

ABSTRACT
Historically, the ubiquitin-proteasome system (UPS) and autophagy pathways were believed to be independent; however, recent data indicate that these pathways engage in crosstalk. To date, the players mediating this crosstalk have been elusive. Here, we show experimentally that EI24 (EI24, autophagy associated transmembrane protein), a key component of basal macroautophagy/autophagy, degrades 14 physiologically important E3 ligases with a RING (really interesting new gene) domain, whereas 5 other ligases were not degraded. Based on the degradation results, we built a statistical model that predicts the RING E3 ligases targeted by EI24 using partial least squares discriminant analysis. Of 381 RING E3 ligases examined computationally, our model predicted 161 EI24 targets. Those targets are primarily involved in transcription, proteolysis, cellular bioenergetics, and apoptosis and regulated by TP53 and MTOR signaling. Collectively, our work demonstrates that EI24 is an essential player in UPS-autophagy crosstalk via degradation of RING E3 ligases. These results indicate a paradigm shift regarding the fate of E3 ligases.
KEYWORDS: autophagy, computational biology, E3 ligase, EI24, proteasome, RING-domain
Introduction
The UPS and autophagy are 2 major protein degradation mechanisms, and they regulate nearly all aspects of cellular physiology.1 The proteasome system specifically recognizes ubiquitinated proteins. By contrast, engulfment of cytosolic contents by a phagophore (the precursor to the autophagosome) was considered to be nonselective.2 However, selective autophagy receptors, such as SQSTM1/p62 (sequestosome 1) and NBR1 (NBR1, autophagy cargo receptor), have recently been identified.3,4 These receptors specifically bind ubiquitin via the ubiquitin-associated domain (UBA) and deliver these ubiquitinated proteins to the phagophore through an interaction between MAP1LC3A/B (microtubule-associated protein 1, light chain 3 α/β; described as LC3 in the text hereafter) and the LC3-interacting region (LIR) motif. This discovery has fundamentally altered the perception that autophagy is a random cytosolic event and has established shared degradation mechanisms with the UPS.3,4 Furthermore, a recent report demonstrates autophagic degradation of the 26S proteasome in Arabidopsis, which supports the hypothesis that there is crosstalk between autophagy and the UPS.5 However, concrete evidence connecting the UPS with autophagy and the molecular players mediating their crosstalk in mammalian system is lacking.
EI24 (EI24, autophagy-associated transmembrane protein) is a target gene of TP53/p53 with tumor suppressor activity that plays an important role in the negative regulation of cell growth.6 We have reported that EI24 suppresses the epithelial-to-mesenchymal transition (EMT) and tumor progression by suppressing RELA/NFKB p65 (RELA proto-oncogene, NF-kB subunit) activity, which induces autophagy-dependent degradation of RING (really interesting new gene) E3 ligases, including TRAF2 (TNF receptor associated factor 2) and TRAF5.7 We have also reported that EI24-induced degradation of a RING E3 ligase, TRIM41/RINCK1 (tripartite motif containing 41), results in PRKCA/PKCα (protein kinase C α) stabilization, and this signaling is important for the development of DMBA-TPA (7,12-dimethylbenz[a]-anthracene-12-O-tetradecanoylphorbol-13-acetate)-induced skin carcinogenesis in mice.8 Based on these studies illustrating EI24-mediated degradation of RING domain E3 ligases and recent reports describing EI24 as an essential autophagy gene in C. elegans9 and mice,10 we hypothesized that EI24 might degrade RING E3 ligases using the autophagy machinery, thus establishing a connection between autophagy and the UPS. This proposed autophagy-UPS crosstalk may be responsible for orchestrating diverse cellular processes.
Results
EI24 degrades TRIM41 by autophagy independent of the proteasome system
The involvement of EI24 in autophagy has first been identified while screening genes required for autophagy in C. elegans,9 and a follow-up study reports this gene as an essential component of basal autophagy in mammals.10 However, the functional role of EI24-induced autophagy remains to be elucidated.
We used appearance of punctate LC3-II, a reliable marker11 for monitoring EI24-mediated autophagy activation. Consistent with previous reports,9,10 ectopic expression of EI24 resulted in the formation of punctate LC3-II (Fig. 1A). Furthermore, EI24 transfection in 293T cells resulted in increased LC3-II formation along with SQSTM1 degradation (Fig. 1B). Autophagy activation, induced by nutrient depletion using HBSS (Hank's balanced salt solution), was dysfunctional when EI24 expression was knocked down by an EI24-specific siRNA (Fig. 1C). Collectively, these data demonstrate that EI24 activates autophagy and is required for the proper execution of autophagic flux.
Figure 1.
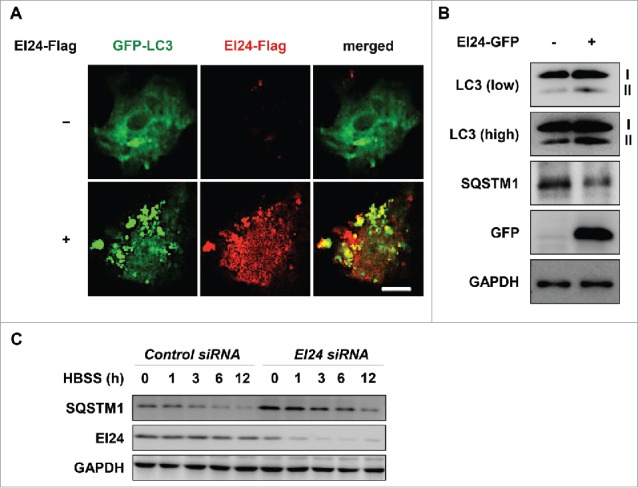
EI24 activates autophagy. (A) H1299 cells, with or without EI24 overexpression, were transfected with GFP-LC3, and immunocytochemistry was performed. LC3, green; EI24, red; nucleus, blue (4′,6-diamidino-2-phenylindole, DAPI). Scale bar: 10 μm. (B) EI24 overexpression results in the formation of LC3-II and degradation of SQSTM1 in 293T cells. (C) SQSTM1 is not degraded when EI24 is knocked down. When EI24 levels decreased in 293T cells, SQSTM1 failed to degrade in HBSS-treated conditions.
We have previously reported that EI24 binds and degrades TRIM41, resulting in PRKCA stabilization.8 However, we were unable to determine the detailed mechanism of TRIM41 degradation by EI24 at that time. Because TRIM41 self-ubiquitinates and undergoes proteasome-dependent degradation,12 we first examined whether EI24-mediated degradation of TRIM41 occurs via the UPS. EI24 overexpression reduced TRIM41-Flag level; however, the ubiquitinated TRIM41 signal also decreased (Fig. 2A). Furthermore, TRIM41 ubiquitination was not rescued when cells were treated with the proteasome inhibitor MG132, suggesting proteasome-independent degradation (Fig. 2A). Because the autophagy pathway is the alternate protein degradation machinery13 and EI24 is essential for basal autophagy,10 we examined the potential role of EI24 in autophagy-dependent TRIM41 degradation. EI24 overexpression induced TRIM41 degradation (Fig. 2B, lane 1 and 2), and TRIM41 ubiquitination and protein level were rescued when autophagy was inhibited using bafilomycin A1 (BAF) (Fig. 2B, lane 3, 4). Therefore, EI24 degrades TRIM41 through autophagy, independent of the UPS.
Figure 2.
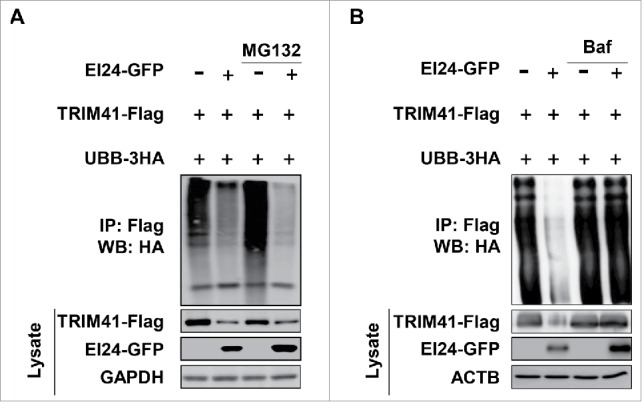
EI24 degrades TRIM41 using autophagy. (A) TRIM41 is degraded by EI24, independent of the proteasome. 293T cells were transfected with TRIM41-Flag and 3HA-ubiquitin B (UBB), with or without EI24-GFP overexpression, and TRIM41 ubiquitination was observed. MG132 (10 μM, 6 h) was used as a proteasome inhibitor. (B) EI24 degrades TRIM41 in an autophagy-dependent manner. TRIM41 ubiquitination, with or without EI24 overexpression, was examined, and autophagy was inhibited using BAF (10 mM, 6 h).
The RING domain is essential for TRIM41 binding and degradation by EI24
To dissect the molecular mechanism of autophagy-dependent TRIM41 degradation by EI24, we investigated which domain in TRIM41 binds EI24. For this purpose, we generated Flag-tagged TRIM41 deletion constructs (Fig. 3A). Immunoprecipitation assays revealed that the B-box, coiled-coil 1 (CC1), CC2, and PTPN13-like protein, Y-linked (PRY) domains were dispensable, whereas the RING domain was required for EI24 binding (Fig. 3B). To functionally validate that the RING domain binds EI24, we examined whether TRIM41 with a deleted RING domain (TRIM41RINGΔ) is resistant to EI24-induced degradation. Consistent with our domain-mapping data, EI24 did not degrade TRIM41RINGΔ (Fig. 3C). To determine the role of endogenous EI24 in TRIM41 degradation, Flag-tagged TRIM41 and TRIM41RINGΔ were expressed in cells transfected with either control or EI24-specific siRNA, and protein level changes were determined by immunoblotting. Reduced EI24 expression increased the protein levels of TRIM41 but not TRIM41RINGΔ (Fig. 3D). Collectively, these results indicate that EI24 may recognize the RING domain, which is a common domain in E3 ubiquitin ligases, and degrade them using the autophagy pathway.
Figure 3.
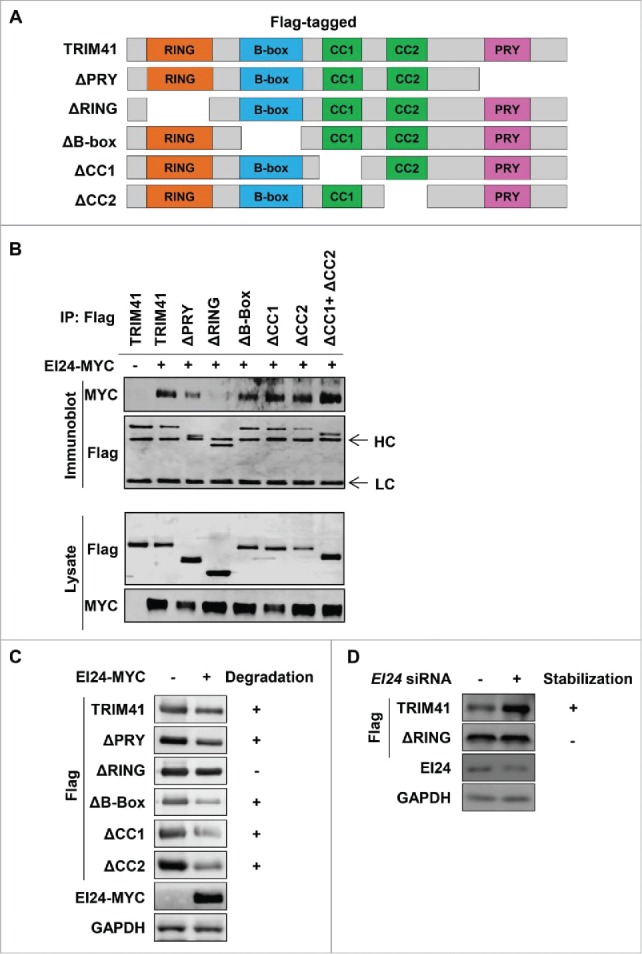
EI24 binds and degrades TRIM41 through the RING domain. (A) Depiction of the Flag-tagged TRIM41 deletion constructs generated in this study. (B) The RING domain facilitates binding between EI24 and TRIM41. The constructs were individually transfected in 293T cells and immunoprecipitation was performed using an anti-Flag antibody. Bound MYC-tagged EI24 was detected by immunoblotting. HC and LC represent immunoglobulin heavy and light chains, respectively. (C, D) TRIM41 lacking the RING domain is resistant to EI24-mediated degradation. The indicated constructs were transfected in 293T cells and immunoblotting was used to determine the protein levels of TRIM41 constructs with or without EI24 overexpression (C) or knockdown (D).
EI24 degrades RING domain-containing E3 ligases
Previously, we have reported that EI24 degrades TRAF2/5 in a RING domain-dependent manner.7 Here, we observed that TRIM41, another E3 ligase, was also degraded by EI24 in a RING domain-dependent manner via the autophagy pathway. Because the RING domain is functional in most E3 ligases, we hypothesized that EI24-induced autophagy may regulate the E3 ligase protein levels. If this hypothesis held true, it would represent a paradigm shift regarding E3 ligase regulation. Currently, E3 ligase regulation is thought to be governed predominantly by self-ubiquitination and degradation by the proteasome and recycling.14 To test this hypothesis, we examined EI24-induced degradation of several E3 ligases in the TRIM (tripartite motif) family, including MID2/TRIM1, TRIM3, TRIM4, TRIM6, and TRIM21. Immunoblotting revealed that all 5 of the TRIMs tested were degraded by EI24 (Fig. 4A). To ensure that the autophagy pathway degraded the target proteins, we examined MID2 degradation by EI24, both with and without BAF, an autophagosome-lysosome fusion inhibitor. EI24-mediated degradation of MID2 was rescued in the presence of BAF, suggesting autophagic degradation (Fig. 4B). FBXO7 (F-Box protein 7) and STUB1/CHIP (STIP1 homology and U-box containing protein 1), which lack RING domains and belong to the F-box and U-box family of E3 ligases, respectively, were EI24-degradation resistant (Fig. 4C). These results indicate that the RING domain is required for degradation of target proteins by EI24.
Figure 4.
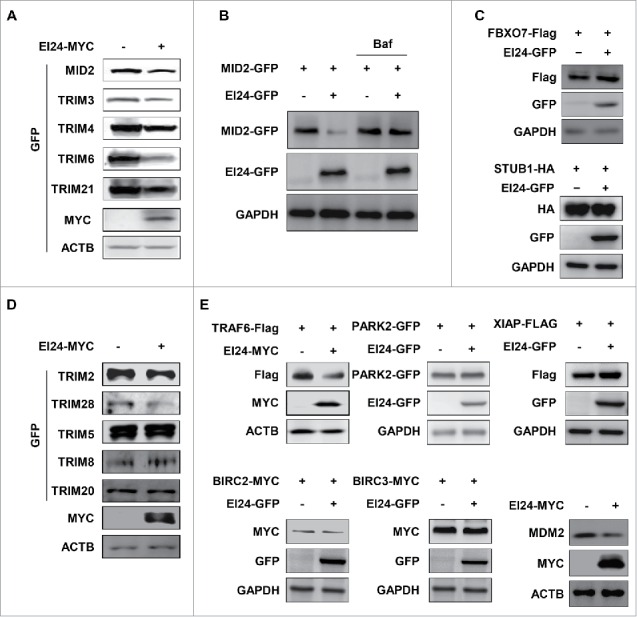
EI24 degrades RING domain-containing proteins. (A, D) EI24-mediated degradation of GFP-tagged TRIM family proteins was detected using immunoblotting. MID2, TRIM2, mouse TRIM3, TRIM4, TRIM5, TRIM6, TRIM8, TRIM20, TRIM21, and mouse TRIM26 were tested. (B) EI24 degrades MID2 using autophagy. EI24-mediated degradation of MID2 could be rescued by treatment with BAF (10 nM, 6 h). (C) Protein levels of FBXO7 and STUB1 lacking RING domains were examined for their susceptibility to EI24-mediated degradation. (E) The susceptibility of the RING domain-containing E3 ligases TRAF6, PARK2, XIAP, BIRC2, BIRC3, and MDM2 to EI24-mediated degradation.
To clearly establish if all RING domain-containing proteins can be degraded by EI24, we examined more TRIM proteins for their susceptibility to EI24-mediated degradation. Interestingly, immunoblotting revealed that TRIM2 and TRIM28 were degraded by EI24, whereas TRIM5 delta (TRIM5δ), TRIM8, and TRIM20 protein levels were unchanged (Fig. 4D).
A previous study demonstrated that EI24 binds to the RING domain of TRAF2/5 and degrades them via the autophagy pathway. Therefore, we expanded our screen to determine whether other RING E3 ligases can be degraded by EI24. Immunoblotting revealed that EI24 overexpression induced the degradation of physiologically important E3 ligases, including TRAF6, BIRC2/CIAP1 (baculoviral IAP repeat containing 2), and MDM2 (MDM2 proto-oncogene), whereas PARK2 (parkin RBR E3 ubiquitin protein ligase), XIAP (X-linked inhibitor of apoptosis), and BIRC3/CIAP2 (baculoviral IAP repeat containing 3) were not degraded (Fig. 4E). These results indicate that the presence of a RING domain is not the only requirement for susceptibility to EI24-mediated degradation.
The RING domain is required for EI24-mediated degradation of E3 ligases
TRIM41 lacking the RING domain is resistant to EI24-mediated degradation (Fig. 3C), which suggests that the RING domain is important for orchestrating the connection between the UPS and autophagy pathways. Given that the RING domain is important for EI24-mediated degradation of E3 ligases, we generated constructs of several E3 ligases that lacked a RING domain and examined if they were resistant to EI24-induced degradation. Full-length TRAF2 and MKRN1 (makorin ring finger protein 1) were degraded by EI24, whereas TRAF2RINGΔ (Fig. 5A) and MKRN1RINGΔ (Fig. 5B) were not. In addition, EI24-induced MRKN1 degradation was rescued with BAF treatment, suggesting that EI24 mediates autophagic MKRN1 degradation (Fig. 5B). These data underscore the fact that the RING domain is essential for recognition and autophagic degradation of EI24-degraded proteins. Thus far, our study has unraveled, for the first time, that the crosstalk between the UPS and autophagy is based on the ability of the basal autophagy protein EI24 to bind and degrade E3 ligases with RING domains.
Figure 5.
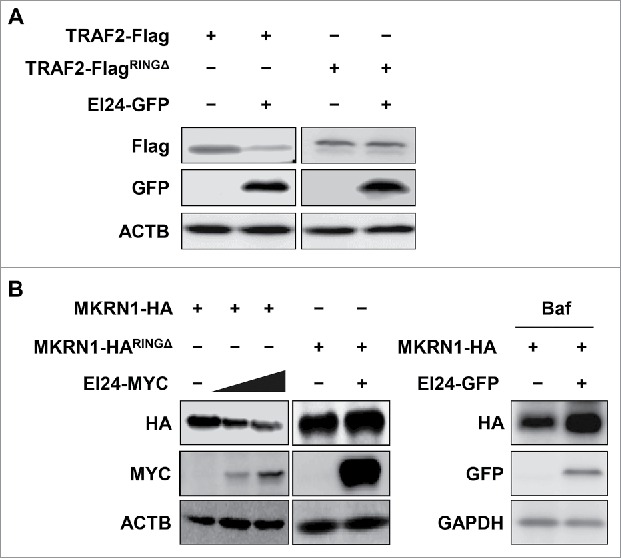
The RING domain is required for EI24-mediated degradation of E3 ligases. Degradation of full-length and RING domain-deficient TRAF2 (A) and MKRN1 (B) by EI24 was examined using immunoblotting. The lack of RING domain confers resistance to EI24-mediated degradation. EI24-induced degradation of MKRN1 was rescued by BAF treatment (10 μM, 6 h).
Functional characterization of EI24-induced, autophagic E3 ligase degradation
The data thus far indicated that something other than the presence of a RING domain may define whether an E3 ligase is targeted by EI24 for autophagy-mediated degradation. To investigate this, we compared the RING domain sequences of the 14 E3 ligases degraded by EI24 (Group 1) with those of the 5 E3 ligases that were not degraded (Group 2). However, the multiple sequence alignment showed no apparent sequence motif related to EI24-mediated degradation susceptibility, because Groups 1 and 2 RING domain sequences have a high degree of similarity (e.g. BIRC2 in Group 1 and BIRC3 in Group 2; Fig. S1A and B).
Another possibility is that the subcellular localization of protein targets could potentially indicate the target's susceptibility for EI24-mediated autophagic degradation. Thus, we compared subcellular localizations of E3 ligases in Groups 1 and 2 based on their GOCCs (gene ontology cellular components). The comparison showed no significant difference in the localization distribution between Groups 1 and 2 (Fig. S1C). Next, we considered if EI24 target proteins have common binding partners with EI24. We analyzed common EI24 binding partners that were also capable of binding E3 ligases in Group 1 and compared them with those in Group 2. To this end, we combined the EI24 interactors in interactome databases15-19 with experimentally verified EI24 binding partners20 to compile a list of EI24 interactors. The interactome analysis comparing Groups 1 and 2 revealed no EI24 interactors that preferentially bound targets in Group 1 compared with those in Group 2 (Fig. S1D).
Gene expression differences between Group 1 and 2 E3 ligases could also potentially contribute to EI24-mediated autophagic degradation susceptibility. To investigate this hypothesis, we examined whether gene expression data can separate Groups 1 and 2 by applying principal component analysis (PCA) to EI24-related gene expression data previously reported and then found that the combined use of the following 2 gene expression datasets can provide effective separation of Groups 1 and 2: 1) GSE52508 in the GEO (gene expression omnibus) database collected from ZR-75-1 breast cancer cells after EI24 knockdown7 and 2) GSE67266 in the GEO database collected from MEF cells after treatment with etoposide, which induces EI24 expression21 (Fig. S2A, B, and C). The use of single data sets showed no separation between Groups 1 and 2 in the PCA space (Fig. S2A and B), but the use of both datasets showed a certain degree of the separation (Fig. S2C). For more effective separation captured by PCA with the 2 data sets, we applied MPLS-DA (multi-block partial least square-discriminant analysis) that can effectively integrate the 2 datasets for classification of Groups 1 and 2 as previously described.22,23 MPLS-DA successfully separated Group 1 from Group 2 (Fig. S2D and E). Using this MPLS-DA model, we then predicted those E3 ligases likely to be susceptible to EI24 degradation. Previous studies identified 689 potential E3 ligases,24,25 381 of which possess RING domains. Those 381 E3 ligases were used as the starting point for our MPLS-DA analysis (Fig. S2F). The MPLS-DA model predicted 161 E3 ligases (predicted Group [pGroup] 1) to be EI24 targets and 64 E3 ligases (pGroup 2) to be nontargets (Fig. 6A; Table S1). The delineation of E3 ligases into targets and nontargets could potentially be used to predict the susceptibility of a particular E3 ligase to EI24-mediated degradation. Notably, the computationally generated pGroups 1 and 2 correctly categorized the previously tested E3 ligases into their respective experimentally identified Groups (Figs. 3 and 4).
Figure 6.

Functional characterization of E3 ligases targeted by EI24. (A) Projected scores (thatc1-3) of predicted EI24 targets (pGroup 1) and nontargets (pGroup 2) for the first 3 MPLS-DA latent variables (LV1-3). Red and blue triangles represent experimentally identified Group 1 (targets) and Group 2 (nontargets) E3 ligases, respectively. Orange and light blue circles denote pGroup 1 (predicted targets) and pGroup 2 (predicted nontargets) E3 ligases, respectively. (B) Correlation coefficient distributions of E3 ligase gene expression levels in pGroups 1 and 2 were evaluated in 2 datasets collected following EI24 knockdown (EI24 KD) and etoposide treatment (EI24 induced). P values represent the significance of the difference between the pGroup 1 and 2 distributions. The GOBPs (C) and GOMFs (D) enriched in pGroups 1 and 2. Enrichment P values for the GOBPs and GOMFs were displayed as -log10(P values). The number of genes with the corresponding GOBP or GOMF in pGroups 1 and 2 is shown in parenthesis (pGroup 1/pGroup 2). Upstream transcription factors (TFs) (E) and kinases (F) enriched in pGroups 1 and 2. P values (P) represent the significance of TF targets and kinase substrates enriched in pGroups 1 and 2.
EI24 target expression is likely to be correlated with EI24 expression. Therefore, we examined the correlation between pGroups and EI24 gene expression in the 2 data sets. Following EI24 knockdown or etoposide treatment, EI24 expression was more strongly correlated with pGroup 1 expression than pGroup 2 expression (Fig. 6B, Fig. S3A).
We could not observe a difference in cellular localizations of proteins in Group 1 and Group 2 (Fig. S1C), which may be attributed to the small size of the samples analyzed (Group 1 sample size = 14, Group 2 sample size = 5). pGroup 1 (n = 161) and pGroup 2 (n = 64) can ensure sufficiently large sample sizes. Thus, we re-examined if there is any difference in the cellular localization between EI24 targets and nontargets using pGroup 1 and pGroup 2. With the varying stringency of probability of a particular E3 ligase belonging to Group 1 or Group 2, we examined GOCCs of the predicted E3 ligases and found that pGroup 1 and pGroup 2 candidates neatly aligned themselves in separate GOCC attributes (Fig. S3B). On the one hand, pGroup 1 members displayed the tendency to be primarily localized to cellular organelles or structures such as endosomes, ubiquitin ligase complexes, vacuoles, lysosomes, chromatin, and the cytoskeleton, most of which are involved in autophagy.26 On the other hand, pGroup 2 was related with perinuclear region of the cytoplasm and Golgi apparatus (Fig. S3B). These results illustrate that in addition to the presence of the RING-domain, the difference in the cellular localization of E3 ligases could be an additional factor that determines the susceptibility of a particular E3 ligase to be degraded by EI24.
To evaluate fairly both sensitivity and specificity in prediction of EI24 targets, we randomly selected 5 predicted EI24 targets and 5 nontargets from pGroups 1 and 2, respectively, which include no E3 ligases in the training set, and then experimentally tested whether the selected E3 ligases are targeted by EI24. Consistent with the MPLS-DA prediction results, all the tested pGroup 1 E3 ligases such as RNF43, RNF6, RNF11, and PML (PML IV: 1–633 amino acids; PML VI: 1–560 amino acids) were truly degraded by EI24, whereas pGroup 2 members such as RNF128, RNF5, ZNF462, and TRIM72 were not (Fig. S4A and B). These experimental results indicate high degrees of sensitivity and specificity to our model. This result provides the credibility to our model in correctly predicting the susceptibility of a particular E3 ligase to EI24-mediated degradation. Interestingly, CBLC (Cbl proto-oncogene C) that belonged to pGroup 2 was also degraded by EI24 (Fig. S4A). When we examined the distribution of casitas B-lineage lymphoma (CBL)-like proteins in our prediction, we found that CBLB and CBLC belonged to pGroup 2 whereas CBLL1 was predicted to be pGroup1. A similar observation was made regarding CIAP isoforms (BIRC2 in Group 1 and BIRC3 in in Group 2, Fig. 4E). Thus, further studies need to focus to elucidate why similar isoforms categorize themselves in separate groups.
To functionally characterize the pGroups, we performed an enrichment analysis of GOBPs (gene ontology biological functions) and GOMFs (molecular functions) on pGroups 1 and 2, and then compared GOBPs and GOMFs between the 2. GOBP analysis showed that proteolysis was enriched in pGroup 1 compared with pGroup 2 (Fig. 6C; Table S2). pGroup 1 also had higher enrichment for apoptosis, cell cycle, histone and chromatin modifications, regulation of RNA metabolic processes, and response to DNA damage stimulus functions. GOMF analysis showed that DNA and chromatin binding, zinc ion binding, and transcription activator/coactivator activities were enriched in pGroup 1 (Fig. 6D; Table S3). These data indicate that EI24 may also be associated with chromatin/histone modifications, RNA metabolism regulation, and transcription.
The GOBP enrichment analysis suggested that EI24 could be functionally linked to cellular physiology regulation by degrading the E3 ligases involved in those processes. Therefore, we investigated upstream regulators that control EI24 targets and their associated cellular processes. First, we analyzed upstream transcriptional regulators of pGroups 1 and 2 using transcription factor enrichment analysis. pGroup 1 was enriched in binding sites for SP1 (Sp1 transcription factor), REPIN1 (replication initiator 1), TP53, and NFYB (nuclear transcription factor Y subunit β) (Fig. 6E). Furthermore, we performed kinase enrichment analysis to examine kinases that were upstream of EI24 targets. E3 ligases phosphorylated by ATM (ataxia telangiectasia mutated), CHEKs (checkpoint kinases), MTOR (mechanistic target of rapamycin [serine/threonine kinase]), and AKT1 (AKT serine/threonine kinase 1) were enriched in pGroup 1 (Fig. 6F). These data show functional and regulatory links between EI24 and physiologically important cellular processes through autophagic degradation of E3 ligases.
EI24-mediated degradation of TRAF2 and MDM2 regulates MTOR and TP53 signaling with implications in cellular bioenergetics and response to genotoxic stress
To elucidate if EI24-mediated degradation of E3 ligases regulates respective downstream signaling, we analyzed the relation of EI24 with the TRAF2 downstream-target MTOR27 and the MDM2 target TP5328 in basal and autophagy-activated conditions.
Previously, we have reported that EI24 mediates autophagy-dependent degradation of TRAF2 resulting in the activation of RELA signaling that is important for the suppression of EMT and metastasis in breast cancers and melanoma.7 Based on this observation, in the current study, we examined if B16F10 cells with stable Ei24 knockdown (Fig. 7A) containing higher levels of TRAF2, retain activated MTOR signaling7. Immunoblotting assay revealed that compared to control cells, Ei24 knockdown cells displayed increased phosphorylation of RPS6KB/p70S6K, a reliable marker of activated MTOR signaling (Fig. 7B). Furthermore, an increased lipidated form of LC3 (LC3-II) and SQSTM1 accumulation indicated impaired autophagy flux in cells containing reduced expression of EI24 (Fig. 7B). Thus, our data are consistent with previous reports demonstrating EI24 as an essential component of basal autophagy10 and also with the well-established fact that activated MTOR-signaling acts as a strong negative regulator of autophagy.29
Figure 7.

Functional validation of the role of EI24 in cellular bioenergetics and response to genotoxic stress by regulating MTOR and TP53 signaling. (A) Validation of Ei24 expression in B16F10 control and Ei24 knockdown cells by real-time qPCR. Ei24 expression was reduced by 90% in knockdown cells. (B, C) Activation of MTOR signaling and suppression of autophagy in Ei24 knockdown B16F10 cells in normal (B) and nutrient-depleted conditions (HBSS-treatment, 6 h (C). p-RPS6KB level was used as a marker of MTOR activity. I and II represent cytosolic and lipidated forms of LC3, respectively. Low indicates low-exposure and high indicates high-exposure of the blot. (D) Knockdown of EI24 stabilizes MDM2 and degradation of TP53 in HCT116 cells. (E) EI24 is required for mounting proper TP53 response in the presence of genotoxic stress in HCT116 cells. Cells were treated with cisplatin (10 μg/ml) for 24 h. CDKN1A protein level was used as a read-out of TP53 transcriptional activity. (F) EI24 is required for replenishing cellular ATP in nutrient-deprived conditions. ATP measurement in control and Ei24 knockdown B16F10 cells grown in basal and nutrient-depleted conditions (HBSS-treatment for 24 h). (G) EI24-induced autophagy is required for cell survival upon nutrient-deficiency. B16F10 control and Ei24 knockdown cells were subjected to nutrient-deprivation for 48 h and cell survival was measured by FACS analysis. X-axis and Y-axis represent FL2-H and events, respectively.
Since MTOR signaling is the master nutrient sensor30 and Ei24 knockdown resulted in accumulation of TRAF2 and activation of MTOR signaling in basal conditions (Fig. 7B), we next examined EI24-TRAF2-MTOR signaling in nutrient-depleted conditions (HBSS treatment). Control cells that were deprived of nutrients for 6 h displayed suppressed MTOR signaling and consequent activation of autophagy as indicated by decreased phosphorylation of the MTOR target RPS6KB (Fig. 7C). However, Ei24 knockdown cells retained higher levels of TRAF2 and activated MTOR signaling and impaired autophagy-flux even in nutrient-depleted conditions (Fig. 7C). Thus, EI24-induced degradation of RING-domain E3 ligase TRAF2 results in the regulation of MTOR signaling in both basal (Fig. 7B) and nutrient-depleted conditions (Fig. 7C).
Next, we sought to determine if EI24-mediated degradation of MDM2 results in the activation of TP53 signaling. For this purpose, we used HCT116 cells retaining functional TP53 (HCT116 TP53 WT cells). Immunoblotting revealed that cells with reduced expression of EI24 displayed accumulated MDM2 that was accompanied with the reduction in the protein levels of TP53 (Fig. 7D). TP53 is a transcription factor that acts as a master tumor suppressor by responding to oncogenic insults and genotoxic stress.31 MDM2 maintains the protein level of TP53 in balance, by its promotion of proteasome-dependent degradation to avert aberrant apoptosis.32 Since EI24 degraded MDM2 that resulted in TP53 accumulation in basal conditions (Fig. 7D), we next examined EI24-MDM2-TP53 signaling in the presence of genotoxic stress. Upon cisplatin treatment, control cells responded by stabilizing TP53 and inducing its transcriptional target CDKN1A whereas EI24 knockdown cells failed to do so (Fig. 7E). These data indicate that EI24-mediated degradation of MDM2 is important for proper TP53 response against genotoxic stress. Collectively, molecular analysis of TP53 and MTOR signaling in basal and autophagy-activated conditions revealed the importance of EI24-mediated degradation of E3 ligases in regulating these pathways with implications in maintaining cellular bioenergetics (through MTOR signaling) and genomic integrity (through TP53 signaling).
Because EI24 regulated MTOR signaling (Fig. 7B and C) and was involved in autophagy-mediated degradation of several E3 ligases, we examined if EI24 regulates cellular bioenergetics. For this purpose, we first determined ATP content in B16F10 control and Ei24 knockdown cells grown in normal media and HBSS-treated conditions. We did not observe significant differences in ATP content in control and Ei24 knockdown cells grown in ad libitum medium. However, total ATP content in Ei24 knockdown cells in HBSS-treated conditions was significantly lower than that of control cells (Fig. 7F). To examine if cells with reduced expression of EI24 display increased vulnerability toward lack of nutrient due to depleted ATP content, we measured cell viability of B16F10 control and Ei24 knockdown cells in nutrient-depleted conditions. Compared to approximately 80% cell-death in Ei24 knockdown cells, only approximately 50% cell-death was observed in control cells when subjected to nutrient deficiency for 48 h (Fig. 7G). This result indicates that EI24-mediated autophagic degradation of target proteins regulates cellular bioenergetics to act as a backup mechanism for replenishing ATP in nutrient-deprived conditions. Our observation is consistent with previous reports demonstrating that end products of the autophagy process are channeled into the tricarboxylic acid cycle to generate energy in nutrient-deprived conditions33 and substantiating the physiological function of autophagy as a cytoprotective mechanism during metabolic stresses.34
Collectively, our work demonstrates that EI24 is an important mediator orchestrating crosstalk between the UPS and autophagy by targeting RING E3 ligases for autophagic degradation. This degradation is functionally linked to the regulation of several cellular processes, which represents a paradigm shift regarding the fate of E3 ligase degradation.
Discussion
In the present study, we showed that the RING domain, which is present in the majority of E3 ligases, acts as an ‘eat-me' signal for EI24-mediated autophagic degradation. We propose the autophagy machinery is integrated with the UPS, indicating that these protein degradation pathways are not as independent as previously suggested.35 The proposed model clearly represents a paradigm shift regarding our understanding of E3 ligase fate-determination.
We also utilized the screening data and combined it with computational methods to determine potential biological functions for the RING domain E3 ligases that are degraded by EI24. Using these methods, we elucidated that EI24 potentially works cooperatively with the master tumor suppressor TP53 and the central nutrient-sensing mediator MTOR to regulate various cellular processes.
Our study revealed that in addition to the presence of RING-domain, cellular localization of E3 ligases could be also a contributing factor to determine the susceptibility to be degraded by EI24. pGroup 1 members were primarily localized to the endosome, ubiquitin ligase complex, vacuole, lysosome, chromatin, and cytoskeleton. Most of these cellular organelles are directly involved in the execution of autophagy process or maintain high-autophagy activity,26 so from the molecular standpoint, it makes sense that pGroup 1 E3-ligases reside in these organelles. One exception is the localization of some of pGroup 1 E3 ligases in the nuclear-compartment, because the autophagy process is largely cytosolic.34 However, exclusivity of the cytosolic nature of the autophagy process has been challenged by recent reports describing the degradation of nuclear proteins36 and degradation of the nucleus itself by autophagy.37 Conversely, pGroup 2 was related with the perinuclear region of the cytoplasm and Golgi apparatus. In addition, we observed that similar protein isoforms aligned themselves in separate groups (BIRC2 in Group 1 and BIRC3 in Group 2, CBLB and CBLC in Group 2 and CBLL1 in Group 1). Although detailed experimental validation has to be done to prove the hypothesis, our data illustrate that in addition to the presence of RING-domain, the difference in the cellular localization of E3 ligases could be an additional factor that determines the susceptibility of a particular E3 ligase to be degraded by EI24.
The data we describe here are consistent with previously reported associations between EI24 and AKT1,38 CHEK,39 TP53,40 and MTOR in autophagy pathways.10 It also suggests potential associations between EI24 and SP1, REPIN1, NFYB, and ATM pathways. Comparative analyses of predicted EI24 targets and nontargets suggest functional and regulatory links between EI24 and physiologically important cellular processes, such as proteolysis, apoptosis, chromatin/histone modifications, and transcription, through autophagic degradation of the E3 ligases involved in these processes (Fig. 6). By combining our analysis results with previously reported molecular interactions in the interactome databases (human protein reference database)41 and the Kyoto encyclopedia of genes and genomes,42 we developed a network model of the functional and regulatory links (Fig. 8). In the network model, the kinase module shows that upstream kinases of EI24 targets (Fig. 6F, Fig. 8) are members of the AKT1-MTOR and ATM-CHEK1/2 pathways, and these pathways crosstalk with each other through a link between AKT1 and CHEK1/2. The transcription factor module shows that TP53 is an upstream transcription factor of EI24 targets (Fig. 6E), and TP53 is regulated negatively and positively by the AKT1-MTOR and ATM-CHECK1/2 pathways, respectively (Fig. 8). Moreover, TP53 transcriptionally regulates the gene expression of both EI24 and its targets. The EI24 target module shows that EI24 degrades E3 ligase targets that are involved in physiologically important cellular processes (Fig. 6C). Furthermore, some of these targets regulate the upstream kinases and transcription factors via feedback mechanisms. For example, EI24 degrades MDM2, which negatively regulates TP53, and it also degrades TRAF2, which positively regulates MTOR (Fig. 8). The feed-forward and feedback links between the upstream regulators and E3 ligase targets of EI24 suggest a complex mechanism regulating EI24-dependent autophagic degradation and E3 ligase-associated cellular processes. Collectively, the numerous interactions represented in the network model demonstrate that EI24 connects all the network modules at the molecular level, indicating that EI24 is a central player in the crosstalk between the UPS and autophagy.
Figure 8.
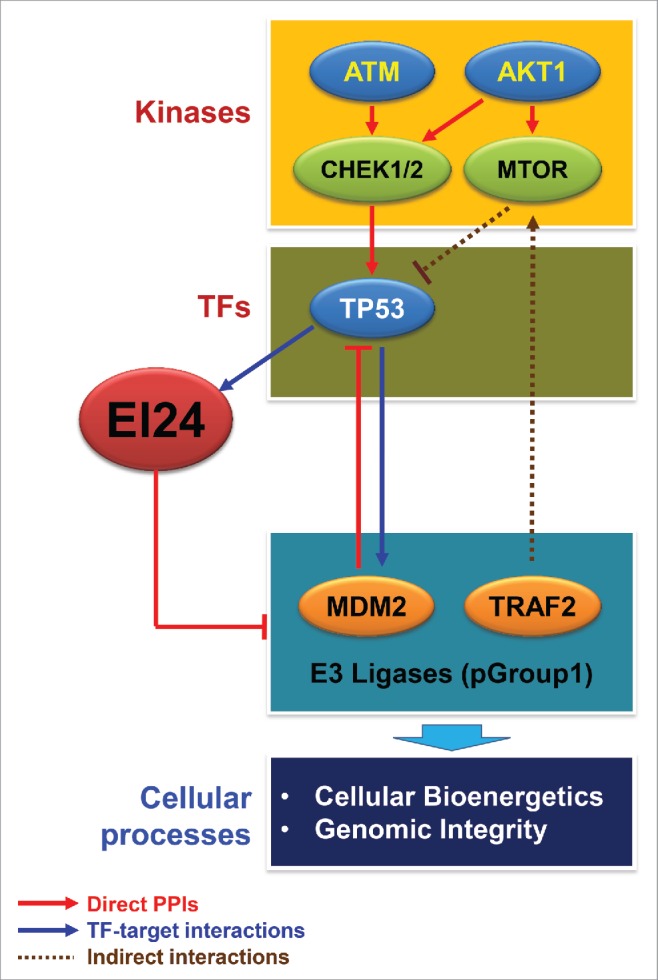
Network model delineating functional and regulatory links between EI24 and kinases, transcription factors (TF), E3 ligase targets, and cellular processes. Kinase and TF modules in the network model include the upstream TFs (Fig. 6E) and kinases (Fig. 6F) whose targets and substrates, respectively, are enriched in pGroup 1 (EI24 targets). The E3 ligase module includes E3 ligase targets of EI24, such as MDM2 and TRAF2. The arrows and repression symbols represent activation and inhibition, respectively, in the regulatory relationships. Solid and dotted lines denote direct and indirect interactions, respectively, in the regulatory reactions. Cellular processes include the GOBPs represented by EI24 target E3 ligases (Fig. 6C). PPIs, protein-protein interactions.
One of the central questions of this study was why do cells need to degrade E3 ligases via autophagy when they are already being degraded by the proteasome. One possible explanation is energy conservation. ATP is essential for various steps during protein degradation via the UPS, and protein unfolding, fueled by ATP hydrolysis, ensures the smooth passage of substrates through the proteasome tunnel.43 The amount of ATP required for autophagy is not well studied; however, the consensus is that autophagy is “cheaper” than the UPS with respect to ATP. From the cellular point of view, E3 ligase protein levels need to be tightly regulated at all times, and autophagic degradation seems to be the economical choice. Another explanation for the crosstalk between autophagy and UPS is that the cell could use autophagy as a backup mechanism for protein degradation. E3 ligases control whether a protein will be degraded or not. Thus, dysfunctional regulation in E3 ligase protein level could have severe ramifications for cells.44 In this context, based on our model, autophagy may serve as a maintenance mechanism for E3 ligase homeostasis in cells.
We previously tested and verified the physiological significance of EI24-mediated degradation of E3 ligases at the functional level using in vivo mouse models. We demonstrated that EI24 binds and degrades TRIM41, an E3 ligase of PRKCA. Thus, loss of EI24 in the mouse resulted in TRIM41 accumulation and reduced PRKCA protein levels. Because PRKCA is required for skin carcinogenesis, we have found that mice with reduced EI24 expression has an attenuated response to DMBA-TPA-induced skin carcinogenesis.8,12 In another study, we report that EI24 degrades TRAF2 and TRAF5 via autophagy based on its recognition of the E3 ligase RING domain. Because TRAF signaling lies upstream of the RELA/NFKB p65 pathway, reduced EI24 expression results in RELA signaling activation, increased expression of proinflammatory cytokines, the emergence of EMT, and tumor metastasis. Thus, the molecular model we propose here has already been verified with respect to skin cancers and tumor metastasis. Furthermore, we have revealed that EI24-induced degradation of TRAF2 suppresses MTOR signaling resulting in the activation of autophagy. Autophagy-mediated proteolysis channels amino acids into the tricarboxylic acid cycle to generate energy that is required for cell survival in nutrient-deprived conditions.33 Consistent with this paradigm, we found that cells with reduced expression of EI24 that cannot mount a proper autophagy response contain decreased ATP levels in HBSS-treated conditions. As a consequence of inability to replenish ATP, Ei24 knockdown cells displayed increased cell death in nutrient-deprived conditions. Increased susceptibility of Ei24 knockdown cells that lack autophagy-inducing activity is consistent with previous reports demonstrating the protective nature of autophagy process during metabolic stress.34
In this study, we used MPLS-DA to predict E3 ligases whose degradation is regulated by EI24. PLS-DA has been applied to mRNA expression data for classification of the samples. In PLS-DA, the data matrixes are commonly auto-scaled. In the sample classification problems, the auto-scaling of mRNA expression data is usually performed across genes or proteins. However, in this study, our goal was to classify E3 ligases (gene classification), not the samples (sample classification). For the gene classification, we first explored whether protein sequences (Fig. S1A and B), cellular localizations (Fig. S1C), known binding partners (Fig. S1D), and mRNA expression levels (Fig. S2A to C) of E3 ligases showed any correlation with Group memberships (i.e., Group 1 [EI24 targets] and Group 2 [nontargets]). We found that only mRNA expression levels showed significant correlation with Group memberships (Fig. S2A to C). Thus, considering E3 ligases as independent observations defined by their mRNA expression levels (attributes), we formed n × m data matrix (X-block) for n E3 ligases and m samples and then auto-scaled the matrix for each column to have zero mean and unit variance.
When the performance of a prediction model is evaluated, it is common not to include the training set in the test set. In this study, however, the training set (16 E3 ligases) used to build the MPLS-DA model was included in the test set (342 array probes for 308 E3 ligases). In our gene classification, we used the mixture modeling and probabilistic prediction of EI24 targets based on the mixture model, both of which require inclusion of the training set to assure accurate estimation of the mixture model and also to reliably determine the cutoff of the probability of being EI24 targets. For the gene classification, we first built the MPLS-DA model using the expression data matrixes (X1, X2, and Y) auto-scaled for the 16 E3 ligases (training set). In the MPLS-DA model, the latent variables (LVs) were determined to optimally separate Groups 1 and 2 (11 and 5 of 16 E3 ligases, respectively) in the training set on the PLS space. Next, to predict whether 308 E3 ligases (342 probes) in the test set are EI24 targets (Y_pred = 1) or not (Y_pred = 0), we auto-scaled the expression data matrixes (X1pred and X2pred) for the 342 probes and then estimated Y_pred values of the 342 probes using the LVs determined for the 16 E3 ligases. The application of the MPLS-DA LVs identified from the 16 E3 ligases to the auto-scaled X1pred and X2pred for the 342 probes will result in a suboptimal separation of EI24 targets (pGroup 1) and nontargets (pGroup 2) on the PLS space. To improve the classification accuracy by resolving this problem, we thus additionally applied a Gaussian mixture modeling to estimate 2 Gaussian distributions for pGroups 1 and 2, respectively, and then statistically determined whether each of 342 probes belongs to pGroup 1 by calculating the probability of each E3 ligase being a EI24 target [P(x ∈ Group 1|Y_pred)] using the 2 Gaussian distributions based on Bayesian rule. During the mixture modeling, the use of 11 EI24 targets (Group 1) and 5 nontargets (Group 2) in the training set enables reliable estimation of the 2 Gaussian distributions.
Moreover, to determine the cutoff of the probability for the targets, the use of Groups 1 and 2 would be important because it can guarantee that pGroups 1 and 2 determined by the cutoff can include Groups 1 and 2 in the training set, respectively. PeptideProphet software45 used a similar approach involving linear discriminant analysis of a training set followed by a mixture modeling of predicted values for a test set for statistical peptide identification. However, to evaluate sensitivity and specificity in the prediction based on the MPLS-DA model, we randomly selected 5 predicted EI24 targets and 5 nontargets from pGroups 1 and 2, respectively, which include no E3 ligases in the training set, and then experimentally tested whether the selected E3 ligases are targeted by EI24. The experimental results showed that all 5 predicted EI24 targets were found to be truly EI24 targets while all 4 predicted nontargets were to be truly nontargets, indicating high degrees of sensitivity and specificity to our model. CBLC that belonged to pGroup 2 was also degraded by EI24 (Fig. S4A). Further studies must focus to elucidate why similar protein isoforms aligned themselves in separate groups (BIRC2 in Group 1 and BIRC3 in Group 2, CBLL1 in Group 1 and CBLB and CBLC in Group 2).
Overall, our study revealed that EI24-mediated autophagic degradation of RING E3 ligases allows crosstalk between autophagy and the UPS. This crosstalk presents the opportunity to manage protein degradation machineries and cellular bioenergetics, both in cancers and other human diseases, when cell physiology goes awry.
Materials and methods
Cell culture and transfection
293T, HeLa, B16F10, and HCT116 cells were cultured in Dulbecco's modified Eagle's medium (Thermo Fisher Scientific, 11965092) supplemented with 10% fetal bovine serum (Hyclone, SH30919.03), 100 units/ml penicillin, and 100 μg/ml streptomycin (Thermo Fisher Scientific, 15070-063). Cells were grown at 37°C in a humidified chamber containing 5% CO2. Cells were transfected with polyethylenimine (Sigma-Aldrich, 764647) at a ratio of 6 µg polyethylenimine/µg DNA. For siRNA transfection, Lipofectamine RNAiMAX (Thermo Fisher Scientific, 13778150) was used. The EI24 knockdown siRNA sequence was: 5′-GCAAGAGAGUGAGCCACGUAUUGUUTT-3′.
Immunocytochemistry and ATP measurement
Immunocytochemistry was performed as described previously with slight modifications.46 Briefly, cells, with or without EI24 overexpression, were transfected with GFP-LC3 plasmids and seeded on gelatin-coated coverslips (Deckglasser, 0111520). Following a 4% paraformaldehyde (Sigma-Aldrich, P6148) in PBS (HyClone, SH30256.01) fixation step, cells were permeabilized with 0.5% Triton X-100 (Sigma-Aldrich, X100). Nonspecific signals were blocked with 1% normal goat serum (Thermo Fisher Scientific, 16210072) in PBS with 0.1% Triton X-100 for 30 min. Cells were then stained for 1 h at room temperature with their respective antibodies. Primary fluorescent signals were detected using Alexa Fluor 488 (Thermo Fisher Scientific, A-11008)- or Alexa Fluor 568 (Thermo Fisher Scientific, A-11008)-conjugated secondary antibodies. The data were imaged as described previously.8 Cellular ATP content in B16F10 control and Ei24 knockdown cells grown in normal media or HBSS (Thermo Fisher Scientific, 14025-092)-treated conditions (12 h) was determined using the Mitochondrial ToxGlo Assay Kit following manufacturer's protocol (Promega, G8000).
FACS analysis
FACS analysis was performed as described previously.47 Briefly, B16F10 control and Ei24 knockdown cells grown in normal medium or in HBSS (48 h) were fixed in 70% ethanol, treated with RNase A (Sigma-Aldrich, R4875), and stained with propidium iodide (Sigma-Aldrich, P4170) to stain DNA. FACS analysis was conducted using a FACS calibur apparatus 342975 (BD Biosciences, San Jose, CA, USA) to generate plots depicting counts (Y-axis) versus FL2-H (X-axis).
Plasmids and constructs
Flag-tagged TRIM41/RINCK1 was provided by Prof. Alexandra Newton (University of California, San Diego)48 and GFP-tagged TRIMs were provided by Profs. Germana Meroni (Cluster in Biomedicine, Italy) and Andrea Ballabio (Telethon Institute of Genetics and Medicine, Italy).49 HA-tagged Ub, MKRN1, MKRN1RINGΔ, and STUB1 as well as Flag-tagged TRAF2, TRAF2RINGΔ, TRAF6, and ZFPL1 were provided by Prof. Jaewhan Song (Yonsei University, Republic of Korea). Flag-tagged FBXO7 and XIAP, MYC-tagged BIRC2 and BIRC3, and GFP-tagged PARK2 were provided by Prof. Kwang Chul Chung (Yonsei University, Republic of Korea). HA-tagged PML ([1-633 amino-acids: PML-IV] and [1-560 amino-acids: PML-VI]) were a kind gift from Prof. Jin Hyun Ahn (Sungkyunkwan University School of Medicine, Republic of Korea). HA-tagged CBLC was provided by Prof. Kwang Youl Lee (Chonnam National University, Republic of Korea). Prof. Young-Gyu Ko (Korea University, Republic of Korea) provided MYC-tagged TRIM72 construct. MYC-tagged EI24 has been reported previously7 and was transferred to a pEGFPN1 vector (Clontech, 6085-1) to generate a GFP-tagged EI24 construct. TRIM41/RINCK1-Flag deletion constructs were generated as described previously.50
Ubiquitination assay
Cells were harvested in PBS containing 2 mM N-ethylmaleimide (NEM; Sigma-Aldrich, E3876) and lysed in Tris-buffered saline (Sigma-Aldrich, T6664) containing 1% SDS (Sigma-Aldrich, L3771) and 20 mM NEM 30 h after transfection. The lysate was boiled, sonicated, and centrifuged at 14,000 × g for 15 min. The supernatant was diluted in NP-40 buffer containing 2 mM NEM, and immunoprecipitation was carried out using standard methods.
Immunoprecipitation and immunoblotting
Immunoprecipitation was performed as described previously with slight modifications.51 Briefly, cell lysates were prepared in NP-40 buffer comprised of 20 mM Tris (Sigma-Aldrich, 252859)-HCl (Sigma-Aldrich, 258148), 137 mM NaCl (Sigma-Aldrich, S9888), 1% NP-40 (Sigma-Aldrich, NP40S), 2 mM EDTA (Sigma-Aldrich, E9884), 10% glycerol (Sigma-Aldrich, G5516), 1 mM PMSF (Sigma-Aldrich, P7626), 2 mM sodium fluoride (Sigma-Aldrich, 201154), 1 mM sodium vanadate (Sigma-Aldrich, S6508), 1 mM β-glycerophosphate (Sigma-Aldrich, G9422), and 20 μg/ml each aprotinin (Sigma-Aldrich, A6106), pepstatin (Sigma-Aldrich, P5318), and leupeptin (Sigma-Aldrich, L2884). After centrifugation at 14,000 ×g for 15 min at 4°C, the supernatant underwent immunoprecipitation using 1 μg of the respective antibodies. After 3 h, 30 μl of protein G agarose beads (Thermo Fisher Scientific, 15920010) were added to the supernatant and incubated for 3 h. The beads were washed in the cell lysis buffer and boiled in an equal volume of 2× SDS sample buffer. For immunoblotting, cells were lysed in a radioimmunoprecipitation assay buffer consisting of 50 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate (Sigma-Aldrich, D6750), 0.1% SDS, and 2 mM EDTA along with protease and phosphatase inhibitors.
Antibodies
The following antibodies were used: HA (Santa Cruz Biotechnology, sc-805), GAPDH (Santa Cruz Biotechnology, sc-25778), ACTB (Santa Cruz Biotechnology, sc-8432), SQSTM1 (Santa Cruz Biotechnology, sc-28359), TP53 (Santa Cruz Biotechnology, sc-126), CDKN1A (Santa Cruz Biotechnology, sc-6246), GFP (Santa Cruz Biotechnology, sc-9996), Flag (Sigma-Aldrich, F1804), MYC (Cell Signaling Technology, 2276), LC3B (Cell Signaling Technology, 2775), TRAF2 (Cell Signaling Technology, 4724S), RPS6KB/p70S6K (Cell Signaling Technology, 2708), p-RPS6KB/p-p70S6K (Cell Signaling Technology, 9205), MDM2 (Cell Signaling Technology, 323), RNF6 (abcam, ab80427), and ZNF462 (abcam, ab117771). The EI24 rabbit polyclonal antibody has been reported previously.8
Multi-block partial least square-discriminatory analysis (MPLS-DA)
We performed MPLS-DA on gene expression levels to build a model that can separate Groups 1 and 2 and predict pGroups 1 and 2 using the previously reported MPLS algorithm.22 Of linear, nonparametric, and nonlinear classification methods, we used MPLS-DA because it effectively integrates multiple different datasets for classification of Groups 1 and 2 and enables us to interpret how different expression data contribute to the separation of Group 1 from Group 2, as previously demonstrated.22,23 For the analysis, we obtained gene expression data previously generated from ZR-75-1 breast cancer cells after EI24 knockdown (GSE52508 in GEO database)7 and MEF cells after etoposide treatment (GSE67266 in GEO database).21 We transformed the measured intensities into log2-intensity and then normalized the log2-intensity data using the quantile normalization.52 Of the 381 E3 ligases with RING domains, 354 were included in the gene expression data, and 354 included 11 of the 14 E3 ligases in Group 1 and all 5 E3 ligases in Group 2. For the 11 targets and 5 nontargets, we generated 2 X-block data matrixes: a 16×8 X1-block (X1) and a 16×6 X2-block (X2) containing mRNA expression levels of the 16 E3 ligases measured in data sets 1 and 2, respectively (Fig. S2D and Table S4). We also generated a 16×1 Y-block data matrix (Y) designating “1” for EI24 targets and “0” for nontargets (Table S4). All data matrixes were auto-scaled such that each column had zero mean and unit standard deviation. MPLS-DA was then applied to the auto-scaled X1, X2, and Y (Fig. S2D). After MPLS-DA, we selected 4 PLS latent variables (LVs) based on leave-one-out cross-validation.53 Before predicting EI24 targets, of 354 E3 ligases (396 array probes), we first selected 308 (342 probes) with MAD>25% to focus on E3 ligases with reasonable information contents. To predict E3 ligases targeted by EI24, we then generated a 342×8 X1-block (X1pred) and a 342×6 X2-block (X2pred) containing mRNA expression levels of all the E3 ligases measured in datasets 1 and 2 (Table S5). We then applied the MPLS-DA model to the X1- and X2-blocks, after auto-scaling the data matrixes (X1pred and X2pred) as described above, and obtained ‘predicted Y' values for the 342 probes (Y_pred in Fig. S2F).53 Finally, a mixture Gaussian model was applied to Y_pred values, and we used the Bayesian rule to calculate the probability of each E3 ligase (x) being a EI24 target [P(x ∈ Group 1|Y_pred)] or a nontargets [P(x ∈ Group 2|Y_pred)] using the Gaussian distributions estimated for EI24 targets and nontargets.54,55 The EI24 targets (pGroup 1) and nontargets (pGroup 2) were identified as E3 ligases with P(x ∈ Group 1|Y_pred) > 0.75 and P(x ∈ Group 2|Y_pred) > 0.75, respectively. See Supplementary Materials and Methods for implementation of MPLS-DA and mixture modeling.
Upstream regulator analysis
We performed TF (transcription factor) and kinase enrichment analyses to identify upstream regulators of EI24 targets. For these analyses, we first collected experimentally verified TF-target interactions56-61 and kinase-substrate interactions.62-64 For each regulator (TF or kinase), the null distributions for the numbers of targets (TF targets or kinase substrates) that overlapped with pGroups 1 and 2 were estimated by randomly sampling the same numbers of proteins in pGroups 1 and 2. To determine the observed numbers of targets for the pGroup 1 or 2 regulator, the significance (P values) of the targets enriched in pGroup 1 or 2 was computed using a one-tailed test of the observed number of targets based on the null distribution for the corresponding group (pGroup 1 or 2). Finally, the regulators with P value < 0.05 and the number of targets ≥ 5 were selected as the upstream regulators whose targets or substrates were enriched in pGroup 1 or 2. Of the upstream TFs, the ones showing similar expression patterns with EI24 could be desired. Thus, for each of the 2 data sets, after normalizing mRNA expression data using quantile normalization,52 we calculated Pearson correlation coefficients between the normalized expression data of each TF and EI24, as well as the significance of the correlation coefficient, using the MATLAB ‘corr.m' function65 (MathWorks Inc., Natick, MA, USA). To generate a final set of enriched TFs, we combined P values from the 2 datasets using a previously reported integrative statistical method66 and then selected the TFs with the combined P value <0.05.
Supplementary Material
Abbreviations
- BAF
bafilomycin A1
- BIRC2/CIAP1
baculoviral IAP repeat containing 2
- BIRC3/CIAP2
baculoviral IAP repeat containing 3
- CBL
casitas B-lineage lymphoma
- EI24
EI24, autophagy associated transmembrane protein
- GEO
gene expression omnibus
- GOBP
gene ontology biological function
- GOCC
gene ontology cellular components
- GOMF
gene ontology molecular function
- HBSS
Hank's balanced salt solution
- LIR
LC3-interacting region
- MAP1LC3A/B
microtubule-associated protein 1, light chain 3 alpha/beta
- MPLS-DA
multi-block partial least square-discriminant analysis
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- NBR1
NBR1, autophagy cargo receptor
- NFKB
nuclear factor kappa B
- PARK2
parkin RBR E3 ubiquitin protein ligase
- PCA
principal component analysis
- PML
promyelocytic leukemia
- RING
really interesting new gene
- SQSTM1/P62
sequestosome 1
- TRIM
tripartite motif
- UBA
ubiquitin-associated domain
- UPS
ubiquitin-proteasome system
- XIAP
X-linked inhibitor of apoptosis
Disclosure of potential conflicts of interest
The authors declare that they have no conflicts of interest.
Acknowledgments
We would like to thank Germana Meroni and Andrea Ballabio for GFP-tagged TRIM constructs.
Funding
The work was supported by the National Research Foundation of Korea (NRF) grants (2015R1A2A1A01003845 and 2010-0020878) and the Institute for Basic Science (IBS-R013-G1-2014-a00) funded by Korean ministry of science, ICT, and future planning and the National R&D Program for Cancer Control, Ministry for Health, Welfare and Family Affairs (MoHWFA), Republic of Korea (1020220). “The Yonsei University Yonsei-SNU Collaborative Research Fund of 2014” also funded this study. M.A. is supported by Higher Education Commission (HEC) of Pakistan government.
References
- [1].Adams J. The proteasome: structure, function, and role in the cell. Cancer Treat Rev 2003; 29 Suppl 1:3-9; PMID:12738238; http://dx.doi.org/ 10.1016/S0305-7372(03)00081-1 [DOI] [PubMed] [Google Scholar]
- [2].Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol 2010; 12:836-41; PMID:20811356; http://dx.doi.org/ 10.1038/ncb0910-836 [DOI] [PubMed] [Google Scholar]
- [3].Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al.. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007; 131:1149-63; PMID:18083104; http://dx.doi.org/ 10.1016/j.cell.2007.10.035 [DOI] [PubMed] [Google Scholar]
- [4].Kirkin V, Lamark T, Johansen T, Dikic I. NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy 2009; 5:732-3; PMID:19398892; http://dx.doi.org/ 10.4161/auto.5.5.8566 [DOI] [PubMed] [Google Scholar]
- [5].Marshall RS, Li F, Gemperline DC, Book AJ, Vierstra RD. Autophagic degradation of the 26S proteasome is mediated by the dual ATG8/Ubiquitin receptor RPN10 in arabidopsis. Mol Cell 2015; 58:1053-66; PMID:26004230; http://dx.doi.org/ 10.1016/j.molcel.2015.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gu Z, Flemington C, Chittenden T, Zambetti GP. ei24, a p53 response gene involved in growth suppression and apoptosis. Molecular and cellular biology 2000; 20:233-41; PMID:10594026; http://dx.doi.org/ 10.1128/mcb.20.1.233-241.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Choi JM, Devkota S, Sung YH, Lee HW. EI24 regulates epithelial-to-mesenchymal transition and tumor progression by suppressing TRAF2-mediated NF-kappaB activity. Oncotarget 2013; 4:2383-96; PMID:24280371; http://dx.doi.org/ 10.18632/oncotarget.1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Devkota S, Sung YH, Choi JM, Lee J, Ha NY, Kim H, Cho BC, Song J, Lee HW. Ei24-deficiency attenuates protein kinase Calpha signaling and skin carcinogenesis in mice. Int J Biochem Cell Biol 2012; 44:1887-96; PMID:22771957; http://dx.doi.org/ 10.1016/j.biocel.2012.06.034 [DOI] [PubMed] [Google Scholar]
- [9].Tian Y, Li Z, Hu W, Ren H, Tian E, Zhao Y, Lu Q, Huang X, Yang P, Li X, et al.. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell 2010; 141:1042-55; PMID:20550938; http://dx.doi.org/ 10.1016/j.cell.2010.04.034 [DOI] [PubMed] [Google Scholar]
- [10].Zhao YG, Zhao H, Miao L, Wang L, Sun F, Zhang H. The p53-induced gene Ei24 is an essential component of the basal autophagy pathway. J Biol Chem 2012; 287:42053-63; PMID:23074225; http://dx.doi.org/ 10.1074/jbc.M112.415968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chen D, Gould C, Garza R, Gao T, Hampton RY, Newton AC. Amplitude control of protein kinase C by RINCK, a novel E3 ubiquitin ligase. J Biol Chem 2007; 282:33776-87; PMID:17893151; http://dx.doi.org/ 10.1074/jbc.m703320200 [DOI] [PubMed] [Google Scholar]
- [13].Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ 2005; 12 Suppl 2:1542-52; PMID:16247502; http://dx.doi.org/ 10.1038/sj.cdd.4401765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bhattacharyya S, Yu H, Mim C, Matouschek A. Regulated protein turnover: snapshots of the proteasome in action. Nat Rev Mol Cell Biol 2014; 15:122-33; PMID:24452470; http://dx.doi.org/ 10.1038/nrm3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chatr-aryamontri A, Ceol A, Palazzi LM, Nardelli G, Schneider MV, Castagnoli L, Cesareni G. MINT: the molecular INTeraction database. Nucleic Acids Res 2007; 35:D572-4; PMID:17135203; http://dx.doi.org/ 10.1093/nar/gkl950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Orchard S, Ammari M, Aranda B, Breuza L, Briganti L, Broackes-Carter F, Campbell NH, Chavali G, Chen C, del-Toro N, et al.. The MIntAct project–IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res 2014; 42:D358-63; PMID:24234451; http://dx.doi.org/ 10.1093/nar/gkt1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Peri S, Navarro JD, Kristiansen TZ, Amanchy R, Surendranath V, Muthusamy B, Gandhi TK, Chandrika KN, Deshpande N, Suresh S, et al.. Human protein reference database as a discovery resource for proteomics. Nucleic Acids Res 2004; 32:D497-501; PMID:14681466; http://dx.doi.org/ 10.1093/nar/gkh070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Salwinski L, Miller CS, Smith AJ, Pettit FK, Bowie JU, Eisenberg D. The Database of Interacting Proteins: 2004 update. Nucleic Acids Res 2004; 32:D449-51; PMID:14681454; 10.1093/nar/gkh086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res 2006; 34:D535-9; PMID:16381927; http://dx.doi.org/ 10.1093/nar/gkj109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bahk YY, Lee J, Cho IH, Lee HW. An analysis of an interactome for apoptosis factor, Ei24/PIG8, using the inducible expression system and shotgun proteomics. J Proteome Res 2010; 9:5270-83; PMID:20731388; http://dx.doi.org/ 10.1021/pr100552y [DOI] [PubMed] [Google Scholar]
- [21].Boucas J, Fritz C, Schmitt A, Riabinska A, Thelen L, Peifer M, Leeser U, Nuernberg P, Altmueller J, Gaestel M, et al.. Label-free protein-RNA interactome analysis identifies Khsrp signaling downstream of the p38/Mk2 kinase complex as a critical modulator of cell cycle progression. PLoS One 2015; 10:e0125745; PMID:25993413; http://dx.doi.org/ 10.1371/journal.pone.0125745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hwang D, Stephanopoulos G, Chan C. Inverse modeling using multi-block PLS to determine the environmental conditions that provide optimal cellular function. Bioinformatics 2004; 20:487-99; PMID:14990444; http://dx.doi.org/ 10.1093/bioinformatics/btg433 [DOI] [PubMed] [Google Scholar]
- [23].Park MC, Jeong H, Son SH, Kim Y, Han D, Goughnour PC, Kang T, Kwon NH, Moon HE, Paek SH, et al.. Novel morphologic and genetic analysis of cancer cells in a 3D microenvironment identifies STAT3 as a regulator of tumor permeability barrier function. Cancer Res 2015; 76:1044-54; PMID:26676754; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-2611 [DOI] [PubMed] [Google Scholar]
- [24].Hou X, Zhang W, Xiao Z, Gan H, Lin X, Liao S, Han C. Mining and characterization of ubiquitin E3 ligases expressed in the mouse testis. BMC Genomics 2012; 13:495; PMID:22992278; http://dx.doi.org/ 10.1186/1471-2164-13-495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Li W, Bengtson MH, Ulbrich A, Matsuda A, Reddy VA, Orth A, Chanda SK, Batalov S, Joazeiro CA. Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle's dynamics and signaling. PLoS One 2008; 3:e1487; PMID:18213395; http://dx.doi.org/ 10.1371/journal.pone.0001487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sica V, Galluzzi L, Bravo-San Pedro JM, Izzo V, Maiuri MC, Kroemer G. Organelle-specific initiation of autophagy. Mol Cell 2015; 59:522-39; PMID:26295960; http://dx.doi.org/ 10.1016/j.molcel.2015.07.021 [DOI] [PubMed] [Google Scholar]
- [27].Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer 2008; 8:851-64; PMID:18846101; http://dx.doi.org/ 10.1038/nrc2501 [DOI] [PubMed] [Google Scholar]
- [28].Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett 1997; 420:25-7; PMID:9450543; http://dx.doi.org/ 10.1016/s0014-5793(97)01480-4 [DOI] [PubMed] [Google Scholar]
- [29].Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132-41; PMID:21258367; http://dx.doi.org/ 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Howell JJ, Manning BD. mTOR couples cellular nutrient sensing to organismal metabolic homeostasis. Trends Endocrinol Metab 2011; 22:94-102; PMID:21269838; http://dx.doi.org/ 10.1016/j.tem.2010.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zilfou JT, Lowe SW. Tumor suppressive functions of p53. Cold Spring Harb Perspect Biol 2009; 1:a001883; PMID:20066118; http://dx.doi.org/ 10.1101/cshperspect.a001883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Iwakuma T, Lozano G. MDM2, an introduction. Mol Cancer Res 2003; 1:993-1000; PMID:14707282; http://dx.doi.org/ 10.1158/1541-7786 [DOI] [PubMed] [Google Scholar]
- [33].Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, et al.. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 2010; 90:1383-435; PMID:20959619; http://dx.doi.org/ 10.1152/physrev.00030.2009 [DOI] [PubMed] [Google Scholar]
- [34].Mizushima N. Autophagy: process and function. Genes Dev 2007; 21:2861-73; PMID:18006683; http://dx.doi.org/ 10.1101/gad.1599207 [DOI] [PubMed] [Google Scholar]
- [35].Lilienbaum A. Relationship between the proteasomal system and autophagy. Int J Biochem Mol Biol 2013; 4:1-26; PMID:23638318 [PMC free article] [PubMed] [Google Scholar]
- [36].Dou Z, Xu C, Donahue G, Shimi T, Pan JA, Zhu J, Ivanov A, Capell BC, Drake AM, Shah PP, et al.. Autophagy mediates degradation of nuclear lamina. Nature 2015; 527:105-9; PMID:26524528; http://dx.doi.org/ 10.1038/nature15548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mijaljica D, Devenish RJ. Nucleophagy at a glance. J Cell Sci 2013; 126:4325-30; PMID:24013549; http://dx.doi.org/ 10.1242/jcs.133090 [DOI] [PubMed] [Google Scholar]
- [38].Humphrey SJ, Yang G, Yang P, Fazakerley DJ, Stockli J, Yang JY, James DE. Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab 2013; 17:1009-20; PMID:23684622; http://dx.doi.org/ 10.1016/j.cmet.2013.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mazumder Indra D, Mitra S, Singh RK, Dutta S, Roy A, Mondal RK, Basu PS, Roychoudhury S, Panda CK. Inactivation of CHEK1 and EI24 is associated with the development of invasive cervical carcinoma: clinical and prognostic implications. Int J Cancer 2011; 129:1859-71; PMID:21154811; http://dx.doi.org/ 10.1002/ijc.25849 [DOI] [PubMed] [Google Scholar]
- [40].Gentile M, Ahnstrom M, Schon F, Wingren S. Candidate tumour suppressor genes at 11q23-q24 in breast cancer: evidence of alterations in PIG8, a gene involved in p53-induced apoptosis. Oncogene 2001; 20:7753-60; PMID:11753653; http://dx.doi.org/ 10.1038/sj.onc.1204993 [DOI] [PubMed] [Google Scholar]
- [41].Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R, Shafreen B, Venugopal A, et al.. Human Protein Reference Database-2009 update. Nucleic Acids Res 2009; 37:D767-72; PMID:18988627; http://dx.doi.org/ 10.1093/nar/gkn892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 2012; 40:D109-14; PMID:22080510; http://dx.doi.org/ 10.1093/nar/gkr988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Liu CW, Li X, Thompson D, Wooding K, Chang TL, Tang Z, Yu H, Thomas PJ, DeMartino GN. ATP binding and ATP hydrolysis play distinct roles in the function of 26S proteasome. Mol Cell 2006; 24:39-50; PMID:17018291; http://dx.doi.org/ 10.1016/j.molcel.2006.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Paul S. Dysfunction of the ubiquitin-proteasome system in multiple disease conditions: therapeutic approaches. Bioessays 2008; 30:1172-84; PMID:18937370; http://dx.doi.org/ 10.1002/bies.20852 [DOI] [PubMed] [Google Scholar]
- [45].Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 1994; 5:976-89; PMID:24226387; http://dx.doi.org/ 10.1016/1044-0305(94)80016-2 [DOI] [PubMed] [Google Scholar]
- [46].Kim YJ, Kim JE, Choi HC, Song HK, Kang TC. Cellular and regional specific changes in multidrug efflux transporter expression during recovery of vasogenic edema in the rat hippocampus and piriform cortex. BMB Rep 2015; 48:348-53; PMID:25388209; http://dx.doi.org/ 10.5483/BMBRep.2015.48.6.237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Sung YH, Jin Y, Kang Y, Devkota S, Lee J, Roh JI, Lee HW. Ei24, a novel E2F target gene, affects p53-independent cell death upon ultraviolet C irradiation. J Biol Chem 2013; 288:31261-7; PMID:24014029; http://dx.doi.org/ 10.1074/jbc.M113.477570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chen D, Gould C, Garza R, Gao T, Hampton RY, Newton AC. Amplitude control of protein kinase C by RINCK, a novel E3 ubiquitin ligase. J Biol Chem 2007; 282:33776-87; PMID:17893151; http://dx.doi.org/ 10.1074/jbc.m703320200 [DOI] [PubMed] [Google Scholar]
- [49].Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, Riganelli D, Zanaria E, Messali S, Cainarca S, et al.. The tripartite motif family identifies cell compartments. EMBO J 2001; 20:2140-51; PMID:11331580; http://dx.doi.org/ 10.1093/emboj/20.9.2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hansson MD, Rzeznicka K, Rosenback M, Hansson M, Sirijovski N. PCR-mediated deletion of plasmid DNA. Anal Biochem 2008; 375:373-5; PMID:18157935; http://dx.doi.org/ 10.1016/j.ab.2007.12.005 [DOI] [PubMed] [Google Scholar]
- [51].Park YM. Oxidized LDL induces phosphorylation of non-muscle myosin IIA heavy chain in macrophages. BMB Rep 2015; 48:48-53; PMID:25322953; http://dx.doi.org/ 10.5483/BMBRep.2015.48.1.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003; 19:185-93; PMID:12538238; http://dx.doi.org/ 10.1093/bioinformatics/19.2.185 [DOI] [PubMed] [Google Scholar]
- [53].Shahlaei M, Fassihi A, Saghaie L, Zare A. Prediction of partition coefficient of some 3-hydroxy pyridine-4-one derivatives using combined partial least square regression and genetic algorithm. Res Pharm Sci 2014; 9:143-53; PMID:25657783 [PMC free article] [PubMed] [Google Scholar]
- [54].Ballabio DCV. Classification tools in chemistry. Part 1: linear models: PLS-DA. Analytical Methods 2013; 5:3790-8; http://dx.doi.org/ 10.1039/C3AY40582F [DOI] [Google Scholar]
- [55].Perez NFFJ, Boque R. Calculation of the relability of classification in discriminant partial least-squares binary classification. Chemometrics and Intelligent Laboratory Systems 2009; 95:122-8; http://dx.doi.org/ 10.1016/j.chemolab.2008.09.005 [DOI] [Google Scholar]
- [56].Bovolenta LA, Acencio ML, Lemke N. HTRIdb: an open-access database for experime ntally verified human transcriptional regulation interactions. BMC Genomics 2012; 13:405; PMID:22900683; http://dx.doi.org/ 10.1186/1471-2164-13-405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Jiang C, Xuan Z, Zhao F, Zhang MQ. TRED: a transcriptional regulatory element database, new entries and other development. Nucleic Acids Res 2007; 35:D137-40; PMID:17202159; http://dx.doi.org/ 10.1093/nar/gkl1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Linhart C, Halperin Y, Shamir R. Transcription factor and microRNA motif discovery: the Amadeus platform and a compendium of metazoan target sets. Genome Res 2008; 18:1180-9; PMID:18411406; http://dx.doi.org/ 10.1101/gr.076117.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ryu T, Jung J, Lee S, Nam HJ, Hong SW, Yoo JW, Lee DK, Lee D. bZIPDB: a database of regulatory information for human bZIP transcription factors. BMC Genomics 2007; 8:136; PMID:17535445; http://dx.doi.org/ 10.1186/1471-2164-8-136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Severin J, Waterhouse AM, Kawaji H, Lassmann T, van Nimwegen E, Balwierz PJ, de Hoon MJ, Hume DA, Carninci P, Hayashizaki Y, et al.. FANTOM4 EdgeExpressDB: an integrated database of promoters, genes, microRNAs, expression dynamics and regulatory interactions. Genome Biol 2009; 10:R39; PMID:19374773; http://dx.doi.org/ 10.1186/gb-2009-10-4-r39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al.. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005; 102:15545-50; PMID:16199517; http://dx.doi.org/ 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Dinkel H, Chica C, Via A, Gould CM, Jensen LJ, Gibson TJ, Diella F. Phospho.ELM: a database of phosphorylation sites-update 2011. Nucleic Acids Res 2011; 39:D261-7; PMID:21062810; http://dx.doi.org/ 10.1093/nar/gkq1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, Latham V, Sullivan M. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res 2012; 40:D261-70;PMID:22135298; http://dx.doi.org/ 10.1093/nar/gkr1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Yang CY, Chang CH, Yu YL, Lin TC, Lee SA, Yen CC, Yang JM, Lai JM, Hong YR, Tseng TL, et al.. PhosphoPOINT: a comprehensive human kinase interactome and phospho-protein database. Bioinformatics 2008; 24:i14-20; PMID:18689816; http://dx.doi.org/ 10.1093/bioinformatics/btn297 [DOI] [PubMed] [Google Scholar]
- [65].JD G. Nonparametric statistical inference. M Dekker 1985 [Google Scholar]
- [66].Hwang D, Rust AG, Ramsey S, Smith JJ, Leslie DM, Weston AD, de Atauri P, Aitchison JD, Hood L, Siegel AF, et al.. A data integration methodology for systems biology. Proc Natl Acad Sci U S A 2005; 102:17296-301; PMID:16301537; http//dx..org/ 10.1073/pnas.0508647102 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.