 cells were immunoblotted with antibody against MYC, HDAC1 and GAPDH. HDAC1 and GAPDH were used as nuclear and cytosolic markers, respectively. Data are representative of three independent experiments. (B) MYC-bound protein complexes were identified by IP-mass analysis. Unstimulated GEN2.2 cells were lysed and nuclear extracts were fractionated as in A and immunoprecipitated with MYC-specific antibody and rabbit IgG control. Then, immune complexes from MYC- IP were separated on a 4–20% Gradient SDS-PAGE gel (Thermo Scientific) and stained with Coomassie Blue (Sigma). All bands were in-gel digested with trypsin, and proteins were identified by nanoflow LC-MS/MS analysis with a nano-LC1000 (Thermo Scientific) coupled to Thermo ORbitrap Velos™ (Thermo Scientific) mass spectrometer. Acquired MS spectra were processed by BioWorks software to convert data into peptide and protein composition information. (C) Co-immunoprecipitation experiments showed direct interaction of MYC and NCOR2 in unstimulated pDC cell lines. Unstimulated GEN2.2 cells were fractionated for nuclear extracts and co-IPed with anti-MYC antibody and immunoblotted with anti-NCOR2. IP with IgG was used as a control. Data are representative of three independent experiments. (D) MYC interacts with HDAC3. Co-IP experiments showed direct interaction of MYC and HDAC3 in unstimulated pDC cell lines. Unstimulated GEN2.2 cells were fractionated for nuclear extracts, co-IPed with anti-MYC antibody and immunoblotted with anti-HDACs. IP with IgG was used as a control. Data are representative of three independent experiments. (E) Reciprocal co-IP showing endogenous binding of NCOR2, HDAC3 and MYC. Nuclear extracts of GEN2.2 cells were subjected to IP with anti-NCOR2 antibody followed by western blot analysis for NCOR2, MYC and HDAC3. Data are representative of three independent experiments. (F) Pharmaceutical inhibition of HDACs recovered IRF7 expression. GEN2.2 cells were treated with 100 μM of HDAC inhibitor, valproic acid (VPA) and mRNA was measured for IRF7. Primers specific for GAPDH mRNA were used to normalize samples. These data are representative of three independent experiments performed in duplicate. (G) HDAC3 depletion enhanced IRF7 expression in GEN2.2 cells. Cell lysates from GEN2.2 cells transfected with control and HDAC3 siRNAs were analyzed by western blot analysis with antibodies against IRF7, HDAC3 and GAPDH. Data are representative of three independent experiments. (H) HDAC3 depletion enhanced IRF7 expression in GEN2.2 cells. GEN2.2 cells were transfected with control and HDAC3 siRNAs for 48 h, followed by stimulation with CpGB (0.5 μM) for 4 h. IRF7, IFNα4 and IFNβ mRNA were analyzed by qRT-PCR. Primers specific for GAPDH mRNA were used to normalize samples. Data are representative of three independent experiments (average of triplicates ± standard deviation). (I) HDAC3 depletion enhanced type I IFN expression in GEN2.2 cells. A human pDC cell line was transfected with siRNAs against HDAC3 for 48 h followed by stimulation with CpGB (0.5 μM) for 4 h in 96-well culture plates. IFNα and IFNβ production was measured by ELISA. These data are representative of three independent experiments performed in duplicate. (J) MYC interacts with NCOR2 and HDAC3 in GEN2.2 cells stimulated with CpGB. Co-IP experiments showed direct interaction of MYC, NCOR2 and HDAC3 in GEN2.2 cells unstimulated and stimulated with CpGB (0.5 μM) for 2 h. GEN2.2 cells were fractionated for nuclear extracts, co-IPed with anti-MYC antibody and immunoblotted with anti-HDAC3 and NCOR2. IP with IgG was used as a control. Data are representative of three independent experiments.
Table 1.
Selected Putative MYC interacting proteins from IP-MASS
| NCBI gi no. | Protein Name | Size (Kda) |
|---|---|---|
| 331284178 | nuclear receptor corepressor 2 isoform 1 | 2525 |
| 192807320 | transcription activator BRG1 isoform F | 1647 |
| 21237808 | SWI/SNF complex subunit SMARCC2 isoform b | 1214 |
| 325651836 | SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A member 5 | 1052 |
| 156523968 | poly [ADP-ribose] polymerase 1 | 1014 |
| 5032179 | transcription intermediary factor 1-beta | 835 |
| 14141170 | metastasis-associated protein MTA2 | 668 |
| 106879206 | bifunctional lysine-specific demethylase and histidyl-hydroxylase NO66 | 641 |
| 223556010 | protein arginine N-methyltransferase 3 isoform 3 | 361 |
Pharmaceutical inhibition of MYC reveals the inhibitory function of MYC on IRF7 expression
To assess the therapeutic potential of reducing type I IFN production in pDC by blocking MYC, we employed a pharmaceutical inhibitor of MYC (c-Myc Inhibitor II, CalBiochem). Interestingly, MYC inhibition caused by the pharmaceutical inhibitor dramatically induced IRF7 mRNA expression (Figure 4A). As a control, CyclinD1 (CCND1, which is known to be positively regulated by MYC) and p21 (negatively regulated by MYC) are differently regulated by the MYC inhibitor as expected (Figure 4B and (46)). The MYC inhibitor induced IFNα/β and proinflammatory cytokine IL8 and IL6 (Figure 4C and D, measurements of mRNA and protein, respectively), further supporting the negative role of MYC in pDC function.
We next sought to investigate if pharmaceutical inhibition of MYC in GEN2.2 pDC cell line could be represented in human primary pDC. To this end, we measured type I IFN productions after pharmaceutical inhibition of MYC using the same inhibitor. Since survival of isolated primary pDC is strictly IL-3-dependent (47), we treated primary pDC with IL-3 to promote viability and activation while treating with MYC inhibitor. The dose of MYC inhibitor was titrated and optimized for primary pDC (data not shown). In agreement with an observation in pDC cell line, type I IFN (IFNα/β) productions were increased when MYC was inhibited in human primary pDC after CpGA and CpGB treatment (Figure 4E and F, respectively). These data describe a molecular basis for the therapeutic activity of anti-MYC agents.
Next, binding of MYC on IRF promoter region shown in pDC cell line GEN2.2 were further validated in human primary pDC by ChIP. The occupancy of MYC in the IRF7 promoter region was confirmed by qRT-PCR using primers against the IRF7 promoter (Figure 4G). Consistent with an observation in GEN2.2 cells, IRF7 promoter was occupied by MYC after CpGB treatment. Moreover, NCOR2- and HDAC3-ChIP showed similar results as MYC-ChIP in human primary pDC further supporting our hypothesis (Figure 4H).
MYC inhibits TLR9-mediated NF-κB/MAPK activation as well as IRF7 nuclear translocation in pDC
Activation of TLR9 in pDC stimulates a complex network of signal transduction pathways (48). To pursue the hypothesis that MYC plays a central role in TLR9-mediated proinflammatory cytokine/chemokine production in pDCs, the impact of depletion of MYC on TLR9 downstream signaling pathways was assessed. Interestingly, consistent with the robust production of type I IFN and proinflammatory cytokines/chemokines (Figure 1C–E), silencing of MYC expression greatly enhances TLR9-mediated IKKα/β phosphorylation and IκBα phosphorylation and degradation, hallmarks of substantial NF-κB activation (Figure 5A). Moreover, mitogen-activated protein kinases (MAPKs), including c-Jun N-terminal kinases (JNK), p38 mitogen-activated protein kinases (p38) and extracellular signal-regulated kinases (ERK) are shown to be activated as evidenced by their phosphorylation status in MYC-depleted GEN2.2 cells.
Figure 5. MYC-depletion enhances TLR9 downstream signaling.
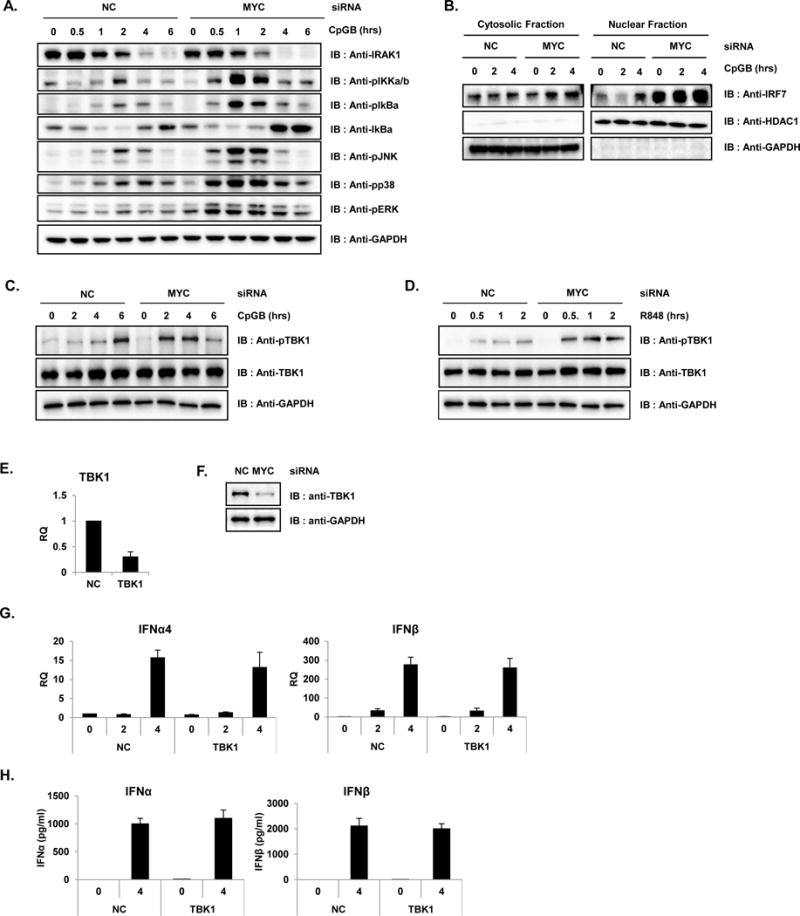
(A) GEN2.2 cells nucelofected with either NC or MYC-specific siRNA was analyzed for TLR9 downstream signaling pathway. Cell lysates from siRNA-transfected GEN2.2 cells, with or without 0.5 μM of CpGB for the indicated times were analyzed by western blot analysis with antibodies against IRAK1, phospho-IKKα/β, phospho-IκBα, IκBα, phospho-JNK, phospho-p38, phospho-ERK and GAPDH. All results are representative of three independent experiments. (B) MYC-depletion enhances TLR9-mediated IRF7 nuclear translocation. Nuclear fractions from GEN2.2 cells transfected with NC and MYC siRNAs for 48 h were stimulated with or without 0.5 μM of CpGB for the indicated times and immunoblotted with anti-IRF7. Anti-HDAC1 and anti-GAPDH were used as nuclear and cytosolic markers, respectively. All results are representative of three independent experiments. (C) MYC-depletion enhances TLR9-mediated TBK1 activation. Cell lysates from NC and MYC siRNA-transfected GEN2.2 cells, with or without 0.5 μM of CpGB for the indicated times were analyzed by western blot analysis with antibodies against pTBK1, TBK1 and GAPDH. All results are representative of three independent experiments. (D) MYC-depletion enhances TLR7-mediated TBK1 activation. Cell lysates from NC and MYC siRNA-transfected GEN2.2 cells, with or without 1 μg/ml of R848 for the indicated times were analyzed by western blot analysis with antibodies against pTBK1, TBK1 and GAPDH. All results are representative of three independent experiments. (E–F) GEN2.2 cells were nucleofected with siRNA against NC or TBK1 and then analyzed for knockdown efficiency by qRT-PCR (E) and by western blot analysis (F). (G) mRNA levels of IFNα4 and IFNβ in TBK1- and NC-siRNA nucleofected GEN2.2 cells are represented at the time points after stimulation with CpGB. Data are representative of two independent experiments (average of triplicates ± standard deviation). (H) GEN2.2 cells treated with siRNA against TBK1 or NC for 48 h were further stimulated with CpGB or a control for the indicated time. IFNα and IFNβ production was measured by ELISA using the supernatant from each experiment. Data are representative of two independent experiments (average of triplicates ± standard deviation).
IRF7 translocation to the nucleus from the cytoplasm after viral infection is the prerequisite for robust type I IFN production in pDC (49, 50). Therefore, to investigate the requirement of MYC for CpG-triggered signaling that culminates in the activation of IRF7, we monitored nuclear localization of IRF7 in GEN2.2 cells after MYC-knockdown (Figure 5B). Interestingly, IRF7 nuclear translocation was greatly induced by knockdown of MYC and further enhanced by CpGB treatment whereas NC had no effect. This result further supports the inhibitory role of MYC in nuclear translocation of IRF7 and resulting type I IFN production in pDC. Taken together, these data further support that MYC directly occupies the promoter region of IRF7 to repress the type I IFN production in pDC.
TBK1 has been implicated in TLR-mediated IRF3 phosphorylation/activation in many tissues by cell- and ligand-specific manner (51, 52). However, the role of TBK1 in IRF7 activation in pDC has been poorly studied. To test whether TBK1 plays any roles in the IFN production in pDCs, expression of TBK1 was measured in Western blot analysis in MYC knockdown cells. Since human pDC predominantly expresses TLR7 and TLR9 (53–55), TLR7- and TLR9- specific ligands (CpGB and R848, respectively) were treated after MYC knockdown. Interestingly, the phosphorylation of TBK1, a hallmark of TBK1 activation was enhanced in the absence of MYC (Figure 5 C and D). We then knocked down TBK1 in pDC cell line (Figure 5E and F), however, TLR7/9-mediated type I IFN production showed no difference compared to NC siRNA indicating that TBK1 may not have a massive role in TLR7/9-mediated type I IFN production (Figure 5 G, H and data not shown).
DISCUSSION
pDC comprise a unique cell type that is dedicated to the production of type I IFN (IFN-α/β) in response to viruses and bacteria. In the present study, we identified a novel central regulatory node that controls maintenance of the pDC response and IFN secretion: MYC. Ablation of MYC in pDC cell lines (GEN2.2) resulted in an enhanced antiviral immune response with remarkable ISG production, including IRF7, a key player in pDC function. ChIP-seq analysis identified that MYC directly bound at the promoter region of IRF7.
While both CpGA and B are known to induce the secretion of IFNα, their downstream genes and mechanism somewhat differ from each other (56). CpGA up-regulates genes that are associated with metabolic functions but CpGB uniquely up-regulates genes that support antibacterial responses such as the NF-kB-dependent pathway. It has been suggested that MYC suppresses NF-kB and its target genes (32–34). Consistent with previous reports, known, direct targets of NF-κB transcription factor were upregulated in MYC knockdown cells in our study, however, detailed mechanism of how MYC suppresses NF-κB activation cannot be fully explained with our present study because of the complexity of NF-κB system and the possibility of many positive or negative feedback regulation. Therefore, further in-depth study is warranted. We utilized CpGB to address global downstream pathways of antiviral activity in pDC. Consistent with the increased production of type I IFN and pro-inflammatory cytokine/chemokine in MYC-depleted cells, activation of TLR9-mediated NF-kB and MAPK as well as IRF7 nuclear translocation were greatly enhanced with MYC depletion. Accordingly, a MYC inhibitor dramatically induced IRF7, IFNα/β and IL-8 expression, further highlighting the regulatory role of MYC in antiviral innate immune responses in pDC. It is important to note that like IRF7, other crucial transcription factors for type I IFN production such as STAT1 and IRF9, which form the ISGF3 complex with STAT2 (57), are also transcriptionally repressed by MYC. Additionally, two important ISGs, ISG15 and ISG20 are also shown as direct targets of MYC. ISG15 is the first ISG identified and is a key regulator of ISGylation, a newly characterized ubiquitinylation-like posttranslational modification in immune responses (58). ISG20 is another important ISG which inhibits RNA and DNA virus replications through its exonuclease activity (59, 60). Moreover, ablation of MYC increased the stability of mRNA and protein expression of IRF7 and STAT1 (unpublished observation). This suggests that multiple layers of type I IFN production pathways are regulated by MYC to ensure the appropriate activation upon viral and bacterial challenge.
Treatment of pDCs with an HDAC inhibitor induced expression of IRF7, indicating the possible role of chromosome remodeling activity of HDACs in controlling the antiviral function of pDCs. HDAC3 is interesting because it appears to be a core component of nuclear receptor co-repressor complexes with NCOR1 and NCOR2 (43). We found that MYC is recruited to the IRF7 promoter with NCOR2 and HDAC3 in pDC, where it represses IRF7 transcription and subsequent type I IFN expression. This is the first mechanism reported, to our knowledge, that the formation of this ternary complex that could possibly repress IRF7 expression and HDAC3 might regulate and induce the epigenetic change of the IRF7 promoter in human pDC. It has been recently established that MYC also controls long non-coding RNAs (lncRNAs) as well as microRNAs, which represent two classes of important non-coding RNAs in eukaryotes (61–63). These non-coding RNAs have fundamental roles in development, immunity and tumorigenesis (64, 65). Therefore, it would be interesting to look at the epigenetic regulation of non-coding RNAs in the transcriptional regulation of pDC by MYC.
IRF7 and its target genes have been implicated as strong inhibitors of metastasis of certain cancers such as breast cancer (66). Several studies suggested that IRF7 could be a fundamental molecule for type I IFN-based anti-tumor therapies (67–69), however 70% of cancers has aberrant expression of MYC (70–72). Therefore, we could hypothesize that the balance between type I IFN production and MYC may be a potential indicator of cancer and autoimmune disease. In fact, very recent research from the international multisite SLE cohort study revealed that breast, endometrial and ovarian cancer risks were significantly decreased in an SLE group compared to a non-SLE group (73). These cancers are sex hormone-dependent cancers that affect only women. However, SLE is also known to predominantly affect women of childbearing age and above. As such, we suspect that the inter-relationship between MYC/IRF7 plays a role in tumorigenesis of these types of cancers in SLE patients. It is intriguing that rates of sex hormone-independent cancers were not significantly altered in SLE patients. Moreover, it is well-established that type I IFN-based anticancer therapies often resulted in exacerbated phenotypes especially for those with an autoimmune disease such as SLE or RA (74, 75).
In summary, our findings highlight a new physiological role for MYC and identify MYC as an important brake for uncontrollable type I IFN production in pDC. Since excessive expression of type I IFN initiates autoimmune disease development and type I IFN is the most potent mediator of anti-tumor activity, we propose that new approaches to manipulate MYC expression in pDC for early therapeutic intervention in autoimmune disease as well as potential anti-cancer therapeutics might be promising.
Supplementary Material
Acknowledgments
We acknowledge Harrod Carson for editing the manuscript.
This work was supported by a grant from the National Institutes of Health awarded to YJL (R01AI097348-03).
Footnotes
Authorship Contributions
T.W. Kim designed research, performed experiments, analyzed data and wrote paper. S. Hong designed research, analyzed data and wrote paper. Y. Lin designed research, analyzed data and wrote paper. H. Joo and E. Murat performed experiments. T. Kim designed research and wrote paper. V. Pascual and Y. Liu designed and supervised research.
Conflict of Interest Disclosures
The authors have no conflicting financial interests.
References
- 1.Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, Colonna M. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nature medicine. 1999;5:919–923. doi: 10.1038/11360. [DOI] [PubMed] [Google Scholar]
- 2.Gilliet M, Cao W, Liu YJ. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nature reviews Immunology. 2008;8:594–606. doi: 10.1038/nri2358. [DOI] [PubMed] [Google Scholar]
- 3.Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, Antonenko S, Liu YJ. The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999;284:1835–1837. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- 4.Biron CA. Interferons alpha and beta as immune regulators–a new look. Immunity. 2001;14:661–664. doi: 10.1016/s1074-7613(01)00154-6. [DOI] [PubMed] [Google Scholar]
- 5.Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity. 2006;25:373–381. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 6.Pestka S. Interferon: a decade of accomplishments, foundations of the future in research and therapy. Seminars in hematology. 1986;23:27–37. [PubMed] [Google Scholar]
- 7.Obermoser G, Pascual V. The interferon-alpha signature of systemic lupus erythematosus. Lupus. 2010;19:1012–1019. doi: 10.1177/0961203310371161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ivashkiv LB. Type I interferon modulation of cellular responses to cytokines and infectious pathogens: potential role in SLE pathogenesis. Autoimmunity. 2003;36:473–479. doi: 10.1080/08916930310001605882. [DOI] [PubMed] [Google Scholar]
- 9.Thurlings RM, Boumans M, Tekstra J, van Roon JA, Vos K, van Westing DM, van Baarsen LG, Bos C, Kirou KA, Gerlag DM, Crow MK, Bijlsma JW, Verweij CL, Tak PP. Relationship between the type I interferon signature and the response to rituximab in rheumatoid arthritis patients. Arthritis and rheumatism. 2010;62:3607–3614. doi: 10.1002/art.27702. [DOI] [PubMed] [Google Scholar]
- 10.Ioannou Y, Isenberg DA. Current evidence for the induction of autoimmune rheumatic manifestations by cytokine therapy. Arthritis and rheumatism. 2000;43:1431–1442. doi: 10.1002/1529-0131(200007)43:7<1431::AID-ANR3>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 11.Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nature reviews Immunology. 2012;12:125–135. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lew W, Bowcock AM, Krueger JG. Psoriasis vulgaris: cutaneous lymphoid tissue supports T-cell activation and “Type 1” inflammatory gene expression. Trends in immunology. 2004;25:295–305. doi: 10.1016/j.it.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 13.Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, Taya C, Taniguchi T. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature. 2005;434:1035–1040. doi: 10.1038/nature03547. [DOI] [PubMed] [Google Scholar]
- 14.Hemmi H, Takeuchi O, Sato S, Yamamoto M, Kaisho T, Sanjo H, Kawai T, Hoshino K, Takeda K, Akira S. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. The Journal of experimental medicine. 2004;199:1641–1650. doi: 10.1084/jem.20040520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uematsu S, Sato S, Yamamoto M, Hirotani T, Kato H, Takeshita F, Matsuda M, Coban C, Ishii KJ, Kawai T, Takeuchi O, Akira S. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-{alpha} induction. The Journal of experimental medicine. 2005;201:915–923. doi: 10.1084/jem.20042372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Litvak V, Ratushny AV, Lampano AE, Schmitz F, Huang AC, Raman A, Rust AG, Bergthaler A, Aitchison JD, Aderem A. A FOXO3-IRF7 gene regulatory circuit limits inflammatory sequelae of antiviral responses. Nature. 2012;490:421–425. doi: 10.1038/nature11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu R, Au WC, Yeow WS, Hageman N, Pitha PM. Regulation of the promoter activity of interferon regulatory factor-7 gene. Activation by interferon snd silencing by hypermethylation. The Journal of biological chemistry. 2000;275:31805–31812. doi: 10.1074/jbc.M005288200. [DOI] [PubMed] [Google Scholar]
- 18.Li Y, Dai J, Song M, Fitzgerald-Bocarsly P, Kiledjian M. Dcp2 decapping protein modulates mRNA stability of the critical interferon regulatory factor (IRF) IRF-7. Molecular and cellular biology. 2012;32:1164–1172. doi: 10.1128/MCB.06328-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee MS, Kim B, Oh GT, Kim YJ. OASL1 inhibits translation of the type I interferon-regulating transcription factor IRF7. Nature immunology. 2013;14:346–355. doi: 10.1038/ni.2535. [DOI] [PubMed] [Google Scholar]
- 20.Colina R, Costa-Mattioli M, Dowling RJ, Jaramillo M, Tai LH, Breitbach CJ, Martineau Y, Larsson O, Rong L, Svitkin YV, Makrigiannis AP, Bell JC, Sonenberg N. Translational control of the innate immune response through IRF-7. Nature. 2008;452:323–328. doi: 10.1038/nature06730. [DOI] [PubMed] [Google Scholar]
- 21.Prakash A, Levy DE. Regulation of IRF7 through cell type-specific protein stability. Biochemical and biophysical research communications. 2006;342:50–56. doi: 10.1016/j.bbrc.2006.01.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nature reviews Molecular cell biology. 2005;6:635–645. doi: 10.1038/nrm1703. [DOI] [PubMed] [Google Scholar]
- 23.Blackwell TK, Kretzner L, Blackwood EM, Eisenman RN, Weintraub H. Sequence-specific DNA binding by the c-Myc protein. Science. 1990;250:1149–1151. doi: 10.1126/science.2251503. [DOI] [PubMed] [Google Scholar]
- 24.Cole MD, McMahon SB. The Myc oncoprotein: a critical evaluation of transactivation and target gene regulation. Oncogene. 1999;18:2916–2924. doi: 10.1038/sj.onc.1202748. [DOI] [PubMed] [Google Scholar]
- 25.Cheng SW, Davies KP, Yung E, Beltran RJ, Yu J, Kalpana GV. c-MYC interacts with INI1/hSNF5 and requires the SWI/SNF complex for transactivation function. Nature genetics. 1999;22:102–105. doi: 10.1038/8811. [DOI] [PubMed] [Google Scholar]
- 26.Frank SR, Schroeder M, Fernandez P, Taubert S, Amati B. Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes & development. 2001;15:2069–2082. doi: 10.1101/gad.906601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gartel AL. A new mode of transcriptional repression by c-myc: methylation. Oncogene. 2006;25:1989–1990. doi: 10.1038/sj.onc.1209101. [DOI] [PubMed] [Google Scholar]
- 28.Satou A, Taira T, Iguchi-Ariga SM, Ariga H. A novel transrepression pathway of c-Myc. Recruitment of a transcriptional corepressor complex to c-Myc by MM-1, a c-Myc-binding protein. The Journal of biological chemistry. 2001;276:46562–46567. doi: 10.1074/jbc.M104937200. [DOI] [PubMed] [Google Scholar]
- 29.Kurland JF, Tansey WP. Myc-mediated transcriptional repression by recruitment of histone deacetylase. Cancer research. 2008;68:3624–3629. doi: 10.1158/0008-5472.CAN-07-6552. [DOI] [PubMed] [Google Scholar]
- 30.Brenner C, Deplus R, Didelot C, Loriot A, Vire E, De Smet C, Gutierrez A, Danovi D, Bernard D, Boon T, Pelicci PG, Amati B, Kouzarides T, de Launoit Y, Di Croce L, Fuks F. Myc represses transcription through recruitment of DNA methyltransferase corepressor. The EMBO journal. 2005;24:336–346. doi: 10.1038/sj.emboj.7600509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Q, Zhang H, Kajino K, Greene MI. BRCA1 binds c-Myc and inhibits its transcriptional and transforming activity in cells. Oncogene. 1998;17:1939–1948. doi: 10.1038/sj.onc.1202403. [DOI] [PubMed] [Google Scholar]
- 32.Schlee M, Holzel M, Bernard S, Mailhammer R, Schuhmacher M, Reschke J, Eick D, Marinkovic D, Wirth T, Rosenwald A, Staudt LM, Eilers M, Baran-Marszak F, Fagard R, Feuillard J, Laux G, Bornkamm GW. C-myc activation impairs the NF-kappaB and the interferon response: implications for the pathogenesis of Burkitt’s lymphoma. International journal of cancer Journal international du cancer. 2007;120:1387–1395. doi: 10.1002/ijc.22372. [DOI] [PubMed] [Google Scholar]
- 33.Tanaka H, Matsumura I, Ezoe S, Satoh Y, Sakamaki T, Albanese C, Machii T, Pestell RG, Kanakura Y. E2F1 and c-Myc potentiate apoptosis through inhibition of NF-kappaB activity that facilitates MnSOD-mediated ROS elimination. Molecular cell. 2002;9:1017–1029. doi: 10.1016/s1097-2765(02)00522-1. [DOI] [PubMed] [Google Scholar]
- 34.You Z, Madrid LV, Saims D, Sedivy J, Wang CY. c-Myc sensitizes cells to tumor necrosis factor-mediated apoptosis by inhibiting nuclear factor kappa B transactivation. The Journal of biological chemistry. 2002;277:36671–36677. doi: 10.1074/jbc.M203213200. [DOI] [PubMed] [Google Scholar]
- 35.Chaperot L, Blum A, Manches O, Lui G, Angel J, Molens JP, Plumas J. Virus or TLR agonists induce TRAIL-mediated cytotoxic activity of plasmacytoid dendritic cells. Journal of immunology. 2006;176:248–255. doi: 10.4049/jimmunol.176.1.248. [DOI] [PubMed] [Google Scholar]
- 36.Talukder AH, Bao M, Kim TW, Facchinetti V, Hanabuchi S, Bover L, Zal T, Liu YJ. Phospholipid scramblase 1 regulates Toll-like receptor 9-mediated type I interferon production in plasmacytoid dendritic cells. Cell research. 2012;22:1129–1139. doi: 10.1038/cr.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghosh HS, Cisse B, Bunin A, Lewis KL, Reizis B. Continuous expression of the transcription factor e2-2 maintains the cell fate of mature plasmacytoid dendritic cells. Immunity. 2010;33:905–916. doi: 10.1016/j.immuni.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harper SE, Qiu Y, Sharp PA. Sin3 corepressor function in Myc-induced transcription and transformation. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:8536–8540. doi: 10.1073/pnas.93.16.8536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perissi V, Jepsen K, Glass CK, Rosenfeld MG. Deconstructing repression: evolving models of co-repressor action. Nature reviews Genetics. 2010;11:109–123. doi: 10.1038/nrg2736. [DOI] [PubMed] [Google Scholar]
- 42.Guenther MG, Barak O, Lazar MA. The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Molecular and cellular biology. 2001;21:6091–6101. doi: 10.1128/MCB.21.18.6091-6101.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ishizuka T, Lazar MA. The N-CoR/histone deacetylase 3 complex is required for repression by thyroid hormone receptor. Molecular and cellular biology. 2003;23:5122–5131. doi: 10.1128/MCB.23.15.5122-5131.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.You SH, Lim HW, Sun Z, Broache M, Won KJ, Lazar MA. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nature structural & molecular biology. 2013;20:182–187. doi: 10.1038/nsmb.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herbst A, Hemann MT, Tworkowski KA, Salghetti SE, Lowe SW, Tansey WP. A conserved element in Myc that negatively regulates its proapoptotic activity. EMBO reports. 2005;6:177–183. doi: 10.1038/sj.embor.7400333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Molecular and cellular biology. 1999;19:1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Drenou B, Amiot L, Setterblad N, Taque S, Guilloux V, Charron D, Fauchet R, Mooney N. MHC class II signaling function is regulated during maturation of plasmacytoid dendritic cells. Journal of leukocyte biology. 2005;77:560–567. doi: 10.1189/jlb.0704423. [DOI] [PubMed] [Google Scholar]
- 48.Perry AK, Chen G, Zheng D, Tang H, Cheng G. The host type I interferon response to viral and bacterial infections. Cell research. 2005;15:407–422. doi: 10.1038/sj.cr.7290309. [DOI] [PubMed] [Google Scholar]
- 49.Wathelet MG, Lin CH, Parekh BS, Ronco LV, Howley PM, Maniatis T. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo. Molecular cell. 1998;1:507–518. doi: 10.1016/s1097-2765(00)80051-9. [DOI] [PubMed] [Google Scholar]
- 50.Yang H, Lin CH, Ma G, Baffi MO, Wathelet MG. Interferon regulatory factor-7 synergizes with other transcription factors through multiple interactions with p300/CBP coactivators. The Journal of biological chemistry. 2003;278:15495–15504. doi: 10.1074/jbc.M212940200. [DOI] [PubMed] [Google Scholar]
- 51.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nature immunology. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 52.McWhirter SM, Fitzgerald KA, Rosains J, Rowe DC, Golenbock DT, Maniatis T. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:233–238. doi: 10.1073/pnas.2237236100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, Endres S, Hartmann G. Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. Journal of immunology. 2002;168:4531–4537. doi: 10.4049/jimmunol.168.9.4531. [DOI] [PubMed] [Google Scholar]
- 54.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 55.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 56.Steinhagen F, Meyer C, Tross D, Gursel M, Maeda T, Klaschik S, Klinman DM. Activation of type I interferon-dependent genes characterizes the “core response” induced by CpG DNA. Journal of leukocyte biology. 2012;92:775–785. doi: 10.1189/jlb.1011522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Matsumoto M, Tanaka N, Harada H, Kimura T, Yokochi T, Kitagawa M, Schindler C, Taniguchi T. Activation of the transcription factor ISGF3 by interferon-gamma. Biological chemistry. 1999;380:699–703. doi: 10.1515/BC.1999.087. [DOI] [PubMed] [Google Scholar]
- 58.Zhang D, Zhang DE. Interferon-stimulated gene 15 and the protein ISGylation system. Journal of interferon & cytokine research: the official journal of the International Society for Interferon and Cytokine Research. 2011;31:119–130. doi: 10.1089/jir.2010.0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Espert L, Degols G, Gongora C, Blondel D, Williams BR, Silverman RH, Mechti N. ISG20, a new interferon-induced RNase specific for single-stranded RNA, defines an alternative antiviral pathway against RNA genomic viruses. The Journal of biological chemistry. 2003;278:16151–16158. doi: 10.1074/jbc.M209628200. [DOI] [PubMed] [Google Scholar]
- 60.Zhou Z, Wang N, Woodson SE, Dong Q, Wang J, Liang Y, Rijnbrand R, Wei L, Nichols JE, Guo JT, Holbrook MR, Lemon SM, Li K. Antiviral activities of ISG20 in positive-strand RNA virus infections. Virology. 2011;409:175–188. doi: 10.1016/j.virol.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hung CL, Wang LY, Yu YL, Chen HW, Srivastava S, Petrovics G, Kung HJ. A long noncoding RNA connects c-Myc to tumor metabolism. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:18697–18702. doi: 10.1073/pnas.1415669112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 63.Sampson VB, Rong NH, Han J, Yang Q, Aris V, Soteropoulos P, Petrelli NJ, Dunn SP, Krueger LJ. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer research. 2007;67:9762–9770. doi: 10.1158/0008-5472.CAN-07-2462. [DOI] [PubMed] [Google Scholar]
- 64.Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nature reviews Genetics. 2009;10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 65.Spizzo R, Almeida MI, Colombatti A, Calin GA. Long non-coding RNAs and cancer: a new frontier of translational research? Oncogene. 2012;31:4577–4587. doi: 10.1038/onc.2011.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bidwell BN, Slaney CY, Withana NP, Forster S, Cao Y, Loi S, Andrews D, Mikeska T, Mangan NE, Samarajiwa SA, de Weerd NA, Gould J, Argani P, Moller A, Smyth MJ, Anderson RL, Hertzog PJ, Parker BS. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nature medicine. 2012;18:1224–1231. doi: 10.1038/nm.2830. [DOI] [PubMed] [Google Scholar]
- 67.Slaney CY, Moller A, Hertzog PJ, Parker BS. The role of Type I interferons in immunoregulation of breast cancer metastasis to the bone. Oncoimmunology. 2013;2:e22339. doi: 10.4161/onci.22339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Repetto L, Giannessi PG, Campora E, Pronzato P, Vigani A, Naso C, Spinelli I, Conte PF, Rosso R. Tamoxifen and interferon-beta for the treatment of metastatic breast cancer. Breast cancer research and treatment. 1996;39:235–238. doi: 10.1007/BF01806190. [DOI] [PubMed] [Google Scholar]
- 69.Takaoka A, Tamura T, Taniguchi T. Interferon regulatory factor family of transcription factors and regulation of oncogenesis. Cancer science. 2008;99:467–478. doi: 10.1111/j.1349-7006.2007.00720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sears R, Leone G, DeGregori J, Nevins JR. Ras enhances Myc protein stability. Molecular cell. 1999;3:169–179. doi: 10.1016/s1097-2765(00)80308-1. [DOI] [PubMed] [Google Scholar]
- 71.Savelyeva L, Schwab M. Amplification of oncogenes revisited: from expression profiling to clinical application. Cancer letters. 2001;167:115–123. doi: 10.1016/s0304-3835(01)00472-4. [DOI] [PubMed] [Google Scholar]
- 72.Popescu NC, Zimonjic DB. Chromosome-mediated alterations of the MYC gene in human cancer. Journal of cellular and molecular medicine. 2002;6:151–159. doi: 10.1111/j.1582-4934.2002.tb00183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bernatsky S, Ramsey-Goldman R, Labrecque J, Joseph L, Boivin JF, Petri M, Zoma A, Manzi S, Urowitz MB, Gladman D, Fortin PR, Ginzler E, Yelin E, Bae SC, Wallace DJ, Edworthy S, Jacobsen S, Gordon C, Dooley MA, Peschken CA, Hanly JG, Alarcon GS, Nived O, Ruiz-Irastorza G, Isenberg D, Rahman A, Witte T, Aranow C, Kamen DL, Steinsson K, Askanase A, Barr S, Criswell LA, Sturfelt G, Patel NM, Senecal JL, Zummer M, Pope JE, Ensworth S, El-Gabalawy H, McCarthy T, Dreyer L, Sibley J, St Pierre Y, Clarke AE. Cancer risk in systemic lupus: an updated international multi-centre cohort study. Journal of autoimmunity. 2013;42:130–135. doi: 10.1016/j.jaut.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ronnblom LE, Alm GV, Oberg KE. Possible induction of systemic lupus erythematosus by interferon-alpha treatment in a patient with a malignant carcinoid tumour. Journal of internal medicine. 1990;227:207–210. doi: 10.1111/j.1365-2796.1990.tb00144.x. [DOI] [PubMed] [Google Scholar]
- 75.Passos de Souza E, Evangelista Segundo PT, Jose FF, Lemaire D, Santiago M. Rheumatoid arthritis induced by alpha-interferon therapy. Clinical rheumatology. 2001;20:297–299. doi: 10.1007/pl00011206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.