

Abstract
Monocytic myeloid-derived suppressor cells (M-MDSCs) were increased during LP-BM5 retroviral infection, and were capable of suppressing not only T-cell, but also B-cell responses. In addition to previously demonstrating iNOS- and VISTA-dependent M-MDSC mechanisms, in this paper, we detail how M-MDSCs utilized soluble mediators, including the reactive oxygen and nitrogen species superoxide, peroxynitrite, and nitric oxide, and TGF-β, to suppress B cells in a predominantly contact-independent manner. Suppression was independent of cysteine-depletion and hydrogen peroxide production. When two major mechanisms of suppression (iNOS and VISTA) were eliminated in double knockout mice, M-MDSCs from LP-BM5-infected mice were able to compensate using other, soluble mechanisms in order to maintain suppression of B cells. The IL-10 producing regulatory B-cell compartment was among the targets of M-MDSC-mediated suppression.
Keywords: Monocytic MDSC, B cell, reactive oxygen species, reactive nitrogen species, iNOS, superoxide, peroxynitrite, nitric oxide, Breg, suppression
Introduction
Myeloid-derived suppressor cells (MDSCs) are an immunosuppressive subset of cells that arise from myeloid progenitors that fail to terminally differentiate (reviewed in (1, 2)). Murine MDSCs are Gr-1+CD11b+ and generally lack expression of mature macrophage and dendritic cell markers (3, 4). In healthy mice, Gr-1+ CD11b+ cells are present in very small numbers in the spleen and in circulation and mainly reside in the bone marrow. Unlike MDSCs, these immature myeloid cells are less suppressive (5). MDSCs quickly proliferate and migrate from the bone marrow in response to soluble mediators such as cytokines and chemokines that are produced at sites of inflammation and in the tumor milieu (6).
MDSCs are heterogeneous, but can be classified into two subsets based on extensive studies of suppression of T cells in tumor models. Granulocytic MDSCs (G-MDSCs) are polymorphonuclear and express high levels of Ly6G and low levels of Ly6C (6–8). G-MDSCs often utilize reactive oxygen species (ROS) and arginase 1 for their immunosuppressive function, and require antigen-specific interactions with their target T cells, although there is system-to-system variability (8–10). Monocytic MDSCs (M-MDSCs), are mononuclear and express low levels of Ly6G and high levels of Ly6C (7, 8, 10). M-MDSCs tend to utilize inducible nitric-oxide synthase (iNOS) for suppression (9, 10), and may also utilize arginase 1 (10), although this can vary in different systems. MDSCs are implicated in a wide range of both murine and human pathologies and have been heavily studied in cancer systems as inhibitors of anti-tumor immunity. Both G-MDSCs and M-MDSCs have been reported to be expanded during oncogenesis, with G-MDSCs often showing a larger expansion (10). Despite their higher numbers, G-MDSCs tend to be less potent suppressors in tumor models than M-MDSCs on a per cell basis (2). MDSC expansion has been correlated with reduced survival among cancer patients (11). In addition to their role in inhibiting anti-tumor immunity, MDSCs have also been suggested to inhibit anti-viral immunity during influenza infection (12), murine models of chronic hepatitis B (13), vesicular stomatitis virus (14), and certain strains of herpes simplex virus (15). Conversely, a protective role for MDSCs has been reported in several autoimmune models, including collagen-induced arthritis (CIA) (16), autoimmune hepatitis (17), inflammatory bowel disease (18), and experimental autoimmune encephalomyelitis (19, 20).
The role of MDSCs in murine AIDS (MAIDS) has recently been explored by our lab (21–24). LP-BM5 retrovirus-induced MAIDS is a murine disease syndrome with many features similar to HIV/AIDS, including early activation parameters such as hypergammaglobulinemia, splenomegaly, and lymphadonopathy; dependence on CD4+ T cells for induction; immunodeficiency characterized by loss of CD4+ T-cell function, decreased T- and B-cell responses, and increased susceptibility to infection and death by opportunistic infections; and development of B-cell lymphomas in late stages (25–27). In susceptible hosts, MAIDS pathogenesis is dependent on binding of CD154 (CD40L) on pathogenic CD4+ T cells with CD40 on B cells (28–33).
LP-BM5 infection results in a suppressive M-MDSCs population characterized by our lab (21). These M-MDSCs suppress T cells ex vivo in an iNOS-dependent manner and also suppress B cells in a partially iNOS-dependent manner (21). Suppression of B cells was also partially dependent on the novel negative checkpoint regulator, V-domain Ig suppressor of activation (VISTA) (23). To our knowledge, our report was the first detailing MDSC (specifically monocytic)-mediated suppression of B cells, in particular in a retroviral system. Previous studies discussed natural suppressor cells from murine bone marrow or neonatal spleens, which were reported to be capable of suppressing B cell responses in an iNOS-dependent manner (34–36). However, whether this population of cells included true MDSCs as now defined, is unknown, as the only phenotypic descriptions indicated that the cells are non-adherent, low-density and Thy-and Ig-negative (34–36). Since then, to our knowledge, three groups have shown MDSC-mediated suppression of B cells in autoimmune models. One group found that MDSCs induced during CIA are also capable of suppressing B cells in an iNOS-dependent manner (16), and yet another reported that MDSCs could inhibit proliferation of B cells in experimental autoimmune myasthenia gravis (EAMG) via iNOS and arginase (37). A third group found that MDSC-injection into lupus mice induced suppression of effector B cell population, including germinal cells and plasma cells, via iNOS while simultaneously increasing the proportion of regulatory B cells (38). Additionally, MDSCs have been identified as inhibitors of B-cell lymphopoiesis in the bone marrow during obesity and aging (39). Although we are unaware of any studies evaluating suppression of B cells by HIV-derived MDSCs, MDSCs capable of suppressing both antigen-specific and non-specific CD8+ T-cell responses were increased in HIV patients, supporting our findings in the LP-BM5 retroviral system, with MDSC-frequencies correlating with clinical parameters such as decreased CD4+ T-cell frequency and increased viral load (40).
Inducible nitric oxide synthase (iNOS) catalyzes the production of nitric oxide (NO) from L-arginine and O2 (41). In addition to its function as a proinflammatory mediator and its ability to inhibit viral replication (41), NO can also inhibit immune responses and promote chronic infection (42). While our previous work indicates that iNOS accounts for approximately half of the M-MDSC-mediated suppression of B cells, and that VISTA also plays a major role in this suppression (23), the existence and identity of other suppressive mechanism(s) active against B-cell targets are unknown (21). In this LP-BM5 retroviral system, suppression of B cells was independent of: arginase 1, another common suppressive mechanism utilized by MDSCs, as well as PD-1/PD-L1 interactions, IL-10, and indoleamine 2,3-dioxygenase (IDO) activity (21, 43). Other mechanisms of suppression utilized by either MDSC subset in their inhibition of T-cell responses in different disease settings can include membrane-bound or soluble transforming growth factor β (TGF-β) (44–46), cysteine depletion (47), ROS production (48–51), prostaglandin-E2 (52–54), induction of regulatory T cells (55–57), and down-regulation of L-selectin expression (7). As MDSC-mediated suppression of B cells is understudied, it is not clear whether these and/or other potential suppressive pathways, such as adenosine production (58) and stimulation of the inhibitory receptors FcγRIIβ (CD32) (59–62) and CD22 (63–67), or CD72 (68), are involved in M-MDSC suppression of B-cell targets.
Given the scarcity of studies examining MDSC-mediated suppression of B cells, we utilized the LP-BM5 retroviral system to characterize the mechanism(s) in addition to NO production and VISTA that are used by M-MDSCs to suppress B cells. The following work began with triaging experiments to determine if suppression was contact-dependent and if soluble mediators were involved. Antioxidants, inhibitors, antibodies, and other methods were utilized to block potential reactive nitrogen or oxygen species, soluble TGF-β, and downstream mediator-dependent mechanisms. The effects of genetic ablation of major suppressive mechanisms, as well as the effect of M-MDSCs on regulatory, IL-10-producing B cells, were also examined. These data, and in particular the concept of MDSC plasticity in molecular mechanisms of inhibition, may have a number of potential clinical implications relevant to MDSCs present in a variety of human disease settings.
Materials and Methods
Mice
7-week-old male C57BL/6 (B6) mice were purchased from the National Cancer Institute (NCI, Bethesda, MD) or Charles River (Wilmington, MA). B6-background 10BiT (Thy1.1 gene under the control of IL-10 promoter) (69) and VISTA knockout mice were generous gifts from the laboratories of Lloyd Kasper and Randolph Noelle, respectively (Geisel School of Medicine at Dartmouth, Lebanon, NH). B6-background iNOS knockout, purchased from Jackson Lab (Farmington, CT), and VISTA knockout mice were crossed to create F2-generation-derived VISTA/iNOS double knockout breeders with assistance by the Dartmouth Immunology COBRE (Geisel School of Medicine at Dartmouth, Lebanon, NH), which were confirmed using PCR and by their inability to produce nitric oxide and lack of VISTA expression (measured by flow cytometry). All mice were bred and/or housed in the Center for Comparative Medicine and Research (CMMR) at the Geisel School of Medicine at Dartmouth, and used at ~8–10 wks of age. All animal experiments were done with the approval of the Institutional Animal Care and Use Committee of Dartmouth College, in conjunction with the Dartmouth CCMR, an AALAC approved animal facility.
Cell Purification
For enrichment of suppressor cell populations, splenocytes from 3–6 LP-BM5-infected B6 mice were pooled 5 wpi and labeled with anti-Ly6G-biotin-coupled beads, and subsequently with anti-biotin-coupled paramagnetic beads, as previously published (21). The negative flow-through was collected from a MACS column (Miltenyi Biotech, Auburn, CA). The flow-through was labeled with anti-CD11b-coupled paramagnetic beads and column purified, leading to the standard enriched M-MDSC preparation previously employed (21–24). Responder cell populations consisted of pooled splenocytes from 3–4 uninfected B6 mice.
LP-BM5 Virus Inoculation
LP-BM5 virus was prepared in the lab using G6 cells as provided and described by Janet Hartley and Herbert Morse (NIH) (70) and as we have adapted (30). Mice were infected intraperitoneally with 5*104 ecotropic PFU as determined by standard retroviral XC plaque assay (71).
[3H] Thymidine Incorporation Proliferation Assays
Responder cells (uninfected, wildtype B6 splenocytes) were plated at 5*105 cells/well (96-well plates) or 3*106 cells/well (24-well transwell plates). Suppressor cells (obtained from 5 w.p.i. LP-BM5-infected mice, and employed as Ly6G-depleted/CD11b-enriched splenocytes) were plated at various suppressor: responder (S:R) cell ratios as indicated in 3–8 wells per condition in supplemented medium (containing 5% fetal calf serum, L-glutamine, and antibiotics). Responder cells were stimulated with 10μg/mL lipopolysaccharide (LPS) (shown) or 50μg/mL anti-CD40 plus 10ng/mL IL-4 (data not shown) at 37*C, 5% CO2. For transwell studies, suppressors were plated either in the bottom well with responders (with contact) or in a 0.4μm Corning 24-well transwell inserts (without contact). As a control, both responders and M-MDSCs were cultured in the bottom well in the presence of the insert. No change in suppression relative to normal co-culture conditions was detected, indicating that lack of cell contact, as opposed to the presence of the insert, affected levels of suppression. Beginning at hour 66, plates were terminally pulsed with 1mCi (96-well plates) or 3.5mCi (24-well transwell plates) [3H] thymidine (PerkinElmer, Waltham, MA). At 72 hrs, plates were harvested and assessed for thymidine incorporation using a scintillation counter. Percent suppression was calculated from the control response, as previously described (21). In brief, percent residual responsiveness (R) was calculated as: R = (cpm of responder cells with suppressor cells)/(cpm of responder cells alone) * 100%. Residual responsiveness was subtracted from the % control responsiveness (defined as the cpm of responders alone, set at 100%) to determine the percent suppression, such that % suppression = 100% - R.
iNOS Blockade Assays
To test for iNOS-dependence, the iNOS/NOS2 inhibitor L-Nil (Enzo Life Sciences, Farmingdale, NY) was added to suppressor cells or control media before culture at 100μM. Blockade of suppression was calculated as 100% - (percent suppression with L-Nil treatment)/(control percent suppression without treatment). As a negative control, L-Nil did not affect the proliferation of responder cells cultured in the absence of M-MDSCs.
Griess’ Assays
Griess’ reagent was used, as previously described (24), to measure cumulative levels of nitrite (a downstream product of the iNOS-pathway) in suppressive cultures set up as described above. Briefly, 50μL supernate was removed from suppressive cultures at 66 hours, and incubated with 50μL Griess’ reagent (Sigma-Aldrich, St. Louis, MO) for 10 min at room temperature. Absorbance at 540nm was compared to a standard curve generated using NaNO2 to calculate concentration of nitrite, as per the manufacturers protocol.
Supernate Transfer Assays
Responder cells alone (R) or responder cells co-cultured with suppressor cells (R+S) at an R:S ratio of 1:0.33, as described above for the [3H] thymidine assays, were polyclonally stimulated for 40 hrs (donor plates) as a source of cell-free supernate. Responder cells were cultured alone with stimulation in parallel for subsequent use as recipient cultures – such that the kinetics were identical to those for the donor plates at the time of transfer. At 40 hrs. (the earliest timepoint where cumulative NO production was consistently significantly above background), supernate from either R or R+S donor wells was transferred to the recipient cultures. Suppression was determined by [3H] thymidine incorporation, as described above.
Cysteine Add-Back Assays
To test for involvement of cysteine depletion, β-mercaptoethanol (2ME, Sigma Aldrich, St. Louis, MO) or N-acetyl cysteine (NAC, MP Biomedicals, Santa Ana, CA) was used to increase extracellular cysteine levels. Blockade of suppression was calculated as described above for L-Nil experiments. As positive controls, Ellman’s reagent (Sigma, St. Louis, MO) was used to spectrophotometrically ensure that 2ME and NAC increased cysteine concentrations, and the AKR-H-2bSL1(72) murine leukemia cell line was used to ensure bioactivity of the 2ME. As a negative control, 2ME and NAC did not affect the proliferation of responder cells cultured in the absence of M-MDSCs at the doses tested.
Antioxidant Assays
To test for involvement of reactive oxygen species, supernates were treated with catalase (MP Biomedicals, Santa Ana, CA), superoxide dismutase (SOD, MP Biomedicals, Santa Ana, CA), carboxy-PTIO (Sigma-Aldrich, St. Louis, MO), uric acid (UA, Pointe Scientific, Canton, MI), or MnTBAP (Enzo Life Sciences, Farmingdale, NY) at indicated concentrations to neutralize/scavenge hydrogen peroxide, superoxide, nitric oxide, or peroxynitrite, respectively. Concentrations depicted represent the final concentration after transfer. Supernate assays were performed as described above (with wells receiving antioxidant-treated supernate from responder cells cultured alone used as a control to ensure lack of effect of antioxidants on baseline proliferation), and percent blockade of suppression was calculated by comparing supernates treated with the antioxidants to untreated supernate controls, as described above. As a positive control for catalase activity, the ability to break down hydrogen peroxide was measured spectrophotometrically.
XTT assay
To test for superoxide production, an XTT assay of the supernate was performed, as previously described (73). Briefly, suppression assays were set up as described above, but using media without phenol red. At 40 hrs, supernates were removed and plated with 0.83μg/mL phenazine methosulfate (PMS, Sigma Aldrich, St. Louis, MO) and 1.3mg/mL 2,3-Bis-(2-Methoxy-4-Nitro-5-Sulfophenyl)-2H-Tetrazolium-5-Carboxanilide (XTT, Sigma Aldrich, St. Louis, MO) and incubated, protected from light, overnight. Subsequently, absorbance was read at 30 min intervals at 470nm (adjusted for non-specific turbidity – i.e. 660nm absorbance) to detect conversion of the XTT tetrazolium salt to formazen by superoxide. Superoxide dismutase was used to ensure superoxide-specific absorbance. The enzyme xanthinine oxidase (Sigma Aldrich, St. Louis, MO) was used as a positive control at concentrations indicated, with 0.2mM pterin (Sigma Aldrich, St. Louis, MO) added as its substrate.
Anti-TGF-β
To test for involvement of TGF-β, supernates were treated with mouse anti-TGF-β mAb (clone #1D11, R&D Systems, Minneapolis, MN) or mouse IgG1 isotype control (clone #11711, R&D Systems, Minneapolis, MN) at indicated concentrations to neutralize TGF-β. Supernate assays were performed as described above. As a negative control, anti-TGF-β did not affect the proliferation of responder cells cultured in the absence of M-MDSC-conditioned supernate.
Arginase Blockade
To test for arginase-dependence, the specific inhibitor NorNOHA (Cayman Chemical Company, Ann Arbor, MI) was added to suppressor cells before culture in a standard suppression assay at 1mM. Blockade of suppression was calculated as in the iNOS blockade experiments detailed above. As a positive control for inhibitor efficacy, reproducibly (including two head-to-head experiments), the same doses of NorNOHA substantially, and in some trials completely, blocked arginase-1-dependent T-cell suppression by ID8 tumor-derived B6 MDSCs, as previously described (74). As a negative control, NorNOHA did not affect the proliferation of responder cells stimulated by LPS cultured in the absence of M-MDSCs.
Flow Cytometry
For cell-surface staining, 5*105 spleen cells were incubated with fluorescein isothiocyanate (FITC-), phycoerythrin (PE−), or allophycocyanin (APC−) antibodies, followed by direct immunofluorescence quantified by log amplification (MACSQuant flow cytometer; Miltenyi Biotec) using mAbs for Thy1.1 (clone HIS51, Ebioscience) and CD19 (clone 6D5, Biolegend); the proliferation dye CFSE (Molecular Probes by Life Technologies); and the viability stain 7-AAD (Ebioscience). Fluorescence minus one (FMO) and isotype controls were used as gating controls.
Statistical Analysis
Standard deviations (SD) of the mean were determined and compared statistically using a Student t test, and the Holm-Bonferroni method was used to correct for multiple testing.
Results
iNOS-dependence of B-cell suppression by M-MDSCs
Despite many studies on the mechanisms utilized by MDSCs to suppress T cells, little is known about MDSC-mediated suppression of B cells, early exceptions being our lab’s study in the LP-BM5 retroviral system (21) and subsequent studies in a few other systems: e.g. in mice with collagen induced arthritis (16) and autoimmune myasthenia gravis (37). In LP-BM5 retrovirus-infected mice, which develop MAIDS, we demonstrated that although T-cell responses were suppressed in a predominantly iNOS-dependent manner, M-MDSC suppression of B-cell responsiveness to LPS (shown) and anti-CD40 plus IL-10 (not shown) was only partially iNOS-dependent (21). To further clarify the role of NO, a Griess’ assay for NO production was performed in the context of M-MDSC-mediated suppression of B cells. LP-BM5 expanded, Miltenyi-bead enriched (see Materials and Methods), Ly6G±/low Ly6C+ CD11b+ M-MDSC produced NO in parallel with their suppression of B cells, and this NO production was fully blocked by the iNOS-specific inhibitor, L-Nil (Fig. 1a). Suppression assays reveal that L-Nil was able to inhibit some, but not all, of the M-MDSC-mediated suppression of B-cell responses to polyclonal stimulation (Fig. 1b). iNOS-inhibitors blocked an average of ~45% of M-MDSC suppression of B cells, with blockade up to 60% but never near 100%, even with L-Nil at concentrations well above that required to fully block NO production (Fig. 1a) and M-MDSC suppression of T-cell responsiveness (data not shown and ref. (21)).
Fig. 1. iNOS-dependence of B-cell suppression by M-MDSCs.


(A) The Griess’ assay was used to measure levels of NO production in cultures of stimulated B-cells alone, or co-cultured with enriched M-MDSCs in the presence or absence of 100μM L-Nil. A similar pattern of results was observed in 3 experiments. (B) A [3H] thymidine incorporation assay was used to assess M-MDSC suppression of B-cell proliferation in response to LPS stimulation in the presence or absence of 100μM L-Nil. Higher doses of L-Nil did not show increased blockade. Data shown are from representative experiments, with similar pattern of results was observed in 3 experiments. Significance levels: **, p<0.01; ***, p<0.001; NS, not significant.
Contact-dependence of B-cell suppression by M-MDSCs
To broadly characterize potential mechanisms used by M-MDSCs in addition to iNOS to suppress B cells, contact-dependence was tested using a transwell system in which the M-MDSCs were segregated from their target responder B cells. M-MDSCs (upper well) retained most, but not all, of their suppressive ability when physically separated from responding B cells (lower well) (Fig. 2a). In the absence of cell contact, M-MDSCs exhibited a mean (in more than 5 experiments) of 78±14% as much suppressive capacity as M-MDSCs in contact with responders in parallel assays. These data indicate that a majority of the suppression was independent of contact, yet a significant portion of suppression required contact between M-MDSCs and their target B cells.
Fig. 2. Contact-dependence of B-cell suppression by M-MDSCs.

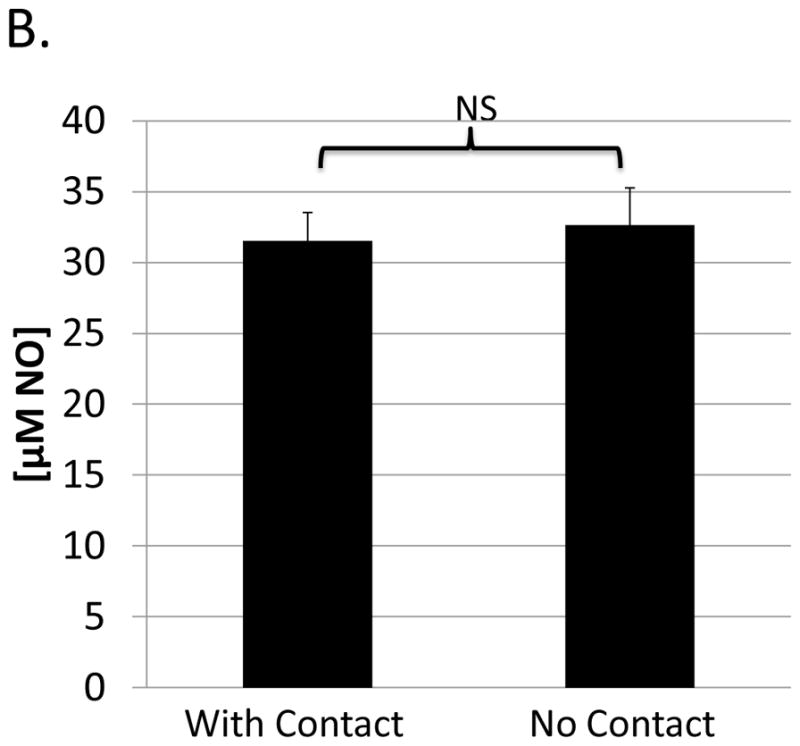
Assays were performed as in Figure 1, specifically for the co-cultures (with contact). In addition: (A) M-MDSCs (upper well) were physically separated from responder cells (lower well) using a transwell system (see Materials and Methods). (B) A Griess’ assay was performed with stimulated B-cells (lower well) co-cultured with M-MDSCs (upper well) in the absence of cell contact. Data shown are from representative experiments, with a similar pattern of results (as shown for both A and B) observed in at least 5 experiments each. Significance levels: *, p<0.05; **, p<0.01; NS, not significant.
As previously established (see Fig. 1a+1b and Green et al. (21)) iNOS contributed substantially (approximately 45% of the M-MDSC-mediated suppression of B-cell responses). A Griess’ assay was used in the context of the transwell system to determine if iNOS-mediated NO production by M-MDSCs required contact with responder cells. M-MDSCs (in the upper well) produced NO (detected at an undiminished level in the lower well (responder cells only)) in the absence of cell contact, indicating sufficient availability to target the responder B cells (Fig. 2b). Given this context, the consistent findings that NO only accounts for approximately half of the M-MDSC-mediated suppression of B cells, yet 75–80% of the suppression was contact independent, collectively suggest at least one other contact-independent mechanism of M-MDSC suppression.
Involvement of soluble mediators in B-cell suppression by M-MDSCs
Because the majority of M-MDSC-mediated suppression was contact-independent, supernate transfer assays were utilized to determine if suppression was the result of soluble mediators. Supernate taken from suppressive co-cultures of M-MDSCs with responder B cells (as set up as in Fig. 1) was directly used as a possible source of such cell-free factors to test for alterations in the response of B cells cultured alone with polyclonal activation (see Materials and Methods). In an attempt to define a timepoint such that the final effector soluble mediators were fully formed, yet the reaction time with responders was sufficient to allow full detection of suppressive effects of the transferred supernate, preliminary kinetic experiments were conducted that lead to choosing 40hrs as the time of transfer. Indeed, supernates were capable of inhibiting B-cell responses, with a mean of ~55% of the suppression observed in parallel, responder B-cell/M-MDSC co-culture assays (Fig. 3a, legend). Thus, soluble factors mediated, in significant part, the M-MDSC suppression of B-cell responsiveness.
Fig. 3. Involvement of soluble mediators in B-cell suppression by M-MDSCs.


(A) Supernates from suppressive cultures of MDSCs and B cells were tested for suppressive capability and showed a mean of 56±15% of the suppression observed in parallel, cell-based assays. A similar pattern of results was observed in 5 experiments. (B) Supernates from suppressive cultures of MDSCs and B cells in the presence or absence of 100μM L-Nil were tested for suppressive capability. Data shown are from representative experiments, with a similar pattern of results was observed in 4 experiments. Significance levels: *, p<0.05; **, p<0.01.
Given the role for iNOS in suppression by these M-MDSCs, a dependency on NO for suppression in the supernate transfer assays was investigated. Pretreatment of M-MDSCs (used as a source of supernate) with the iNOS inhibitor L-Nil showed substantial, but only partial, blockade of the suppressive capability of the supernate (despite complete inhibition by the same L-Nil concentration of NO production as measured by Griess’ assay (data not shown)) (Fig. 3b). Of note, the percentage blockade of suppression by L-Nil was roughly equivalent in these supernate transfer experiments when compared to the cell-based suppression assay (see Fig. 1b). Collectively, these data indicate that though iNOS-generated NO was an important component of the suppression of B cells by soluble mediators derived from M-MDSCs, there appear to be other additional soluble mediators utilized.
Role of cysteine-depletion in B-cell suppression by M-MDSCs
Given (a) soluble mediator(s) were shown to be involved, potential final effector molecules were investigated. One soluble mechanism utilized by MDSCs to suppress T cells in tumor systems is cysteine depletion (47). To test whether increasing cysteine concentrations affected M-MDSC-mediated suppression of B cells, beta-mercaptoethanol (2ME), a reducing agent that converts extracellular cystine to cysteine, and N-acetylcysteine (NAC), a form of cysteine that is resistant to extracellular oxidation, were used to counter possible cysteine limitation. As controls for reagent bio-activity, both 2ME and NAC increased cysteine concentration in the cultures, as measured by Ellman’s reagent; and 2ME was able to support the growth of a 2ME-dependent cancer cell line (data not shown, see Materials and Methods). Even using concentrations of NAC up to 5-fold greater and 2ME concentrations up to 25-fold greater than previously published in other systems to counter MDSC inhibition of T cells (47), suppression of B cells was not blocked, indicating that LP-BM5-expanded M-MDSCs did not utilize cysteine-depletion as a substantial mechanism to suppress B cells (data not shown).
Role of ROS production in B-cell suppression by M-MDSCs
Another class of soluble mediators used by MDSCs in many systems to suppress T-cell responses is reactive oxygen species (ROS), such as hydrogen peroxide (48). Thus, the involvement of hydrogen peroxide in M-MDSC-mediated suppression here was tested using catalase, which converts hydrogen peroxide to water and molecular oxygen. Even at concentrations up to 5-fold greater than published as effective in blocking MDSC function (48), suppression by the transferred supernates was not blocked, despite our independent confirmation of the ability of the catalase used to break down hydrogen peroxide (data not shown, see Materials and Methods).
Involvement of another ROS, superoxide, was tested using superoxide dismutase (SOD), which converts superoxide to hydrogen peroxide (75). SOD treatment partially blocked M-MDSC-supernate-mediated suppression of B-cell responses, with 5 out of 6 experiments showing significance (p=0.01 to 1*10−7), and an average dose-dependent maximum blockade of approximately 30% (Fig. 4a). To confirm an involvement of superoxide as a final mediator of suppression, an XTT assay was performed on supernates in parallel with a supernate transfer suppression assay, based on the ability of XTT to be reduced by superoxide. Superoxide was indeed detectable in the supernate conditioned by M-MDSCs (but not in control supernate from stimulated responder cells alone), corroborating data from the SOD blocking experiments (Fig. 4b).
Fig. 4. Role of ROS production in B-cell suppression by M-MDSCs.



Suppression of B-cell proliferation was assessed using supernates from suppressive cultures of MDSCs and B cells as in Figure 3. Supernates were treated with increasing doses of: (A) superoxide dismutase (SOD), (C) uric acid (UA), and (D) MnTBAP. Higher doses than those shown were tested, but did not show additional blockade and began to induce toxicity. (B) Superoxide levels in supernates were tested using the XTT assay; XO represents the positive control. Data shown are from representative experiments, these patterns of results were representative of at least 3 experiments for each panel. Significance levels:*, p<0.05; ***, p<0.001. p-values are versus untreated.
As superoxide readily reacts with NO resulting in the generation of peroxynitrite (ONOO−) (76), and we previously demonstrated the involvement of iNOS generated NO in M-MDSC-mediated suppression of B cells (Figs. 1 and 3b and ref. (21)), peroxynitrite formation was considered as an additional final soluble mediator of suppression. Using uric acid (UA), a peroxynitrite scavenger, suppression in the supernate transfer system was reliably blocked, with an average dose-dependent maximum of blockade at approximately 20% (Fig 4c). This level of blockade was verified using a second peroxynitrite scavenger, MnTBAP (see Fig. 4d). Taken together, these data indicate that M-MDSC-mediated suppression of B cells was partially dependent on reactive oxygen species, specifically the end effectors superoxide and peroxynitrite, mechanisms more commonly associated with G-MDSCs (48, 49, 77), but not hydrogen peroxide.
Role of nitric oxide in the absence of peroxynitrite formation
The NO-specific scavenger, carboxy-PTIO, which neutralizes NO while not affecting peroxynitrite, was also used in supernate transfer experiments. Carboxy-PTIO reproducibly blocked in a dose-dependent manner ~20% of the suppressive capability of M-MDSC-conditioned supernates (with significant blockade in 6 out of 7 experiments, p=0.04 to 1*10−7) (Fig. 5), indicating that nitric oxide was suppressive on its own, in addition to forming suppressive peroxynitrite (Fig. 4c, d).
Fig. 5. Role of nitric oxide in the absence of peroxynitrite formation.

Suppression of B-cell proliferation by supernate from suppressive cultures of MDSCs and B cells was assessed as in Fig. 3. Supernates were treated with increasing doses of carboxy-PTIO (PTIO). Higher doses were tested, but did not show additional blockade and began to induce toxicity. Data shown are from a representative experiment. This pattern of results is representative of 7 experiments. Significance levels:*, p<0.05; **, p<0.01. p-values are versus untreated.
Together, these data indicate that M-MDSCs expanded after LP-BM5 retroviral infection utilized superoxide (Fig. 4a, b), peroxynitrite (Fig. 4c, d), and nitric oxide (Fig. 5) soluble mediators, when summed up, as responsible for a major portion of the total soluble M-MDSC-mediated suppression of B cells.
Additive nature of superoxide and iNOS/NO blockade
As just shown (Figs. 4 and 5), superoxide and nitric oxide as end-products both individually and collectively (peroxynitrite formation) contribute to the suppressive capacity of M-MDSC supernates. However, at the level of generation of these ROS/RNS species, either independent or overlapping pathways could be involved. To address these questions, including whether the contributions of NO and superoxide were additive, M-MDSCs were treated with L-Nil to block iNOS, and supernates from these treated M-MDSCs were also treated with SOD. The level of suppression from this dual inhibitor blocking schema was compared to that of each individual treatment performed in parallel. Suppression by the SOD and L-Nil combination treated M-MDSC-supernate was significantly less than suppression by supernates treated with either inhibitor independently in 3 of 4 experiments (representative experiment shown in Fig. 6a). Further, when the levels of blockade for all 4 experiments were compiled, blockade of suppression by M-MDSC supernates following the combination treatment exceeded the blockade for either treatment applied individually (Fig. 6b). The levels of combined blockade were roughly additive, indicating that the pathways generating superoxide and nitric oxide were sufficiently non-overlapping that substantial cross-competition and diminishment of the individual blocking effect was not observed.
Fig. 6. Additive nature of superoxide and iNOS/NO blockade.


Suppression of B-cell proliferation by supernates from suppressive cultures of MDSCs treated with L-Nil and SOD either alone, or in combination, was assessed as in Fig. 3 and Fig. 4. (A) Data shown are from a representative experiment. This pattern of results was observed in 3 of 4 experiments. (B) Compiled blockade values from all 4 experiments. Significance levels:*, p<0.05; **, p<0.01; ***, p<0.001.
Role of soluble TGF-β in B-cell suppression by M-MDSCs
Because the above results suggested that NO, superoxide, and peroxynitirite could together account for perhaps only 70% of the suppression by supernates (Figs. 4–5), TGF-β as a known inhibitor of B-cell proliferation was assessed next. Although membrane-bound TGF-β may also be employed by MDSCs in other systems (see Discussion and ref.(44)), to determine if soluble TGF-β was involved, M-MDSC supernates were treated with mouse anti-TGF-β mAb. Anti-TGF-β, but not the mouse IgG1 isotype control, consistently blocked suppression, with average blockade at ~25% of the soluble suppression (with significant blockade in 5/6 experiments, p=0.026 to 0.007; and an average blockade of 26±4%). These results indicated that most of the previously unaccounted for soluble suppression appeared to be dependent on soluble TGF-β (Fig. 7).
Fig. 7. Role of soluble TGF-β in B-cell suppression by M-MDSCs.

Suppression of B-cell proliferation by supernate from suppressive cultures of MDSCs and B cells was assessed as in Fig. 3. Supernates were treated with increasing doses of anti-TGF-β or IgG1 control. Higher doses were tested, but did not show additional blockade and began to induce toxicity. Data shown are from a representative experiments. This pattern of results is representative of four experiments. Significance levels: **, p<0.01. p-values are versus untreated.
Suppressive ability of M-MDSCs after eliminating two of the major suppressive mechanisms
As iNOS and its downstream products account for approximately half of the M-MDSC suppression of B cells (see Fig. 1 and ref. (21)), and our lab has recently demonstrated that the new negative checkpoint regulator VISTA is required for an additional substantial portion of the M-MDSC mechanisms providing suppression of B cells (23), we next considered whether M-MDSCs from VISTA/iNOS double knockout mice (genotype confirmed, see Materials and Methods) were still able to suppress in the absence of these two dominant mechanisms. M-MDSCs from such double knockout mice were robustly suppressive of polyclonal B-cell responses, with similar levels of suppression (not statistically different) as seen with wild-type M-MDSC (Fig. 8a). Importantly, as a control and as expected, addition of L-Nil had no effect on the suppressive capability of VISTA/iNOS double knockout M-MDSCs (data not shown). These data indicate that these M-MDSCs are able to compensate with expanded roles by the more minor mechanisms, as described above, and/or with novel additional inhibitory mechanisms when the two major suppressive mechanisms are removed.
Fig. 8. Suppressive ability of M-MDSCs after eliminating two of the major suppressive mechanism.


(A) Suppression of B-cell proliferation by MDSCs from LP-BM5-infected wild-type or VISTA/iNOS double knockout MDSCs. (B) Supernate transfer suppression assays (as in Fig. 3) left untreated or treated with superoxide dismutase (SOD) showed that soluble suppression by double knockout MDSCs accounted for about 2/3rds of total suppression, as opposed to with WT M-MDSCs, where soluble mediators account for approximately 55% the suppression (see Fig. 3a). Data shown are from a representative experiments. A similar pattern of results was observed in at least 3 experiments for each panel. Significance levels: **, p<0.01; NS, not significant.
M-MDSC supernate transfer assays, including double knockout M-MDSCs, lead to substantial suppression of B-cell responsiveness (Fig. 8b). In fact, supernates conditioned by VISTA/iNOS double knockout M-MDSCs had statistically more suppressive capability than supernate conditioned by wild-type M-MDSCs in 3 (p=5.5*10−4 to 4*10−5) out of 4 experiments. In parallel experiments, superoxide dismutase or the anti-TGF-β were used to demonstrate that, like wild-type M-MDSCs, suppression by double knockout M-MDSCs is partially superoxide-and TGF-β-dependent. Increased dependence on these mechanisms for suppression was not observed (Fig. 8b and data not shown). Additionally, the arginase inhibitor NorNOHA was unable to block suppression by the double knockout M-MDSCs, despite our independent confirmation of its ability to block suppression by an arginase-1-dependent tumor-derived MDSC population (data not shown, see Materials and Methods), consistent with our previously published results of lack of an effect by NorNOHA on suppression by wild-type M-MDSCs (data not shown) (21).
Ability of M-MDSCs to suppress regulatory B cells
Previously, our lab showed that M-MDSCs expanded during LP-BM5 induced pathogenesis are capable of suppressing IL-10 production by CD4+ regulatory T cells (Treg), both in total percentage of cells and on a per-cell basis (22). Given that these M-MDSCs also suppress the B-cell compartment, IL-10-reporter mice (which express Thy1.1 under the IL-10 promoter) were utilized here to determine if IL-10-production by regulatory B cells was also susceptible to M-MDSC regulation. The proportion of B cells producing IL-10 in response to LPS-stimulation was significantly decreased in the presence of M-MDSCs (Fig. 9a and 9b). In contrast, co-culture with M-MDSCs did not affect the level of IL-10 produced on a per B cell basis (as determined by the Thy1.1 MFI of the remaining Breg cells) (Fig. 9c). CFSE-dilution data corroborated the thymidine incorporation data in general, and additionally specifically demonstrated that proliferation of IL-10 producing B cells is decreased by their co-culture with M-MDSCs (Fig. 9d). This decrease in proliferation can be quantified using mean or median fluorescence intensities, and in either case suppression is significant (p = 0.02 and 9*10−7, respectively).
Fig. 9. Ability of M-MDSCs to suppress regulatory B cells.




Suppression assays were set up as above with w.t. MDSCs but using 10BiT reporter mice as a source of responder B cells. Splenocytes were gated on the 7-AAD− live population and were stained for CD19 and the IL-10 reporter, Thy1.1. (A) Representative flow plots showing the percent of B cells producing IL-10. (B) Quantification of results shown in panel A with means and standard deviations from replicate samples. (C) Mean Thy1.1 fluorescence intensities. (D) Proliferation of IL-10-producing B cells stained with CFSE gated on 7-AAD−, CD19+, IL-10+. A representative flow plot is shown on the left, with mean and standard deviations from replicate samples shown on the right. Data shown are from a representative experiments. These patterns of results (in panels A-D) are representative of at least 3 experiments. Significance levels: ***, p<0.001. Error bars are from replicate samples.
Discussion
Based on the results presented above (Figs. 1, 4, 5, and 7), we show that M-MDSCs from LP-BM5 retrovirus-infected mice suppressed B-cell responsiveness in a contact-independent manner that was partially dependent on soluble mediators (Figs. 2a and 3a), including superoxide, peroxynitrite, nitric oxide, and TGF-β (Figs. 4, 5, and 7). We previously demonstrated that the novel checkpoint regulator VISTA was importantly involved in M-MDSC-mediated suppression of B-cell responsiveness and likely acts in both iNOS/NO-dependent and independent manners, as indicated by studies herein and our previous use of VISTA-Ig in the absence of MDSCs (and therefore in the absence of M-MDSC produced iNOS/NO) (23). VISTA and ROS-production, as well as VISTA and TGF-β-production, are likely in predominantly different inhibitory pathways, as suppression by supernates from VISTA/iNOS double knockout M-MDSCs was still partially blocked by SOD and anti-TGF-β mAb (Fig. 8b and data not shown). The presence of superoxide-mediated suppression by double knockout M-MDSCs indicates that superoxide is capable of suppressing in the absence of peroxynitrite formation. Further, treatment of M-MDSC-conditioned supernate with SOD and L-Nil resulted in essentially additive blockade (Fig. 6). Using β-mercaptoethanol, N-acetyl cysteine, and catalase, no evidence was found to support a role for cysteine-depletion or hydrogen peroxide production. As shown in previous work from our lab utilizing wild-type versus knockout mouse strains in standard M-MDSC B-cell responder suppression assays, suppression was also independent of arginase 1, PD-1/PD-L1 interactions, IL-10 production, and IDO activity (21, 43).
In addition to showing partial iNOS-dependence using inhibitors (L-Nil in this study (Fig. 1b), and LNMMA, previously by our lab (21)), iNOS/NO dependence was shown to broadly effect the M-MDSC suppression of B-cell responses by use of two different polyclonal activators, LPS and anti-CD40 + IL-4. NO can readily pass through cell membranes (42), where it can have a variety of suppressive functions including inhibition of DNA synthesis (78, 79), deamination of DNA (80, 81), inhibition of MHC class II expression (82), nitrosylation and activation or deactivation of signaling proteins (especially those involved in IL-2 receptor signaling and downstream STAT5 signaling) (83–88), and other pro-apoptotic effects (89–91). Given the novelty of MDSC-mediated suppression of B cells, while many of these inhibitory effects have been discovered in the context of T-cell suppression, analogous and other experimental approaches are needed to determine if these or other inhibitory effects are responsible as the final effector mechanism for iNOS/NO-dependent B-cell suppression. This question is particularly relevant in the LP-BM5 retroviral system, as the VISTA-dependent portion of M-MDSC suppression appears to include both iNOS/NO-dependent and independent mechanisms.
ROS generation is frequently associated with G-MDSC-mediated suppression of T cells. The Gabrilovich lab found that G-MDSCs in a murine fibrosarcoma model produced hydrogen peroxide that suppressed T cells (48). In the same study, these authors found that ROS production blocked differentiation of immature myeloid cells resulting in increased MDSC numbers (48). Whether a similar feed-back loop is in play in the LP-BM5 retroviral system has not been determined. The fact that M-MDSCs from LP-BM5-infected mice utilize ROS-generation, which is more typically attributed to G-MDSCs, underscores the heterogeneity of MDSCs in different microenvironments or pathologies (16).
An important area where previous studies of G-MDSCs may inform the less well studied M-MDSCs is the source of ROS production. Potential sites of ROS production include the mitochondria (through the intracellular electron-transport-chain) (92), the endoplasmic reticulum (through cytochrome P450) (93), the plasma membrane (through NADPH oxidases, NOXs) (94), and/or the cytosol (through xanthine oxidase) (95). In mature myeloid cells, the primary source of superoxide is through to be NADPH oxidase 1 (NOX1) (76), however, studies in tumor-bearing mice indicate that NOX2 is the primary source of G-MDSC-produced ROS (49). Further, under low L-arginine conditions, iNOS can switch from primarily producing nitric oxide to superoxide (96, 97). However, data from M-MDSC supernates treated with both L-Nil and SOD indicate that here, iNOS is likely not responsible for superoxide generation (Fig. 6).
Superoxide and NO readily react to form peroxynitrite (76), which has been shown to cross cell membranes (98–100). Peroxynitrite is capable of nitrating and/or oxidizing a variety of targets leading to loss of function or structural integrity (101), including nucleotides (102–105), proteins (106–108), and lipids (100, 109–111). Nitrated lipids and fatty acids can be anti-inflammatory (101, 112). Interestingly, peroxynitrite is able to inactivate Mn superoxide dismutase (MnSOD), resulting in increased sensitivity of target cells to superoxide-mediated suppression, indicating that these two mechanisms may work together in a cooperative manner (113–115). Peroxynitrite can also induce cell death, either through necrosis at high, or apoptosis at lower, peroxynitrite levels (116–118). Which of these effects also occur in B cells subject to M-MDSC suppression is currently unknown.
Suppression was also found to be partially dependent on soluble TGF-β (Fig. 7). Previous published work corroborates that MDSCs are capable of producing TGF-β. Several reports indicate that in breast cancer models, MDSCs produce soluble TGF-β that inhibits CD8+ T-cell cytotoxic responses (45, 46). One report found that tumor-derived MDSC-membrane-bound TGF-β induces anergy of NK cells (44). While involvement for membrane-bound TGF-β was not investigated in this study (which focused on soluble mechanisms), our current and previous (23) results using blocking anti-VISTA mAb and iNOS inhibitors indicate that these two pathways are both the most dominant employed by wildtype M-MDSCs and together appear to be near fully responsible for the contact-dependent component of M-MDSC suppression. As such, it seems unlikely that membrane-bound TGF-β has a significant role. Although none of these MDSC studies looked at the effects of TGF-β on B cells, TGF-β produced by other cell types is known to induce apoptosis of resting B cells and to inhibit B-cell proliferation (119).
Relative to the potential importance of inhibition of both B- and T-cell responses in disease, mice deficient in CD4+ T cells or B cells are resistant to LP-BM5-induced pathology (120), due in substantial part to the dependence of disease on binding of CD154 (CD40L) on T cells with CD40 on B cells (28–33). M-MDSCs may serve a protective role in MAIDS by limiting proliferation or function of these pathogenic CD4+ T cells, as we and others have described and/or by targeting CD40+ B cells, and thus preventing their interaction with CD154+ CD4+ T cells. Alternatively, M-MDSC-mediated suppression may limit protective, antiviral immunity, enhancing disease. Further, cytokines produced by these pathogenic B- and T-cells may be required for MDSC-induction and disease-associated immunosuppression. Additional studies are needed to determine if, in MAIDS, HIV/AIDS, and other disease contexts, MDSCs are protective, deleterious, or active in opposing ways.
Previous studies in our lab have revealed that LP-BM5-expanded M-MDSCs suppress IL-10 production by regulatory CD4+ T cells (22). In this paper we demonstrate that these M-MDSCs also suppress regulatory B cells (Fig. 9a, b, and d). To our knowledge, the only other studies examining relationships of MDSCs and Bregs were correlative studies from human samples (121), which showed that numbers of IL-10-producing Bregs correlated with MDSC numbers, and a study in a murine lupus model, in which injection of MDSCs increased the number of IL-10 producing Bregs (38), making the direct ex vivo suppression (rather than enhancement) of Breg cells by M-MDSCs observed here a novel finding, and indicating that the effect of MDSC on Bregs, whether it be enhancement or suppression, may be dependent on the system. LP-BM5-expanded M-MDSCs were capable of decreasing the frequency of both IL-10-producing Tregs (22) and Bregs (Fig. 9a and b), and were able to inhibit IL-10 production on a per cell basis for Tregs (22). However, IL-10-production by Bregs was unchanged on a per cell basis (as determined by MFI) (Fig 9c). CFSE-dilution data indicate that M-MDSCs inhibit IL-10-producing regulatory B-cell proliferation (Fig. 9d).
Known mechanisms of LP-BM5-expanded wild-type B6 M-MDSC-mediated suppression of B cells are summarized in Fig. 10, which attempts to also provide a rough estimate of the relative contributions by the listed molecular mechanisms. M-MDSCs from LP-BM5-infected mice suppressed B cells in a manner substantially dependent on VISTA, with some but not all of the suppression replicated by VISTA-Ig, strongly suggesting direct VISTA-initiated target cell signaling (23). M-MDSC mediated suppression is also effected in significant part by soluble mediators including nitric oxide, superoxide, peroxynitrite, and TGF-β (Figs. 1, 4, 5, and 7). Importantly, by adding the percentages of blocking by the individual inhibitors, the combined contributions of ROS, RNS, and TGF-β can be viewed as accounting for approximately 95% of all of the suppression by M-MDSC-conditioned supernates. It is well understood that for both the supernate components of suppression, as well as the overall suppression including contact-dependent mechanism(s), there may be nuances of overlap that complicate this simple adding up. The data from the combination treatments in Figure 6, which show rough additivity, underscore the feasibility of the summation of mechanisms in Figure 10, and are useful to frame additional questions for future experimentation. That said, other, unknown soluble mediators, or contact-mediated receptor/ligand events, may also be involved especially if one or more mechanisms are not available by M-MDSC of a particular setting or subpopulation (24), or if a certain B-cell subset is resistant to that suppressive mechanism. Alternatively, there is recognition that it is also difficult to accurately sum up the proportionality of effector suppressive mechanisms due to the likelihood for plasticity of mechanisms and compensation. For example, there is evidence that VISTA has both iNOS/NO-dependent and independent mechanisms of suppression, perhaps with NO as a downstream mediator of VISTA ligation to its receptor on B cells, as suggested by CFSE-tracking of suppression by wild-type vs. iNOS−/− M-MDSCs (Fig. 10, ref. (23), and K. Green unpublished data).
Fig. 10. M-MDSC mechanisms of B-cell suppression.

Based on the studies presented in this paper and in ref. (21), mechanisms utilized by M-MDSCs from LP-BM5-infected wild-type mice are summarized with estimates of their relative contributions to their suppression of B-cell responses.
MDSCs have been implicated in human diseases such as inflammatory bowel disease, HIV/AIDS, and various cancers (18, 40, 122–125) indicating that they represent a potential therapeutic target. Adoptive MDSC-therapy, or induction or activation of MDSCs, may be able to suppress immune responses in the context of transplantation, autoimmunity, and other cases of immunopathology (1, 126). Our findings regarding the mechanisms of M-MDSC-mediated suppression of B cells could be especially relevant, as B cells are key players in a number of autoimmune disorders (127–129). The evidence here suggests that M-MDSCs use a variety of mechanisms to suppress B cells, and that when one or more of these mechanism(s) is removed, other mechanisms compensate and the cells remain suppressive. Indeed, genetic ablation of the VISTA and iNOS-dependent suppressive mechanisms resulted in compensation, such that for these double knockout M-MDSCs suppression of B-cell responsiveness there was no evidence for overall quantitative or qualitative differences compared to that seen in wild-type M-MDSCs (Fig. 9a), other than increased reliance on soluble mediators (Fig. 9b). Results using superoxide dismutase (Fig. 9b) and anti-TGF-β mAb (data not shown) indicated that (an)other, yet to be defined, mechanism(s) appears to compensate for the lack of VISTA and iNOS (and therefore nitric oxide and peroxynitrite), in the face of stable contributions of superoxide and TGF-β. This finding indicates that rather than blocking individual mechanisms of suppression, inhibition of MDSC induction or activation may prove to be a more successful therapeutic strategy. Pharmacological inhibition of MDSCs by neutralizing some of the pro-inflammatory signals that promote induction or activation of MDSCs, such as IFN-γ (9, 10, 17), IL-6(130–132)b, IL-1β (133, 134), complement component C5a (135), SA100A8/A9 (136), granulocyte-macrophage colony-stimulating factor (GM-CSF) (5), or vascular endothelial growth factor (VEGF) (4, 137, 138), could be helpful when MDSC suppression is directly or indirectly pathological, such as during cancer or chronic viral infection. Inducing differentiation of MDSCs to mature M1 macrophages could also stimulate protective immunity in these settings. On the other hand, in cases of immunopathology due to overzealous immune responses associated with inflammation or infection, adoptive transfer of MDSCs, or stimulation of endogenous MDSC responses, could be beneficial.
Highlights.
M-MDSCs from LP-BM5-infected mice utilized soluble mediators including nitric oxide, superoxide, peroxynitrite, and TGF-β to suppress B cells
When two major mechanisms of suppression were eliminated in double knockout mice, M-MDSCs maintained equivalent overall levels of suppression
M-MDSCs from LP-BM5-infected mice decreased proliferation of IL-10 producing regulatory B cells
Acknowledgments
We would like to thank David Leib, Mary Jo Turk, Kathy Green, Megan O’Connor, and Brent Berwin for technical assistance and helpful discussions.
Funding Information
This work was supported by Public Health Service Grants from the National Institute of Health: CA-50157 and a pilot grant fr9p”om P30 GM-10345, both to WRG as well as NIH grant T32 AI007363 (to Dr. Charles Sentman) which provided support to JLR. All animal experiments were done with the approval of the Institutional Animal Care and Use Committee of Dartmouth College, in conjunction with the Dartmouth Center for Comparative Medicine and Research, an AALAC approved animal facility.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Youn J-I, Gabrilovich DI. The biology of myeloid-derived suppressor cells: the blessing and the curse of morphological and functional heterogeneity. Eur J Immunol. 2010;40:2969–2975. doi: 10.1002/eji.201040895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bronte V, Wang M, Overwijk WW, Surman DR, Pericle F, Rosenberg SA, Restifo NP. Apoptotic Death of CD8+ T Lymphocytes After Immunization: Induction of a Suppressive Population of Mac-1+/Gr-1+ Cells. J Immunol. 1998;161:5313–5320. [PMC free article] [PubMed] [Google Scholar]
- 4.Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, Carbone DP. Vascular Endothelial Growth Factor Inhibits the Development of Dendritic Cells and Dramatically Affects the Differentiation of Multiple Hematopoietic Lineages In Vivo. Blood. 1998;92:4150–4166. [PubMed] [Google Scholar]
- 5.Bronte V, Apolloni E, Cabrelle A, Ronca R, Serafini P, Zamboni P, Restifo NP, Zanovello P. Identification of a CD11b+/Gr-1+/CD31+ myeloid progenitor capable of activating or suppressing CD8+ T cells. Blood. 2000;96:3838–3846. [PMC free article] [PubMed] [Google Scholar]
- 6.LaFace D, Talmadge J. Meeting report: Regulatory myeloid cells. Int Immunopharmacol. 2011;11:780–782. doi: 10.1016/j.intimp.2011.01.031. [DOI] [PubMed] [Google Scholar]
- 7.Ostrand-Rosenberg S. Myeloid-derived suppressor cells: more mechanisms for inhibiting antitumor immunity. Cancer Immunol Immunother. 2010;59:1593–1600. doi: 10.1007/s00262-010-0855-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol. 2010;22:238–244. doi: 10.1016/j.coi.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 9.Movahedi K, Guilliams M, denBossche JV, denBergh RV, Gysemans C, Beschin A, Baetselier PD, Ginderachter JAV. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell–suppressive activity. Blood. 2008;111:4233–4244. doi: 10.1182/blood-2007-07-099226. [DOI] [PubMed] [Google Scholar]
- 10.Youn J-I, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of Myeloid-Derived Suppressor Cells in Tumor-Bearing Mice. J Immunol. 2008;181:5791–5802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother. 2011;60:1419–1430. doi: 10.1007/s00262-011-1028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeisy-Scott V, Davis WG, Patel JR, Bowzard JB, Shieh W-J, Zaki SR, Katz JM, Sambhara S. Increased MDSC Accumulation and Th2 Biased Response to Influenza A Virus Infection in the Absence of TLR7 in Mice. PLoS ONE. 2011;6:e25242. doi: 10.1371/journal.pone.0025242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen S, Akbar SMF, Abe M, Hiasa Y, Onji M. Immunosuppressive functions of hepatic myeloid-derived suppressor cells of normal mice and in a murine model of chronic hepatitis B virus. Clin Exp Immunol. 2011;166:134–142. doi: 10.1111/j.1365-2249.2011.04445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Willmon C, Diaz RM, Wongthida P, Galivo F, Kottke T, Thompson J, Albelda S, Harrington K, Melcher A, Vile R. Vesicular Stomatitis Virus-induced Immune Suppressor Cells Generate Antagonism Between Intratumoral Oncolytic Virus and Cyclophosphamide. Mol Ther. 2011;19:140–149. doi: 10.1038/mt.2010.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walker JD, Sehgal I, Kousoulas KG. Oncolytic Herpes Simplex Virus 1 Encoding 15-Prostaglandin Dehydrogenase Mitigates Immune Suppression and Reduces Ectopic Primary and Metastatic Breast Cancer in Mice. J Virol. 2011;85:7363–7371. doi: 10.1128/JVI.00098-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crook KR, Jin M, Weeks MF, Rampersad RR, Baldi RM, Glekas AS, Shen Y, Esserman DA, Little P, Schwartz TA, Liu P. Myeloid-derived suppressor cells regulate T cell and B cell responses during autoimmune disease. J Leukoc Biol. 2015 doi: 10.1189/jlb.4A0314-139R. jlb.4A0314–139R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cripps JG, Wang J, Maria A, Blumenthal I, Gorham JD. Th1 cells induce the accumulation of myeloid derived suppressor cells in the inflamed Tgfb1 knockout mouse liver. Hepatol Baltim Md. 2010;52:1350–1359. doi: 10.1002/hep.23841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haile LA, von Wasielewski R, Gamrekelashvili J, Krüger C, Bachmann O, Westendorf AM, Buer J, Liblau R, Manns MP, Korangy F, Greten TF. Myeloid-Derived Suppressor Cells in Inflammatory Bowel Disease: A New Immunoregulatory Pathway. Gastroenterology. 2008;135:871–881. e5. doi: 10.1053/j.gastro.2008.06.032. [DOI] [PubMed] [Google Scholar]
- 19.Ioannou M, Alissafi T, Lazaridis I, Deraos G, Matsoukas J, Gravanis A, Mastorodemos V, Plaitakis A, Sharpe A, Boumpas D, Verginis P. Crucial Role of Granulocytic Myeloid-Derived Suppressor Cells in the Regulation of Central Nervous System Autoimmune Disease. J Immunol. 2012;188:1136–1146. doi: 10.4049/jimmunol.1101816. [DOI] [PubMed] [Google Scholar]
- 20.Zhu B, Kennedy JK, Wang Y, Sandoval-Garcia C, Cao L, Xiao S, Wu C, Elyaman W, Khoury SJ. Plasticity of Ly-6Chi Myeloid Cells in T Cell Regulation. J Immunol. 2011;187:2418–2432. doi: 10.4049/jimmunol.1100403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Green KA, Cook WJ, Green WR. Myeloid-Derived Suppressor Cells in Murine Retrovirus-Induced AIDS Inhibit T- and B-Cell Responses In Vitro That Are Used To Define the Immunodeficiency. J Virol. 2013;87:2058–2071. doi: 10.1128/JVI.01547-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Connor MA, Vella JL, Green WR. Reciprocal relationship of T regulatory cells and monocytic myeloid-derived suppressor cells in LP-BM5 murine retrovirus-induced immunodeficiency. J Gen Virol. 2015 doi: 10.1099/jgv.0.000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Green KA, Wang L, Noelle RJ, Green WR. Selective Involvement of the Checkpoint Regulator VISTA in Suppression of B-Cell, but Not T-Cell, Responsiveness by Monocytic Myeloid-Derived Suppressor Cells from Mice Infected with an Immunodeficiency-Causing Retrovirus. J Virol. 2015;89:9693–9698. doi: 10.1128/JVI.00888-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Connor MA, Fu WW, Green KA, Green WR. Subpopulations of M-MDSCs from mice infected by an immunodeficiency-causing retrovirus and their differential suppression of T- vs B-cell responses. Virology. 2015;485:263–273. doi: 10.1016/j.virol.2015.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aziz DC, Hanna Z, Jolicoeur P. Severe immunodeficiency disease induced by a defective murine leukaemia virus. Nature. 1989;338:505–508. doi: 10.1038/338505a0. [DOI] [PubMed] [Google Scholar]
- 26.Casabianca A, Orlandi C, Fraternale A, Magnani M. A new one-step RT-PCR method for virus quantitation in murine AIDS. J Virol Methods. 2003;110:81–90. doi: 10.1016/s0166-0934(03)00104-6. [DOI] [PubMed] [Google Scholar]
- 27.Tayar L, Higo K, Kubo Y, Wang Y, Lu L-M, Zhang F, Iwatani Y, Wang L, Ono T, Maeda M, Sakai H, Ishimoto A. Induction of B-Cell Lymphoma in BALB/c Nude Mice with an Ecotropic, B-Tropic Helper Virus Present in the Murine AIDS Virus Stock. J Virol. 1999;73:1640–1644. doi: 10.1128/jvi.73.2.1640-1644.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li W, Green WR. The Role of CD4 T Cells in the Pathogenesis of Murine AIDS. J Virol. 2006;80:5777–5789. doi: 10.1128/JVI.02711-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Green KA, Crassi KM, Laman JD, Schoneveld A, Strawbridge RR, Foy TM, Noelle RJ, Green WR. Antibody to the ligand for CD40 (gp39) inhibits murine AIDS-associated splenomegaly, hypergammaglobulinemia, and immunodeficiency in disease-susceptible C57BL/6 mice. J Virol. 1996;70:2569–2575. doi: 10.1128/jvi.70.4.2569-2575.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Green KA, Noelle RJ, Green WR. Evidence for a Continued Requirement for CD40/CD40 Ligand (CD154) Interactions in the Progression of LP-BM5 Retrovirus-Induced Murine AIDS. Virology. 1998;241:260–268. doi: 10.1006/viro.1997.8970. [DOI] [PubMed] [Google Scholar]
- 31.Yu P, Morawetz RA, Chattopadhyay S, Makino M, Kishimoto T, Kikutani H. CD40-deficient mice infected with the defective murine leukemia virus LP-BM5def do not develop murine AIDS but produce IgE and IgG1 in vivo. Eur J Immunol. 1999;29:615–625. doi: 10.1002/(SICI)1521-4141(199902)29:02<615::AID-IMMU615>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 32.Green KA, Ahonen CL, Cook WJ, Green WR. CD40-Associated TRAF 6 Signaling Is Required for Disease Induction in a Retrovirus-Induced Murine Immunodeficiency. J Virol. 2004;78:6055–6060. doi: 10.1128/JVI.78.11.6055-6060.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Green KA, Noelle RJ, Durell BG, Green WR. Characterization of the CD154-Positive and CD40-Positive Cellular Subsets Required for Pathogenesis in Retrovirus-Induced Murine Immunodeficiency. J Virol. 2001;75:3581–3589. doi: 10.1128/JVI.75.8.3581-3589.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schreiber KL, Forman J. Effects of bone marrow-derived natural suppressor activity on B cell responses to lipopolysaccharide. Transplantation. 1993;56:700–708. doi: 10.1097/00007890-199309000-00038. [DOI] [PubMed] [Google Scholar]
- 35.Angulo I, Rodríguez R, García B, Medina M, Navarro J, Subiza JL. Involvement of nitric oxide in bone marrow-derived natural suppressor activity. Its dependence on IFN-gamma. J Immunol Baltim Md 1950. 1995;155:15–26. [PubMed] [Google Scholar]
- 36.Maier T, Holda JH. Natural suppressor (NS) activity from murine neonatal spleen is responsive to IFN-gamma. J Immunol Baltim Md 1950. 1987;138:4075–4084. [PubMed] [Google Scholar]
- 37.Li Y, Tu Z, Qian S, Fung JJ, Markowitz SD, Kusner LL, Kaminski HJ, Lu L, Lin F. Myeloid-derived suppressor cells as a potential therapy for experimental autoimmune myasthenia gravis. J Immunol Baltim Md 1950. 2014;193:2127–2134. doi: 10.4049/jimmunol.1400857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park M-J, Lee S-H, Kim E-K, Lee E-J, Park S-H, Kwok S-K, Cho M-L. Myeloid-derived suppressor cells induce the expansion of regulatory B cells and ameliorate autoimmunity in Roquinsan/san mice, one of murine models of SLE. Arthritis Rheumatol. 2016:n/a–n/a. doi: 10.1002/art.39767. [DOI] [PubMed] [Google Scholar]
- 39.Kennedy DE, Knight KL. Inhibition of B Lymphopoiesis by Adipocytes and IL-1–Producing Myeloid-Derived Suppressor Cells. J Immunol. 2015;195:2666–2674. doi: 10.4049/jimmunol.1500957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vollbrecht T, Stirner R, Tufman A, Roider J, Huber RM, Bogner JR, Lechner A, Bourquin C, Draenert R. Chronic progressive HIV-1 infection is associated with elevated levels of myeloid-derived suppressor cells. AIDS Lond Engl. 2012;26:F31–37. doi: 10.1097/QAD.0b013e328354b43f. [DOI] [PubMed] [Google Scholar]
- 41.iNOS Signaling. SABiosciences; 2012. [Google Scholar]
- 42.Burrack KS, Morrison TE. The Role of Myeloid Cell Activation and Arginine Metabolism in the Pathogenesis of Virus-Induced Diseases. Front Immunol. 2014:5. doi: 10.3389/fimmu.2014.00428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Connor MA, Green WR. The role of indoleamine 2, 3-dioxygenase in LP-BPM5 murine retroviral disease progression. Virol J. 2013;10:154. doi: 10.1186/1743-422X-10-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-Expanded Myeloid-Derived Suppressor Cells Induce Anergy of NK Cells through Membrane-Bound TGF-β1. J Immunol. 2009;182:240–249. doi: 10.4049/jimmunol.182.1.240. [DOI] [PubMed] [Google Scholar]
- 45.Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, Carbone DP, Matrisian LM, Richmond A, Lin PC, Moses HL. Abrogation of TGFβ signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13:23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Z, Pang Y, Gara SK, Achyut BR, Heger C, Goldsmith PK, Lonning S, Yang L. Gr-1+CD11b+ cells are responsible for tumor promoting effect of TGF-β in breast cancer progression. Int J Cancer J Int Cancer. 2012;131:2584–2595. doi: 10.1002/ijc.27572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-Derived Suppressor Cells Inhibit T-Cell Activation by Depleting Cystine and Cysteine. Cancer Res. 2010;70:68–77. doi: 10.1158/0008-5472.CAN-09-2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-Specific Inhibition of CD8+ T Cell Response by Immature Myeloid Cells in Cancer Is Mediated by Reactive Oxygen Species. J Immunol. 2004;172:989–999. doi: 10.4049/jimmunol.172.2.989. [DOI] [PubMed] [Google Scholar]
- 49.Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI. Mechanism Regulating Reactive Oxygen Species in Tumor-Induced Myeloid-Derived Suppressor Cells. J Immunol. 2009;182:5693–5701. doi: 10.4049/jimmunol.0900092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res. 2001;61:4756–4760. [PubMed] [Google Scholar]
- 51.Szuster-Ciesielska A, Hryciuk-Umer E, Stepulak A, Kupisz K, Kandefer-Szerszeń M. Reactive oxygen species production by blood neutrophils of patients with laryngeal carcinoma and antioxidative enzyme activity in their blood. Acta Oncol Stockh Swed. 2004;43:252–258. doi: 10.1080/02841860410029708. [DOI] [PubMed] [Google Scholar]
- 52.Sarkar FH, Adsule S, Li Y, Padhye S. Back to the future: COX-2 inhibitors for chemoprevention and cancer therapy. Mini Rev Med Chem. 2007;7:599–608. doi: 10.2174/138955707780859431. [DOI] [PubMed] [Google Scholar]
- 53.Sharma S, Yang S-C, Zhu L, Reckamp K, Gardner B, Baratelli F, Huang M, Batra RK, Dubinett SM. Tumor Cyclooxygenase-2/Prostaglandin E2–Dependent Promotion of FOXP3 Expression and CD4+CD25+ T Regulatory Cell Activities in Lung Cancer. Cancer Res. 2005;65:5211–5220. doi: 10.1158/0008-5472.CAN-05-0141. [DOI] [PubMed] [Google Scholar]
- 54.Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, Gilbert J, Ochoa AC. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med. 2005;202:931–939. doi: 10.1084/jem.20050715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-Derived Suppressor Cells Promote Cross-Tolerance in B-Cell Lymphoma by Expanding Regulatory T Cells. Cancer Res. 2008;68:5439–5449. doi: 10.1158/0008-5472.CAN-07-6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang B, Pan P-Y, Li Q, Sato AI, Levy DE, Bromberg J, Divino CM, Chen S-H. Gr-1+CD115+ Immature Myeloid Suppressor Cells Mediate the Development of Tumor-Induced T Regulatory Cells and T-Cell Anergy in Tumor-Bearing Host. Cancer Res. 2006;66:1123–1131. doi: 10.1158/0008-5472.CAN-05-1299. [DOI] [PubMed] [Google Scholar]
- 57.Pan P-Y, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, Divino CM, Chen S-H. Immune Stimulatory Receptor CD40 Is Required for T-Cell Suppression and T Regulatory Cell Activation Mediated by Myeloid-Derived Suppressor Cells in Cancer. Cancer Res. 2010;70:99–108. doi: 10.1158/0008-5472.CAN-09-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kumar V, Sharma A. Adenosine: An endogenous modulator of innate immune system with therapeutic potential. Eur J Pharmacol. 2009;616:7–15. doi: 10.1016/j.ejphar.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 59.Malbec O, Fridman WH, Daëron M. Negative regulation of hematopoietic cell activation and proliferation by Fc gamma RIIB. Curr Top Microbiol Immunol. 1999;244:13–27. [PubMed] [Google Scholar]
- 60.Amigorena S, Bonnerot C, Drake JR, Choquet D, Hunziker W, Guillet JG, Webster P, Sautes C, Mellman I, Fridman WH. Cytoplasmic domain heterogeneity and functions of IgG Fc receptors in B lymphocytes. Science. 1992;256:1808–1812. doi: 10.1126/science.1535455. [DOI] [PubMed] [Google Scholar]
- 61.Muta T, Kurosaki T, Misulovin Z, Sanchez M, Nussenzweig MC, Ravetch JV. A 13-amino-acid motif in the cytoplasmic domain of Fc gamma RIIB modulates B-cell receptor signalling. Nature. 1994;368:70–73. doi: 10.1038/368070a0. [DOI] [PubMed] [Google Scholar]
- 62.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 63.Hanasaki K, Varki A, Stamenkovic I, Bevilacqua MP. Cytokine-induced beta-galactoside alpha-2,6-sialyltransferase in human endothelial cells mediates alpha 2,6-sialylation of adhesion molecules and CD22 ligands. J Biol Chem. 1994;269:10637–10643. [PubMed] [Google Scholar]
- 64.Rudge EU, Cutler AJ, Pritchard NR, Smith KGC. Interleukin 4 reduces expression of inhibitory receptors on B cells and abolishes CD22 and Fc gamma RII-mediated B cell suppression. J Exp Med. 2002;195:1079–1085. doi: 10.1084/jem.20011435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smith KG, Tarlinton DM, Doody GM, Hibbs ML, Fearon DT. Inhibition of the B cell by CD22: a requirement for Lyn. J Exp Med. 1998;187:807–811. doi: 10.1084/jem.187.5.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Doody GM, Justement LB, Delibrias CC, Matthews RJ, Lin J, Thomas ML, Fearon DT. A role in B cell activation for CD22 and the protein tyrosine phosphatase SHP. Science. 1995;269:242–244. doi: 10.1126/science.7618087. [DOI] [PubMed] [Google Scholar]
- 67.Chan VW, Lowell CA, DeFranco AL. Defective negative regulation of antigen receptor signaling in Lyn-deficient B lymphocytes. Curr Biol CB. 1998;8:545–553. doi: 10.1016/s0960-9822(98)70223-4. [DOI] [PubMed] [Google Scholar]
- 68.Parnes JR, Pan C. CD72, a negative regulator of B-cell responsiveness. Immunol Rev. 2000;176:75–85. doi: 10.1034/j.1600-065x.2000.00608.x. [DOI] [PubMed] [Google Scholar]
- 69.Maynard CL, Harrington LE, Janowski KM, Oliver JR, Zindl CL, Rudensky AY, Weaver CT. Regulatory T cells expressing interleukin 10 develop from Foxp3+ and Foxp3- precursor cells in the absence of interleukin 10. Nat Immunol. 2007;8:931–941. doi: 10.1038/ni1504. [DOI] [PubMed] [Google Scholar]
- 70.Klinken SP, Fredrickson TN, Hartley JW, Yetter RA, Morse HC. Evolution of B cell lineage lymphomas in mice with a retrovirus-induced immunodeficiency syndrome, MAIDS. J Immunol Baltim Md 1950. 1988;140:1123–1131. [PubMed] [Google Scholar]
- 71.Rowe WP, Pugh WE, Hartley JW. Plaque assay techniques for murine leukemia viruses. Virology. 1970;42:1136–1139. doi: 10.1016/0042-6822(70)90362-4. [DOI] [PubMed] [Google Scholar]
- 72.Green WR, Nowinski RC, Henney CS. The generation and specificity of cytotoxic T cells raised against syngeneic tumor cells bearing AKR/gross murine leukemia virus antigens. J Exp Med. 1979;150:51–66. doi: 10.1084/jem.150.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ukeda H, Maeda S, Ishii T, Sawamura M. Spectrophotometric Assay for Superoxide Dismutase Based on Tetrazolium Salt 3′-{1-[(Phenylamino)-carbonyl]-3,4-tetrazolium}-bis(4-methoxy-6-nitro)benzenesulfonic Acid Hydrate Reduction by Xanthine–Xanthine Oxidase. Anal Biochem. 1997;251:206–209. doi: 10.1006/abio.1997.2273. [DOI] [PubMed] [Google Scholar]
- 74.Bak SP, Alonso A, Jo Turk M, Berwin B. MURINE OVARIAN CANCER VASCULAR LEUKOCYTES REQUIRE ARGINASE-1 ACTIVITY FOR T CELL SUPPRESSION. Mol Immunol. 2008;46:258–268. doi: 10.1016/j.molimm.2008.08.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Andersen HR, Nielsen JB, Nielsen F, Grandjean P. Antioxidative enzyme activities in human erythrocytes. Clin Chem. 1997;43:562–568. [PubMed] [Google Scholar]
- 76.Lu T, Gabrilovich DI. Molecular Pathways: Tumor-Infiltrating Myeloid Cells and Reactive Oxygen Species in Regulation of Tumor Microenvironment. Clin Cancer Res. 2012;18:4877–4882. doi: 10.1158/1078-0432.CCR-11-2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nagaraj S, Gabrilovich DI. Tumor escape mechanism governed by myeloid-derived suppressor cells. Cancer Res. 2008;68:2561–2563. doi: 10.1158/0008-5472.CAN-07-6229. [DOI] [PubMed] [Google Scholar]
- 78.Lepoivre M, Chenais B, Yapo A, Lemaire G, Thelander L, Tenu JP. Alterations of ribonucleotide reductase activity following induction of the nitrite-generating pathway in adenocarcinoma cells. J Biol Chem. 1990;265:14143–14149. [PubMed] [Google Scholar]
- 79.Kwon NS, Stuehr DJ, Nathan CF. Inhibition of tumor cell ribonucleotide reductase by macrophage-derived nitric oxide. J Exp Med. 1991;174:761–767. doi: 10.1084/jem.174.4.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wink DA, Kasprzak KS, Maragos CM, Elespuru RK, Misra M, Dunams TM, Cebula TA, Koch WH, Andrews AW, Allen JS. DNA deaminating ability and genotoxicity of nitric oxide and its progenitors. Science. 1991;254:1001–1003. doi: 10.1126/science.1948068. [DOI] [PubMed] [Google Scholar]
- 81.Nguyen T, Brunson D, Crespi CL, Penman BW, Wishnok JS, Tannenbaum SR. DNA damage and mutation in human cells exposed to nitric oxide in vitro. Proc Natl Acad Sci U S A. 1992;89:3030–3034. doi: 10.1073/pnas.89.7.3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Harari O, Liao JK. Inhibition of MHC II Gene Transcription by Nitric Oxide and Antioxidants. Curr Pharm Des. 2004;10:893–898. doi: 10.2174/1381612043452893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fischer TA, Palmetshofer A, Gambaryan S, Butt E, Jassoy C, Walter U, Sopper S, Lohmann SM. Activation of cGMP-dependent Protein Kinase Iβ Inhibits Interleukin 2 Release and Proliferation of T Cell Receptor-stimulated Human Peripheral T Cells. J Biol Chem. 2001;276:5967–5974. doi: 10.1074/jbc.M009781200. [DOI] [PubMed] [Google Scholar]
- 84.Duhé RJ, Evans GA, Erwin RA, Kirken RA, Cox GW, Farrar WL. Nitric oxide and thiol redox regulation of Janus kinase activity. Proc Natl Acad Sci U S A. 1998;95:126–131. doi: 10.1073/pnas.95.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bingisser RM, Tilbrook PA, Holt PG, Kees UR. Macrophage-Derived Nitric Oxide Regulates T Cell Activation via Reversible Disruption of the Jak3/STAT5 Signaling Pathway. J Immunol. 1998;160:5729–5734. [PubMed] [Google Scholar]
- 86.Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, Zanovello P, Segal DM. Myeloid Suppressor Lines Inhibit T Cell Responses by an NO-Dependent Mechanism. J Immunol. 2002;168:689–695. doi: 10.4049/jimmunol.168.2.689. [DOI] [PubMed] [Google Scholar]
- 87.Pericle F, Pinto LA, Hicks S, Kirken RA, Sconocchia G, Rusnak J, Dolan MJ, Shearer GM, Segal DM. Cutting Edge: HIV-1 Infection Induces a Selective Reduction in STAT5 Protein Expression. J Immunol. 1998;160:28–31. [PubMed] [Google Scholar]
- 88.Pericle F, Kirken RA, Bronte V, Sconocchia G, DaSilva L, Segal DM. Immunocompromised tumor-bearing mice show a selective loss of STAT5a/b expression in T and B lymphocytes. J Immunol. 1997;159:2580–2585. [PubMed] [Google Scholar]
- 89.Macphail SE, Gibney CA, Brooks BM, Booth CG, Flanagan BF, Coleman JW. Nitric Oxide Regulation of Human Peripheral Blood Mononuclear Cells: Critical Time Dependence and Selectivity for Cytokine versus Chemokine Expression. J Immunol. 2003;171:4809–4815. doi: 10.4049/jimmunol.171.9.4809. [DOI] [PubMed] [Google Scholar]
- 90.Mannick JB, Hausladen A, Liu L, Hess DT, Zeng M, Miao QX, Kane LS, Gow AJ, Stamler JS. Fas-induced caspase denitrosylation. Science. 1999;284:651–654. doi: 10.1126/science.284.5414.651. [DOI] [PubMed] [Google Scholar]
- 91.Rivoltini L, Carrabba M, Huber V, Castelli C, Novellino L, Dalerba P, Mortarini R, Arancia G, Anichini A, Fais S, Parmiani G. Immunity to cancer: attack and escape in T lymphocyte-tumor cell interaction. Immunol Rev. 2002;188:97–113. doi: 10.1034/j.1600-065x.2002.18809.x. [DOI] [PubMed] [Google Scholar]
- 92.Eruslanov E, Kusmartsev S. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol Biol Clifton NJ. 2010;594:57–72. doi: 10.1007/978-1-60761-411-1_4. [DOI] [PubMed] [Google Scholar]
- 93.Suzuki Y, Ono Y, Hirabayashi Y. Rapid and specific reactive oxygen species generation via NADPH oxidase activation during Fas-mediated apoptosis. FEBS Lett. 1998;425:209–212. doi: 10.1016/s0014-5793(98)00228-2. [DOI] [PubMed] [Google Scholar]
- 94.Tammariello Steven P, Quinn MT, Estus S. t NADPH Oxidase Contributes Directly to Oxidative Stress and Apoptosis in Nerve Growth Factor-Deprived Sympathetic Neurons. J Neurosci. 2000;20:RC53–RC53. doi: 10.1523/JNEUROSCI.20-01-j0006.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Finkel T. Redox-dependent signal transduction. FEBS Lett. 2000;476:52–54. doi: 10.1016/s0014-5793(00)01669-0. [DOI] [PubMed] [Google Scholar]
- 96.Bronte V, Serafini P, Santo CD, Marigo I, Tosello V, Mazzoni A, Segal DM, Staib C, Lowel M, Sutter G, Colombo MP, Zanovello P. IL-4-Induced Arginase 1 Suppresses Alloreactive T Cells in Tumor-Bearing Mice. J Immunol. 2003;170:270–278. doi: 10.4049/jimmunol.170.1.270. [DOI] [PubMed] [Google Scholar]
- 97.Xia Y, Roman LJ, Masters BSS, Zweier JL. Inducible Nitric-oxide Synthase Generates Superoxide from the Reductase Domain. J Biol Chem. 1998;273:22635–22639. doi: 10.1074/jbc.273.35.22635. [DOI] [PubMed] [Google Scholar]
- 98.Denicola A, Souza JM, Radi R. Diffusion of peroxynitrite across erythrocyte membranes. Proc Natl Acad Sci U S A. 1998;95:3566–3571. doi: 10.1073/pnas.95.7.3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Marla SS, Lee J, Groves JT. Peroxynitrite rapidly permeates phospholipid membranes. Proc Natl Acad Sci U S A. 1997;94:14243–14248. doi: 10.1073/pnas.94.26.14243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J Biol Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- 101.Szabó C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 102.Salgo MG, Bermudez E, Squadrito GL, Pryor WA. DNA Damage and Oxidation of Thiols Peroxynitrite Causes in Rat Thymocytes. Arch Biochem Biophys. 1995;322:500–505. doi: 10.1006/abbi.1995.1493. [DOI] [PubMed] [Google Scholar]
- 103.Szabó C, Ohshima H. DNA Damage Induced by Peroxynitrite: Subsequent Biological Effects. Nitric Oxide. 1997;1:373–385. doi: 10.1006/niox.1997.0143. [DOI] [PubMed] [Google Scholar]
- 104.Kennedy LJ, Moore K, Caulfield JL, Tannenbaum SR, Dedon PC. Quantitation of 8-oxoguanine and strand breaks produced by four oxidizing agents. Chem Res Toxicol. 1997;10:386–392. doi: 10.1021/tx960102w. [DOI] [PubMed] [Google Scholar]
- 105.Niles JC, Wishnok JS, Tannenbaum SR. Peroxynitrite-induced oxidation and nitration products of guanine and 8-oxoguanine: Structures and mechanisms of product formation. Nitric Oxide. 2006;14:109–121. doi: 10.1016/j.niox.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 106.Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5:641–654. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- 107.Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, Herber D, Schneck J, Gabrilovich DI. Altered recognition of antigen is a novel mechanism of CD8+ T cell tolerance in cancer. Nat Med. 2007;13:828–835. doi: 10.1038/nm1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ekmekcioglu S, Ellerhorst J, Smid CM, Prieto VG, Munsell M, Buzaid AC, Grimm EA. Inducible Nitric Oxide Synthase and Nitrotyrosine in Human Metastatic Melanoma Tumors Correlate with Poor Survival. Clin Cancer Res. 2000;6:4768–4775. [PubMed] [Google Scholar]
- 109.Villa LM, Salas E, Darley-Usmar VM, Radomski MW, Moncada S. Peroxynitrite induces both vasodilatation and impaired vascular relaxation in the isolated perfused rat heart. Proc Natl Acad Sci U S A. 1994;91:12383–12387. doi: 10.1073/pnas.91.26.12383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rubbo H, Radi R, Trujillo M, Telleri R, Kalyanaraman B, Barnes S, Kirk M, Freeman BA. Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation. Formation of novel nitrogen-containing oxidized lipid derivatives. J Biol Chem. 1994;269:26066–26075. [PubMed] [Google Scholar]
- 111.Violi F, Marino R, Milite MT, Loffredo L. Nitric oxide and its role in lipid peroxidation. Diabetes Metab Res Rev. 1999;15:283–288. doi: 10.1002/(sici)1520-7560(199907/08)15:4<283::aid-dmrr42>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 112.Wright MM, Schopfer FJ, Baker PRS, Vidyasagar V, Powell P, Chumley P, Iles KE, Freeman BA, Agarwal A. Fatty acid transduction of nitric oxide signaling: Nitrolinoleic acid potently activates endothelial heme oxygenase 1 expression. Proc Natl Acad Sci U S A. 2006;103:4299–4304. doi: 10.1073/pnas.0506541103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.MacMillan-Crow LA, Crow JP, Kerby JD, Beckman JS, Thompson JA. Nitration and inactivation of manganese superoxide dismutase in chronic rejection of human renal allografts. Proc Natl Acad Sci U S A. 1996;93:11853–11858. doi: 10.1073/pnas.93.21.11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yamakura F, Taka H, Fujimura T, Murayama K. Inactivation of human manganese-superoxide dismutase by peroxynitrite is caused by exclusive nitration of tyrosine 34 to 3-nitrotyrosine. J Biol Chem. 1998;273:14085–14089. doi: 10.1074/jbc.273.23.14085. [DOI] [PubMed] [Google Scholar]
- 115.Quijano C, Hernandez-Saavedra D, Castro L, McCord JM, Freeman BA, Radi R. Reaction of Peroxynitrite with Mn-Superoxide Dismutase ROLE OF THE METAL CENTER IN DECOMPOSITION KINETICS AND NITRATION. J Biol Chem. 2001;276:11631–11638. doi: 10.1074/jbc.M009429200. [DOI] [PubMed] [Google Scholar]
- 116.Szabó C. Multiple pathways of peroxynitrite cytotoxicity. Toxicol Lett. 2003;140–141:105–112. doi: 10.1016/s0378-4274(02)00507-6. [DOI] [PubMed] [Google Scholar]
- 117.Jagtap P, Szabó C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005;4:421–440. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- 118.Virág L, Szabó É, Gergely P, Szabó C. Peroxynitrite-induced cytotoxicity: mechanism and opportunities for intervention. Toxicol Lett. 2003;140–141:113–124. doi: 10.1016/s0378-4274(02)00508-8. [DOI] [PubMed] [Google Scholar]
- 119.Li MO, Wan YY, Sanjabi S, Robertson A-KL, Flavell RA. TRANSFORMING GROWTH FACTOR-β REGULATION OF IMMUNE RESPONSES. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 120.Cerny A, Hügin AW, Hardy RR, Hayakawa K, Zinkernagel RM, Makino M, Morse HC. B cells are required for induction of T cell abnormalities in a murine retrovirus-induced immunodeficiency syndrome. J Exp Med. 1990;171:315–320. doi: 10.1084/jem.171.1.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liu J, Wang H, Yu Q, Zheng S, Jiang Y, Liu Y, Yuan G, Qiu L. Aberrant frequency of IL-10-producing B cells and its association with Treg and MDSC cells in Non Small Cell Lung Carcinoma patients. Hum Immunol. 2016;77:84–89. doi: 10.1016/j.humimm.2015.10.015. [DOI] [PubMed] [Google Scholar]
- 122.Almand B, Clark JI, Nikitina E, Beynen J, van English NR, Knight SC, Carbone DP, Gabrilovich DI. Increased Production of Immature Myeloid Cells in Cancer Patients: A Mechanism of Immunosuppression in Cancer. J Immunol. 2001;166:678–689. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- 123.Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, McDermott D, Quiceno D, Youmans A, O’Neill A, Mier J, Ochoa AC. Arginase-Producing Myeloid Suppressor Cells in Renal Cell Carcinoma Patients: A Mechanism of Tumor Evasion. Cancer Res. 2005;65:3044–3048. doi: 10.1158/0008-5472.CAN-04-4505. [DOI] [PubMed] [Google Scholar]
- 124.Liu C-Y, Wang Y-M, Wang C-L, Feng P-H, Ko H-W, Liu Y-H, Wu Y-C, Chu Y, Chung F-T, Kuo C-H, Lee K-Y, Lin S-M, Lin H-C, Wang C-H, Yu C-T, Kuo H-P. Population alterations of L-arginase- and inducible nitric oxide synthase-expressed CD11b+/CD14−/CD15+/CD33+ myeloid-derived suppressor cells and CD8+ T lymphocytes in patients with advanced-stage non-small cell lung cancer. J Cancer Res Clin Oncol. 2010;136:35–45. doi: 10.1007/s00432-009-0634-0. [DOI] [PubMed] [Google Scholar]
- 125.Poschke I, Mougiakakos D, Hansson J, Masucci GV, Kiessling R. Immature Immunosuppressive CD14+HLA-DR−/low Cells in Melanoma Patients Are Stat3hi and Overexpress CD80, CD83, and DC-Sign. Cancer Res. 2010;70:4335–4345. doi: 10.1158/0008-5472.CAN-09-3767. [DOI] [PubMed] [Google Scholar]
- 126.Cripps JG, Gorham JD. MDSC in Autoimmunity. Int Immunopharmacol. 2011;11:789–793. doi: 10.1016/j.intimp.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dörner T. Crossroads of B cell activation in autoimmunity: rationale of targeting B cells. J Rheumatol. 2006;77:3–11. [PubMed] [Google Scholar]
- 128.Burmester GR, Feist E, Dörner T. Emerging cell and cytokine targets in rheumatoid arthritis. Nat Rev Rheumatol. 2014;10:77–88. doi: 10.1038/nrrheum.2013.168. [DOI] [PubMed] [Google Scholar]
- 129.Wahren-Herlenius M, Dörner T. Immunopathogenic mechanisms of systemic autoimmune disease. The Lancet. 2013;382:819–831. doi: 10.1016/S0140-6736(13)60954-X. [DOI] [PubMed] [Google Scholar]
- 130.Mace TA, Bloomston M, Lesinski GB. Pancreatic cancer-associated stellate cells: A viable target for reducing immunosuppression in the tumor microenvironment. Oncoimmunology. 2013;2:e24891. doi: 10.4161/onci.24891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mantovani A, Sica A, Allavena P, Garlanda C, Locati M. Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Hum Immunol. 2009;70:325–330. doi: 10.1016/j.humimm.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 132.Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, Ugel S, Sonda N, Bicciato S, Falisi E, Calabrese F, Basso G, Zanovello P, Cozzi E, Mandruzzato S, Bronte V. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity. 2010;32:790–802. doi: 10.1016/j.immuni.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 133.Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Inflammation Induces Myeloid-Derived Suppressor Cells that Facilitate Tumor Progression. J Immunol. 2006;176:284–290. doi: 10.4049/jimmunol.176.1.284. [DOI] [PubMed] [Google Scholar]
- 134.Song X, Krelin Y, Dvorkin T, Bjorkdahl O, Segal S, Dinarello CA, Voronov E, Apte RN. CD11b+/Gr-1+ Immature Myeloid Cells Mediate Suppression of T Cells in Mice Bearing Tumors of IL-1β-Secreting Cells. J Immunol. 2005;175:8200–8208. doi: 10.4049/jimmunol.175.12.8200. [DOI] [PubMed] [Google Scholar]
- 135.Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, Coukos G, Lambris JD. Modulation of the anti-tumor immune response by complement. Nat Immunol. 2008;9:1225–1235. doi: 10.1038/ni.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, Ortiz M, Nacken W, Sorg C, Vogl T, Roth J, Gabrilovich DI. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008;205:2235–2249. doi: 10.1084/jem.20080132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ellis LM, Takahashi Y, Liu W, Shaheen RM. Vascular Endothelial Growth Factor in Human Colon Cancer: Biology and Therapeutic Implications. The Oncologist. 2000;5:11–15. doi: 10.1634/theoncologist.5-suppl_1-11. [DOI] [PubMed] [Google Scholar]
- 138.Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, Kavanaugh D, Carbone DP. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 1996;2:1096–1103. doi: 10.1038/nm1096-1096. [DOI] [PubMed] [Google Scholar]