

Abstract
The cancer chemo-preventive effects of equol have been demonstrated for a wide variety of experimental tumours. In a previous study, we found that equol inhibited proliferation and induced apoptotic death of human gastric cancer MGC-803 cells. However, the mechanisms underlying equol-mediated apoptosis have not been well understood. In the present study, the dual AO (acridine orange)/EB (ethidium bromide) fluorescent assay, the comet assay, MTS, western blotting and flow cytometric assays were performed to further investigate the pro-apoptotic effect of equol and its associated mechanisms in MGC-803 cells. The results demonstrated that equol induced an apoptotic nuclear morphology revealed by AO/EB staining, the presence of a comet tail, the cleavage of caspase-3 and PARP and the depletion of cIAP1, indicating its pro-apoptotic effect. In addition, equol-induced apoptosis involves the mitochondria-dependent cell-death pathway, evidenced by the depolarization of the mitochondrial membrane potential, the cleavage of caspase-9 and the depletion of Bcl-xL and full-length Bid. Moreover, treating MGC-803 cells with equol induced the sustained activation of extracellular signal-regulated kinase (ERK), and inhibiting ERK by U0126, a MEK/ERK pathway inhibitor, significantly attenuated the equol-induced cell apoptosis. These results suggest that equol induces mitochondria-dependent apoptosis in human gastric cancer MGC-803 cells via the sustained activation of the ERK1/2 pathway. Therefore, equol may be a novel candidate for the chemoprevention and therapy of gastric cancer.
Keywords: ERK signalling pathway, equol, gastric cancer, mitochondria-dependent apoptosis
INTRODUCTION
Gastric cancer, one of the most common types of cancer and a leading cause of cancer-related deaths worldwide, is a serious threat to human health (Hartgrink et al., 2009). Therefore, developing an effective treatment with minor side effects for gastric cancer is greatly important.
It is encouraging that cancer chemoprevention has received increasing attention in recent years, and the realization that many dietary components possess cancer chemo-preventive activity is particularly significant (Kelloff et al., 2000). It has been suggested that a high rate of consumption of fresh vegetables, fruits, and soy products is associated with a decreased risk of gastric cancer (Hoshiyama and Sasaba, 1992; Kim et al., 2002; Wu et al., 2000). The isoflavones such as genistein and daidzein, which are found in fruits, soybeans and soy-based products, have been widely studied due to the anti-tumour activities they exert against gastric carcinoma. Equol is a bioactive metabolite of daidzein that is produced by gut bacteria. Recently, several reports have suggested that equol has the greatest level of bioactivity among all of the isoflavones that have evaluated in vitro (Hwang et al., 2003; Rufer and Kulling, 2006). However, its anti-tumour effect on gastric carcinoma is not as well understood as that of its precursor daidzein. An epidemiologic study (Ko et al., 2010) found that a higher concentration of equol was associated with a decreased risk of gastric cancer. In our previous in vitro study (Yang et al., 2015), we found that equol induced the cleavage of PARP and caspase-3 in gastric cancer MGC-803 cells, which suggested that the anti-cancer effect of equol on MGC-803 cells might be exerted through its apoptosis-inducing ability. However, the details of the mechanisms that equol induces cell apoptosis have not yet been fully elucidated. The aim of the present study was to elucidate whether death receptor- and/or mitochondria-mediated signalling pathways are involved in equol-induced apoptosis and the underlying mechanisms in human gastric cancer cells.
MATERIALS AND METHODS
Chemicals and reagents
Equol (racemic mixture, 99% pure) was purchased from LC Laboratories (USA). Acridine orange (AO), ethidium bromide (EB) and U0126 were purchased from Sigma-Aldrich (USA). Rabbit polyclonal antibodies specific to cleaved caspase-3, cleaved caspase-9, P-ERK, PARP, P-JNK, P-P38, Bid, Bax, Bcl-xL and caspase-8 were obtained from Cell Signaling Technology (USA). Rabbit monoclonal antibodies specific to GAPDH, Bcl-2, Bak1, cIAP1, ERK, JNK and P-38 and all of the secondary antibodies were purchased from Santa Cruz Biotechnology (USA). The CellTiter 96® Aqueous One Solution Cell Proliferation Assay Kit (MTS) was purchased from Promega (USA). The JC-1-based mitochondrial membrane-potential assay kit was obtained from Beyotime (China). The Annexin V/PI apoptosis kit was obtained from BD Biosciences (USA) and the Cell Cycle Staining Kit was purchased from MultiScience Biotech (China).
Cell culture and treatment
The MGC-803 human gastric cancer cell line (Cell Bank of the Chinese Academy of Sciences, China) was maintained and subcultured at 37°C in RPMI 1640 medium supplemented with 10% FBS, 1% penicillin/streptomycin stock and 2% L-glutamine in a humidified atmosphere of 95% O2 and 5% CO2. For the experiments, the concentration of FBS in the treatment medium was reduced to 2%. The equol stock solutions were prepared by dissolving equol in DMSO, and the final concentration of the DMSO vehicle in the treatment medium was 0.1%.
Dual AO/EB fluorescent staining
The apoptotic morphology of the cells was determined by dual AO/EB fluorescent staining. Briefly, MGC-803 cells that were treated for 48 h with 20 μM equol or the vehicle were harvested and were re-suspended in PBS to a concentration of 1 × 106 cells/ml. To prepare a slide, 1 μl of AO/EB (100 mg/ml) staining solution was mixed with 25 μl of a cell suspension on a clean glass slide, and the cells were then covered with a coverslip. The slide was immediately viewed using fluorescence microscopy (IX70, SIF2 Olympus). In the analysis, the cells with nuclei that were uniformly green, green with bright dots and orange with obvious condensation were identified as healthy, early apoptotic and late apoptotic cells, respectively.
Comet assay
DNA fragmentation associated with apoptosis was determined by comet assay. MGC-803 cells were treated with the vehicle. or with 20 μM equol for 48 h. The treated cells were then collected and were diluted with PBS to 5 × 105 cells/ml. To prepare the slides, a mixture of 10 μl of a cell suspension and 90 μl of 0.8% low temperature melting agarose was pipetted onto frosted slides that had been precoated with a layer of 100 μl 1% normal temperature melting agarose. After gelling, the coverslip was gently removed from each slide. The slides were then placed in a lysis solution (2 M NaCl, 30 mM Na2EDTA, 10 mM Tris, 1% N-lauroylsarcosine sodium salt, pH 8.2–8.5, supplemented with 10% DMSO and 1% Triton X-100) at 4°C for 1 h, and then were incubated in fresh alkaline buffer for 30 min at room temperature to allow the DNA to unwind. After electrophoresis in ice-cold alkaline electrophoresis buffer, the slides were stained with an EB solution (20μg/ml) and were then examined using a fluorescence microscope (IX70, SIF2 Olympus).
Annexin V/PI apoptosis assay
Cells were seeded into 60-mm plates and treated with different doses of equol for desired period. The negative control cells were treated with DMSO under similar conditions. At the end of incubation, the cells were harvested and stained with Annexin-V and PI as described in the instructions for use of the Annexin V/PI apoptosis kit. The samples were analysed using flow cytometry as previously described (Yang et al., 2015).
Mitochondrial membrane potential (MMP) assay
A mitochondrial membrane potential assay kit (containing JC-1) was used according to the manufacturer’s protocol to detect changes in the MMP of cells. Briefly, MGC-803 cells were seeded into 6-well plates and treated with the vehicle or 20μM equol for 48 h. Then the treated cells were harvested and resuspended in 0.5 ml of RPMI1640 medium supplemented with 10% FBS, after which they were stained with 0.5 ml JC-1 (1 ×) for 20 min at 37°C. After being rinsed, the cells were resuspended in 0.5 ml of buffer and were analysed using flow cytometry (BD FACSCalibur, USA).
Cell cycle assay
The treated cells (1 × 106) were harvested and fixed in ice-cold 70% ethanol at 4°C for 2 h. Then, the cells were resuspended with PBS and stained with propidium iodide for 30 min in the dark at 37°C, utilizing a Cell Cycle Staining Kit. Cell cycle analysis was performed using a FACScan flow cytometer (BD FACSCalibur), as previously described (Yang et al., 2015).
Cell viability assay
MGC-803 cell viability was examined using the MTS assay, utilizing the CellTiter 96β Aqueous One Solution. Briefly, cells were seeded into 96-well plates (4 × 103 cells/well) and pretreated with 10 μM U0126 for 1 h, after which equol was added to the media with or without the inhibitor. After 48 h of incubation, the MTS reagent was added. Detailed information regarding the MTS assay was provided in our previous report (Yang et al., 2015).
Western blotting analysis
The cells were seeded into 60-mm plates and treated with equol for the desired period. The treated cells were lysed on ice using RIPA Lysing Buffer, and then the total protein concentration was quantified utilizing a BCA protein assay kit. Equal amounts of proteins (20–80 μg/lane) were separated on 8–12% SDS-PAGE gels and electrotransferred to PVDF membranes. The subsequent steps involved in western blotting analysis were conducted as described previously (Yang et al., 2015).
Statistical analysis
The statistical analyses were conducted utilizing SPSS 17.0 software. All of the values are expressed as the mean values ± S.E. Significant differences between two groups were determined using Student’s t test, and in cases with more than two treatment groups analysed, one-way analysis of variance (ANOVA) followed by Turkey’s post hoc test was performed, with a 0.05 significance level. Each experiment was repeated at least three times.
RESULTS
Equol induces apoptotic cell death in MGC-803 cells
To determine whether equol could induce apoptosis, alterations in the nuclear morphology of equol-treated MGC-803 cells were studied. The AO/EB double staining results (Figs. 1A and 2B) demonstrated that cells treated with 20 μM equol for 48 h were undergoing both early apoptosis (exhibiting green nuclei containing bright green dots) and late apoptosis (exhibiting orange nuclei with obvious condensation). However, most of the control cells that had been treated with 0.1% DMSO were identified as healthy (exhibiting nuclei stained uniformly green), and very few apoptotic cells were observed among them. Therefore, we concluded that equol induces apoptosis in MGC-803 cells.
Fig. 1.
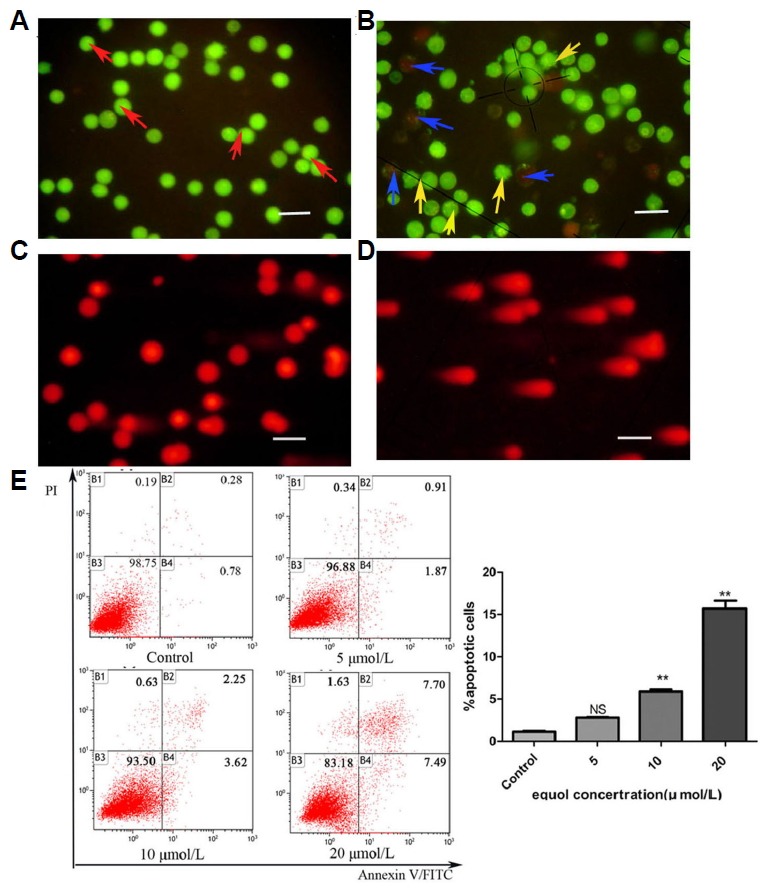
Equol induces MGC-803 cell apoptosis. Harvested cells treated with the vehicle or 20 μM equol were subjected to dual AO/EB fluorescent staining (A) and (B) and the comet assay (C) and (D), and then observed under a fluorescence microscope (200×): (A) Representative healthy control cells with an intact green-stained nucleus (red arrows); (B) Representative 20 μM of equol-treated cells that were undergoing early apoptosis (yellow arrows) and late apoptosis (blue arrows). (C) Representative control cells that did not exhibit a comet tail; (D) Cells treated with 20 μM equol for 48 h exhibited a comet tail. (E) Harvested cells treated with different concentrations of equol for 24 h were subjected to Annexin V/PI apoptosis assay (**P < 0.01 vs control).
Fig. 2.
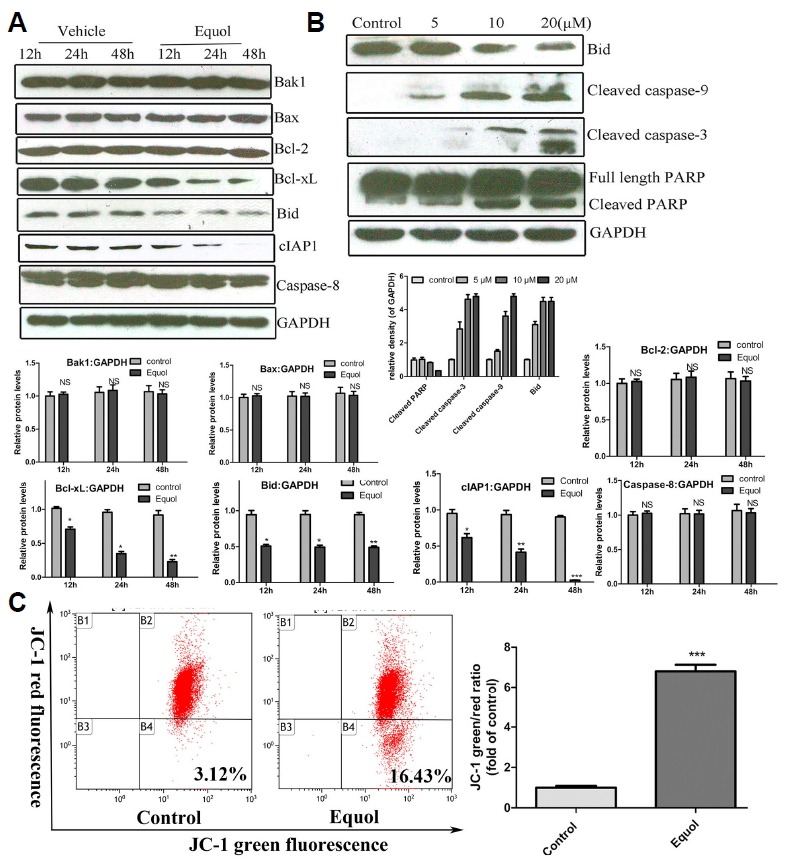
Equol induced the apoptosis of MGC-803 cells through the mitochondria-mediated pathway. (A and B) The levels of proteins associated with apoptosis and caspase cleavage were determined by western blotting using specific antibodies: (A) Cells were treated with 20 μM equol for 12, 24 and 48 h; (B) Cells were treated with equol at various concentrations for 24 h. GAPDH was utilized as the loading control. (C) Cells treated with the vehicle or 20 μM equol were harvested, and their mitochondrial membrane potentials were analysed according to JC-1 staining using flow cytometry (*P < 0.05; **P < 0.01, ***P < 0.001 vs control).
To evaluate the pro-apoptotic effect of equol on MGC-803 cells, the degree of equol-induced DNA damage was determined by the comet assay. The results demonstrated that equol induced DNA damage, as indicated by the presence of a DNA-containing comet tail, whereas the control cells did not exhibit a comet tail (Figs. 1C and 1D). These results showed that equol induced DNA damage in MGC-803 cells, which might activate the apoptotic pathway.
Moreover, the quantification of equol-induced apoptotic cells was determined by Annexin V/PI apoptosis assay. As shown in Fig. 1E, after 24 h incubation, equol dose-dependently induced MGC-803 cells apoptosis. The percentages of apoptotic cells, including the early and late apoptotic cells, were 1.06%, 2.18%, 5.87%, and 15.19% in response to control, 5, 10, and 20 μmol/L of equol treatment, respectively.
Equol induces apoptosis via the mitochondria-mediated pathway
To determine whether equol-induced apoptosis is mediated through the intrinsic mitochondria-dependent pathway or the extrinsic death receptor-dependent pathway, the cleavage of caspase-3/9/8 and PARP in treated cells was evaluated using western blotting. As shown in Figs. 2A and 2B, equol significantly induced the cleavage of caspase-3/9 and subsequently increased the level of cleaved PARP, without affecting the level of caspase-8 (no cleavage was observed), which indicated the involvement of the mitochondria-mediated apoptotic pathway rather than extrinsic death receptor-mediated apoptotic pathway (Figs. 2A and 2B).
To further investigate whether equol-induced apoptosis involves the mitochondria-dependent pathway, JC-1, a cationic fluorescent dye, was used to determine the mitochondrial membrane potential of cells. As shown in Fig. 2C, equol treatment significantly elevated the level of green fluorescence to 16.45%, compared with the level of 3.12% in the vehicle-treated control cells (P < 0.05). However, the intensity of red fluorescence was decreased after equol treatment. These results demonstrated that equol effectively induced the depolarization of the mitochondrial membrane potential and that equol-induced apoptosis involves the mitochondria-mediated pathway.
Effects of equol on the proteins involved in mitochondria-mediated apoptosis
Furthermore, we investigated the effect of equol on the levels of proteins involved in mitochondria-mediated apoptosis, including those of pro-apoptotic proteins (Bax, Bak1, Bid) and anti-apoptotic proteins (Bcl-2, Bcl-xL), using Western blotting assay. As shown in Fig. 2A, equol treatment dramatically decreased the expression of Bcl-xL. However, there was no significant change in the Bcl-2 level in equol-treated MGC-803 cells. In addition, equol treatment did not affect the levels of Bax and Bak1. Moreover, equol-induced cell apoptosis was accompanied by the depletion of full-length Bid, which indicated its cleavage. Our study also demonstrated that equol treatment decreased the level of cIAP1, an inhibitor of pro-apoptotic proteins, which further supported the apoptosis-promoting effect of equol.
Equol induces cell apoptosis through the sustained activation of the ERK1/2 pathway
MAPK-signalling cascades have been shown to play an important role in apoptosis induced by chemotherapeutic drugs (Miyoshi et al., 2004; Remacle-Bonnet et al., 2000). We therefore determined the activation levels of JNK, ERK and p38 in cells treated with equol. As shown in Fig. 3, the phosphorylation level of ERK in response to equol treatment was apparently elevated compared with that of the vehicle-treated control. However, equol treatment did not affect the phosphorylation levels of p38 and JNK at all time points examined. These results indicate that sustained ERK activation might be involved in the equol-induced apoptosis of MGC-803 cells.
Fig. 3.
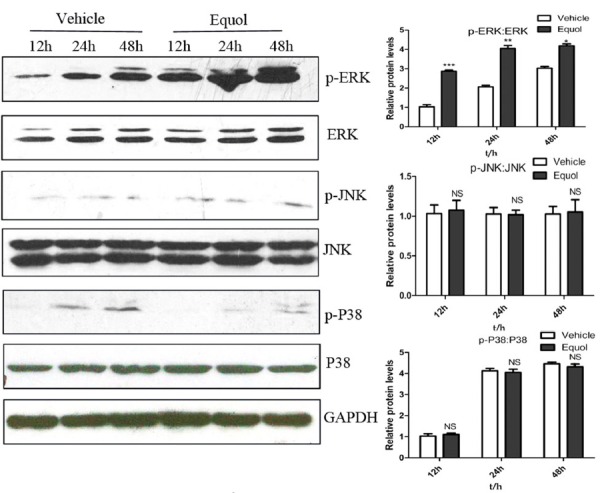
Effect of equol on MAPK activation. Cells were treated with the vehicle or 20 μM of equol for 12, 24 and 48 h. The levels of phosphorylated and total ERK1/2, JNK1/2 and p38 were determined by Western blotting using specific antibodies (*P< 0.05, **P < 0.01, ***P < 0.001 vs control).
To define the role of ERK1/2 in equol-induced cell-growth inhibition and apoptosis, MGC-803 cells were pretreated with the ERK1/2 inhibitor U0126 (10 μM) for 1 h. As shown in Fig. 4, the results of the Annexin V/PI apoptotic assay showed that U0126 treatment alone did not change the percentages of apoptotic cells significantly. Treatment with equol at a concentration of 20 μM for 36 h resulted in 18% apoptotic cells. However, only 5% of the cells were apoptotic when equol (20 μM) was coadministered with U0126 (10 μM). We next evaluated whether U0126 reversed the equol-induced ERK1/2 activation and PARP cleavage, and we found that the levels of phosphorylated ERK1/2 and cleaved PARP were significantly decreased when equol was administered in combination with U0126 (Fig. 5). Furthermore, we observed that the equol-induced depletion of Bcl-xL was partially reversed when equol was coadministered with U0126 (Fig. 5).
Fig. 4.
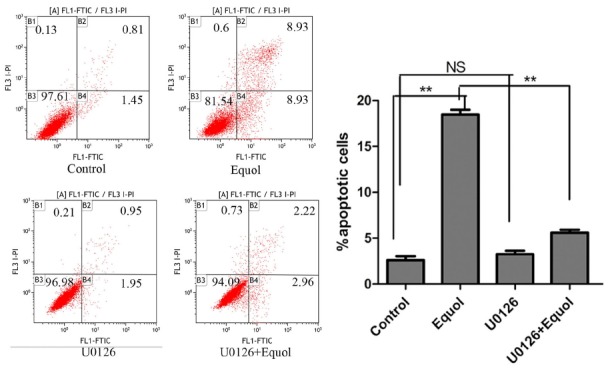
Effect of ERK inhibition on equol-induced cell apoptosis. MGC-803 cells were pretreated with 10 μM U0126 for 1 h prior to treatment with 20 μM equol for 36 h, and the content of apoptotic cells was evaluated using flow cytometry (**P < 0.01 vs control).
Fig. 5.
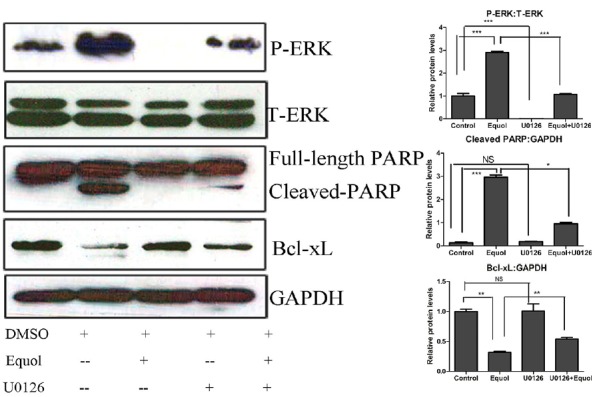
Inhibition of ERK phosphorylation and its effect on PARP cleavage and Bcl-xL expression. Cells were pretreated with 10 μM U0126 for 1 h prior to treatment with 20 μM equol for 48 h. The levels of phosphorylated ERK, total ERK, cleaved-PARP and Bcl-xL were determined using Western blotting (**P < 0.01, ***P < 0.001 vs control).
The results of our previous study (Yang et al., 2015) demonstrated that equol treatment effectively reduced the viability of MGC-803 cells and triggered cell cycle arrest at the G0/G1 phase. To assess whether these effects are mediated by ERK1/2 activation, we next performed MTS and cell-cycle assays. As shown in Fig. 6B, blocking the activation of ERK1/2 with its inhibitor U0126 resulted in an increased level of cell viability (43%) compared with that of cells treated with only equol (33%) (P < 0.05), which indicated that U0126 partially reversed the equol-induced reduction in cell viability. However, we observed that U0126 did not prevent but enhanced the level of equol-induced G0/G1 cell cycle arrest, with 54% and 61% when equol was alone or administered in combination with U0126, respectively (Fig. 6A), which indicated that the equol-induced sustained activation of ERK1/2 was not involved in the induction of G1 phase cell cycle arrest.
Fig. 6.
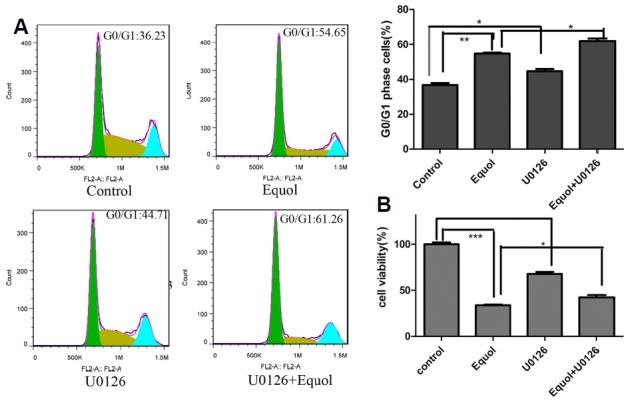
Effect of ERK inhibition on ameliorating the equol-induced G0/G1 phase arrest and reduction in cell viability. MGC-803 cells were pretreated with 10 μM U0126 for 1 h prior to treatment with 20 μM equol. (A) After 24 h, the cell cycle distribution was examined using flow cytometry; (B) After 48 h, the cell viability was determined using the MTS assay (*P < 0.05, **P < 0.01, ***P < 0.001 vs control).
DISCUSSION
In this study, we demonstrated for the first time that equol, a metabolite of the isoflavone daidzein, induces the apoptotic death of human gastric cancer MGC-803 cells via the mitochondria-dependent pathway, which was evidenced by the equol-mediated cleavage of caspase-9 and depolarization of the mitochondrial membrane potential. Furthermore, equol-triggered apoptosis is regulated by apoptosis-related proteins, such as Bcl-xL, Bid and cIAP1. Our study also clearly demonstrated that the sustained activation of ERK1/2 induced by equol is involved in its induction of mitochondria-dependent apoptotic death.
Apoptosis, a fundamental process essential for normal development and the maintenance of tissue homeostasis, plays a critical role in the elimination of cancer cells (Li et al., 2010). Thus, targeting signalling intermediates in apoptosis-inducing pathways is an effective strategy for cancer prevention and therapy (Sun et al., 2004). Several hallmarks of apoptosis, such as significant increase in cells exhibiting comet tails and cells with AO/EB- or Annexin V/PI -positive, were observed among gastric cancer MGC-803 cells after they were treated with equol. These results clearly demonstrated that equol treatment promoted the apoptotic death of MGC-803 cells. In addition, equol treatment resulted in a significantly decreased level of cIAP1, a member of the IAP family of proteins that exert their anti-apoptotic activity by inhibiting caspase-3 and caspase-9 activation (Choi et al., 2009; Kulathila et al., 2009), supporting the apoptosis-promoting activity of equol. In general, apoptosis is mediated through two major pathways, the extrinsic and intrinsic pathways (Sun et al., 2004). The extrinsic pathway is triggered at the plasma membrane by the activation of the death receptor, which leads to the cleavage of caspase-8, whereas the intrinsic pathway is triggered by various apoptotic stress signals and is characterized by mitochondrial dysfunction and the cleavage of caspase-3 and -9. The results of our study supported the conclusion that equol induced MGC-803 cell apoptosis through activation of the intrinsic pathway rather than the extrinsic pathway, as shown by the depolarization of the mitochondrial membrane potential and increase in the levels of cleaved caspases-3 and -9, without a change in the level of cleaved caspase-8, in equol-treated cells (Fig. 7). Furthermore, our results demonstrated that equol treatment decreased the level of Bcl-xL, an anti-apoptotic protein associated with mitochondria-mediated apoptosis, which further verified the involvement of mitochondrial-mediated apoptosis upon equol treatment. In addition, we also found that equol decreased the level of Bid, another protein that plays a vital role in the mitochondrial-mediated pathways, which in contrast, exerts proapoptotic effect. In general, full-length Bid is a relatively poor inducer of cytochrome-c release, whereas the c-terminal portion of Bid (tBid) that is generated upon its cleavage is very potent in this regard. In our study, equol-induced cell death was accompanied by the depletion of full-length Bid, which indicated that equol induced the cleavage of Bid. It has been reported that Bid is cleaved upon caspase-8 activation in response to multiple death-inducing stimuli of the extrinsic apoptotic pathway. However, in other cases, Bid cleavage is catalysed by caspase-3 as a consequence of cytochrome-c release and participates in a feedback loop that amplifies the death signal (Slee et al., 2000). In the present study, equol induced Bid depletion and caspase-3 cleavage without altering the caspase-8 level, thus favouring Bid cleavage via caspase-3 rather than caspase-8 (Fig. 7).
Fig. 7.
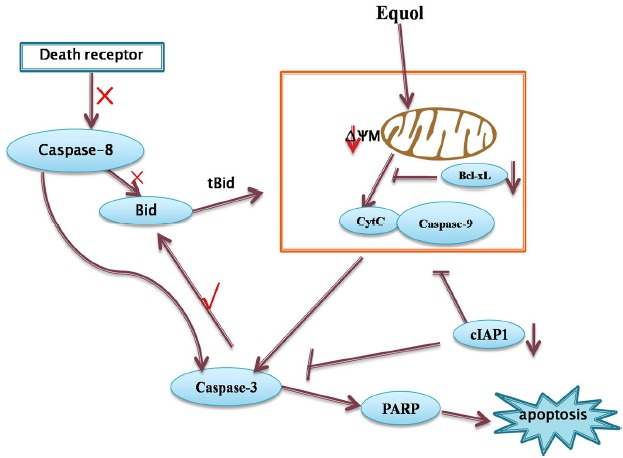
Overview of the pathways for equol-induced apoptosis in MGC-803 cells
The MAPKs comprise a family of serine/threonine protein kinases that are involved in many cellular processes, including proliferation, differentiation, cellular stress responses, and apoptosis (Pearson et al., 2001). Activation of the members of a major MAPK subfamily (JNK, P38, and ERK1/2) has been implicated in the activities of numerous chemotherapeutic and genotoxic drugs. In this study, we found that JNK and P38 were slightly phosphorylated in control MGC-803 cells and that equol treatment did not affect their levels of phosphorylation. However, there was a dramatic increase in the level of phosphorylated ERK in equol-treated cells compared with that of vehicle-treated control cells. ERK activation has been shown to stimulate cellular proliferation (Xia et al., 1995). However, increasing evidence suggests that the activation of ERK promotes cell cycle arrest and apoptosis (Elder et al., 2002; Galve-Roperh et al., 2000; Pumiglia and Decker, 1997; Stanciu et al., 2000). Thus, the ERK pathway, once activated, can exert either proapoptotic or anti-apoptotic effects, depending upon the stimuli to which the cells are responding and the cell type (Elder et al., 2002). In general, transient ERK activation leads to cell proliferation, whereas sustained ERK activation is associated with apoptosis (Hsu et al., 2005; Zhu et al., 2004). In this study, we found that equol treatment induced sustained ERK activation lasting from 12 h to 48 h and that ERK inhibition by U0126 attenuated equol-induced apoptosis, suggesting that the proapoptotic effects of equol on MGC-803 cells are mediated by the sustained activation of the ERK1/2 signalling pathway. In addition, the sustained activation of ERK has also been reported to be involved in cell cycle arrest (Pumiglia and Decker, 1997; Sarfaraz et al., 2006; Yang et al., 2011; Zhu et al., 2004). However, in our study, inhibiting ERK activity did not prevent but rather enhanced the G0/G1 phase cell cycle arrest induced by equol treatment, which suggested that the equol-induced sustained activation of ERK1/2 was not involved in the induction of G0/G1 phase cell cycle arrest. This result does not contradict the results of our previous study (Yang et al., 2015), which suggested that equol induces G0/G1 phase cell cycle arrest via modulating the Akt pathway. Increasing evidence suggests that the sustained activation of ERK affects apoptotic cell death by regulating its downstream targets, including Cox-2, caspase-8, Bax, Bcl-2, and other molecules (DeHaan et al., 2001; Elder et al., 2002; Goillot et al., 1997; Kim et al., 2008). A lot of studies (DeHaan et al., 2001; Li et al., 2007) have demonstrated that sustained ERK activation induced apoptosis by inhibiting the expression of Bcl-xL. Consistent with those findings, our results demonstrated that equol induced the apoptotic death of MGC-803 cells, accompanied by sustained ERK activation and down-regulation of the Bcl-xL level. Furthermore, inhibiting ERK activity with U0126, a specific MEK/ERK-pathway inhibitor, partially rescued the equol-induced apoptosis and Bcl-xL down-regulation, which suggested that the activation of the ERK/Bcl-xL signalling pathway plays a critical role in equol-induced apoptosis of MGC-803 cells (Fig. 7).
Taken together, these findings indicate that equol induces apoptosis in a mitochondria-dependent manner that might be mediated by the sustained activation of ERK. The present study also demonstrated the involvement of apoptosis-associated proteins and end-point markers of apoptosis, including Bid, Bcl-xL, cIAP1, caspase-3 and PARP, in equol-induced apoptosis. The present findings strongly indicate that equol may be a novel candidate for chemo-preventive and chemotherapeutic treatments aimed at gastric cancer.
ACKNOWLEDGEMENTS
The reported work was supported by Special Project on the Integration of Industry, Education and Research of Fujian Province, China, No.2012N0014.
REFERENCES
- Choi Y.E., Butterworth M., Malladi S., Duckett C.S., Cohen G.M., Bratton S.B. The E3 ubiquitin ligase cIAP1 binds and ubiquitinates caspase-3 and -7, via unique mechanisms at distinct steps in their processing. J. Biol. Chem. 2009;284:12772–12782. doi: 10.1074/jbc.M807550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeHaan R.D., Yazlovitskaya E.M., Persons D.L. Regulation of p53 target gene expression by cisplatin-induced extracellular signal-regulated kinase. Cancer Chemother Pharmacol. 2001;48:383–388. doi: 10.1007/s002800100318. [DOI] [PubMed] [Google Scholar]
- Elder D.J., Halton D.E., Playle L.C., Paraskeva C. The MEK/ERK pathway mediates COX-2-selective NSAID-induced apoptosis and induced COX-2 protein expression in colorectal carcinoma cells. Int. J Cancer. 2002;99:323–327. doi: 10.1002/ijc.10330. [DOI] [PubMed] [Google Scholar]
- Galve-Roperh I., Sanchez C., Cortes M.L., Gomez del Pulgar T., Izquierdo M., Guzman M. Anti-tumoral action of cannabinoids: involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nat. Med. 2000;6:313–319. doi: 10.1038/73171. [DOI] [PubMed] [Google Scholar]
- Goillot E., Raingeaud J., Ranger A., Tepper R.I., Davis R.J., Harlow E., Sanchez I. Mitogen-activated protein kinase-mediated Fas apoptotic signaling pathway. Proc. Natl. Acad. Sci USA. 1997;94:3302–3307. doi: 10.1073/pnas.94.7.3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartgrink H.H., Jansen E.P., van Grieken N.C., van de Velde C.J. Gastric cancer. Lancet. 2009;374:477–490. doi: 10.1016/S0140-6736(09)60617-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshiyama Y., Sasaba T. A case-control study of stomach cancer and its relation to diet, cigarettes, and alcohol consumption in Saitama Prefecture, Japan. Cancer Causes Control. 1992;3:441–448. doi: 10.1007/BF00051357. [DOI] [PubMed] [Google Scholar]
- Hsu Y.L., Kuo P.L., Lin L.T., Lin C.C. Asiatic acid, a triterpene, induces apoptosis and cell cycle arrest through activation of extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways in human breast cancer cells. J. Pharmacol. Exp. Ther. 2005;313:333–344. doi: 10.1124/jpet.104.078808. [DOI] [PubMed] [Google Scholar]
- Hwang J., Wang J., Morazzoni P., Hodis H.N., Sevanian A. The phytoestrogen equol increases nitric oxide availability by inhibiting superoxide production: an antioxidant mechanism for cell-mediated LDL modification. Free Radic. Biol. Med. 2003;34:1271–1282. doi: 10.1016/s0891-5849(03)00104-7. [DOI] [PubMed] [Google Scholar]
- Kelloff G.J., Crowell J.A., Steele V.E., Lubet R.A., Malone W.A., Boone C.W., Kopelovich L., Hawk E.T., Lieberman R., Lawrence J.A., et al. Progress in cancer chemoprevention: development of diet-derived chemopreventive agents. J. Nutr. 2000;130:467S–471S. doi: 10.1093/jn/130.2.467S. [DOI] [PubMed] [Google Scholar]
- Kim H.J., Chang W.K., Kim M.K., Lee S.S., Choi B.Y. Dietary factors and gastric cancer in Korea: a case-control study. Int. J Cancer. 2002;97:531–535. doi: 10.1002/ijc.10111. [DOI] [PubMed] [Google Scholar]
- Kim Y.H., Lee D.H., Jeong J.H., Guo Z.S., Lee Y.J. Quercetin augments TRAIL-induced apoptotic death: involvement of the ERK signal transduction pathway. Biochem. Pharmacol. 2008;75:1946–1958. doi: 10.1016/j.bcp.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko K.P., Park S.K., Park B., Yang J.J., Cho L.Y., Kang C., Kim C.S., Gwack J., Shin A., Kim Y., et al. Isoflavones from phytoestrogens and gastric cancer risk: a nested case-control study within the Korean Multicenter Cancer Cohort. Cancer Epidemiol. Biomarkers Prev. 2010;19:1292–1300. doi: 10.1158/1055-9965.EPI-09-1004. [DOI] [PubMed] [Google Scholar]
- Kulathila R., Vash B., Sage D., Cornell-Kennon S., Wright K., Koehn J., Stams T., Clark K., Price A. The structure of the BIR3 domain of cIAP1 in complex with the N-terminal peptides of SMAC and caspase-9. Acta Crystallogr. D Biol. Crystallogr. 2009;65:58–66. doi: 10.1107/S0907444908039243. [DOI] [PubMed] [Google Scholar]
- Li H., Wang X., Li N., Qiu J., Zhang Y., Cao X. hPEBP4 resists TRAIL-induced apoptosis of human prostate cancer cells by activating Akt and deactivating ERK1/2 pathways. J. Biol. Chem. 2007;282:4943–4950. doi: 10.1074/jbc.M609494200. [DOI] [PubMed] [Google Scholar]
- Li S., Dong P., Wang J., Zhang J., Gu J., Wu X., Wu W., Fei X., Zhang Z., Wang Y., et al. Icariin, a natural flavonol glycoside, induces apoptosis in human hepatoma SMMC-7721 cells via a ROS/JNK-dependent mitochondrial pathway. Cancer Lett. 2010;298:222–230. doi: 10.1016/j.canlet.2010.07.009. [DOI] [PubMed] [Google Scholar]
- Miyoshi N., Uchida K., Osawa T., Nakamura Y. A link between benzyl isothiocyanate-induced cell cycle arrest and apoptosis: involvement of mitogen-activated protein kinases in the Bcl-2 phosphorylation. Cancer Res. 2004;64:2134–2142. doi: 10.1158/0008-5472.can-03-2296. [DOI] [PubMed] [Google Scholar]
- Pearson G., Robinson F., Beers Gibson T., Xu B.E., Karandikar M., Berman K., Cobb M.H. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr. Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- Pumiglia K.M., Decker S.J. Cell cycle arrest mediated by the MEK/mitogen-activated protein kinase pathway. Proc. Natl. Acad. Sci USA. 1997;94:448–452. doi: 10.1073/pnas.94.2.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remacle-Bonnet M.M., Garrouste F.L., Heller S., Andre F., Marvaldi J.L., Pommier G.J. Insulin-like growth factor-I protects colon cancer cells from death factor-induced apoptosis by potentiating tumor necrosis factor alpha-induced mitogen-activated protein kinase and nuclear factor kappaB signaling pathways. Cancer Res. 2000;60:2007–2017. [PubMed] [Google Scholar]
- Rufer C.E., Kulling S.E. Antioxidant activity of isoflavones and their major metabolites using different in vitro assays. J. Agric. Food Chem. 2006;54:2926–2931. doi: 10.1021/jf053112o. [DOI] [PubMed] [Google Scholar]
- Sarfaraz S., Afaq F., Adhami V.M., Malik A., Mukhtar H. Cannabinoid receptor agonist-induced apoptosis of human prostate cancer cells LNCaP proceeds through sustained activation of ERK1/2 leading to G1 cell cycle arrest. J. Biol. Chem. 2006;281:39480–39491. doi: 10.1074/jbc.M603495200. [DOI] [PubMed] [Google Scholar]
- Slee E.A., Keogh S.A., Martin S.J. Cleavage of BID during cytotoxic drug and UV radiation-induced apoptosis occurs downstream of the point of Bcl-2 action and is catalysed by caspase-3: a potential feedback loop for amplification of apoptosis-associated mitochondrial cytochrome c release. Cell Death Differ. 2000;7:556–565. doi: 10.1038/sj.cdd.4400689. [DOI] [PubMed] [Google Scholar]
- Stanciu M., Wang Y., Kentor R., Burke N., Watkins S., Kress G., Reynolds I., Klann E., Angiolieri M.R., Johnson J.W., et al. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J. Biol. Chem. 2000;275:12200–12206. doi: 10.1074/jbc.275.16.12200. [DOI] [PubMed] [Google Scholar]
- Sun S.Y., Hail N., Jr., Lotan R. Apoptosis as a novel target for cancer chemoprevention. J. Natl. Cancer Inst. 2004;96:662–672. doi: 10.1093/jnci/djh123. [DOI] [PubMed] [Google Scholar]
- Wu A.H., Yang D., Pike M.C. A meta-analysis of soyfoods and risk of stomach cancer: the problem of potential confounders. Cancer Epidemiol. Biomarkers Prev. 2000;9:1051–1058. [PubMed] [Google Scholar]
- Xia Z., Dickens M., Raingeaud J., Davis R.J., Greenberg M.E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Yang T.Y., Chang G.C., Chen K.C., Hung H.W., Hsu K.H., Sheu G.T., Hsu S.L. Sustained activation of ERK and Cdk2/cyclin-A signaling pathway by pemetrexed leading to Sphase arrest and apoptosis in human non-small cell lung cancer A549 cells. Eur. J. Pharmacol. 2011;663:17–26. doi: 10.1016/j.ejphar.2011.04.057. [DOI] [PubMed] [Google Scholar]
- Yang Z.P., Zhao Y., Huang F., Chen J., Yao Y.H., Li J., Wu X.N. Equol inhibits proliferation of human gastric carcinoma cells via modulating Akt pathway. World J. Gastroenterol. 2015;21:10385–10399. doi: 10.3748/wjg.v21.i36.10385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H., Zhang L., Wu S., Teraishi F., Davis J.J., Jacob D., Fang B. Induction of S-phase arrest and p21 overexpression by a small molecule 2[[3-(2,3-dichlorophenoxy)propyl] amino]ethanol in correlation with activation of ERK. Oncogene. 2004;23:4984–4992. doi: 10.1038/sj.onc.1207645. [DOI] [PubMed] [Google Scholar]