

Abstract
Osteoprotegerin (OPG), a decoy receptor secreted by osteoblasts, is a major negative regulator of bone resorption. It functions by neutralizing the receptor activator of nuclear factor κB ligand (RANKL), which plays a central role in promoting osteoclastogenesis. OPG is known to be a high-affinity heparan sulfate (HS)-binding protein. Presumably, HS could regulate the function of OPG and affect how it inhibits RANKL. However, the molecular detail of HS-OPG interaction remains poorly understood, which hinders our understanding of how HS functions in osteoclastogenesis. Here we report mapping of the HS-binding site of OPG. The HS-binding site, identified by mutagenesis study, consists of eight basic residues that are located mostly at the junction of the second death domain and the C-terminal domain. We further show that heparin-derived dodecasaccharide is able to induce dimerization of OPG monomers with a stoichiometry of 1:1. Small-angle X-ray scattering analysis revealed that upon binding of HS, OPG undergoes a dramatic conformational change, resulting in a more compact and less flexible structure. Importantly, we present here three lines of evidence that HS, OPG, and RANKL form a stable ternary complex. Using a HS binding-deficient OPG mutant, we further show that in an osteoblast/bone marrow macrophage co-culture system, immobilization of OPG by HS at the osteoblast cell surface substantially lowers the inhibitory threshold of OPG toward RANKL. These discoveries strongly suggest that HS plays an active role in regulating OPG-RANKL interaction and osteoclastogenesis.
Keywords: conformational change, dimerization, heparan sulfate, osteoblast, small-angle X-ray scattering (SAXS), RANKL, osteoclastogenesis, osteoprotegerin, ternary complex
Introduction
Bone is a dynamic tissue that constantly undergoes turnover. This task is undertaken by the concerted work of osteoclasts, which resorb old bone, and osteoblasts, which form new bone. Interestingly, osteoclastogenesis is largely controlled by osteoblast-expressed factors, and the most important of which are RANKL and OPG. Osteoblasts express receptor activator of nuclear factor κB ligand (RANKL)3 as a membrane-associated juxtacrine factor that drives osteoclastogenesis by interacting with its receptor RANK on osteoclast precursors (1, 2). To keep osteoclastogenesis in check, osteoblasts secrete OPG as a decoy receptor that neutralizes the activity of RANKL (3, 4). It is a consensus now that maintaining optimal RANKL/OPG ratio is crucial in regulating bone remodeling. When the balance is tipped toward RANKL, as in the case of diseases like osteoporosis and rheumatoid arthritis, unrestrained osteoclastogenesis leads to excessive bone loss (5).
Although OPG is known to bind heparin with low nanomolar affinity (6), very little is known regarding the structural details of HS-OPG interactions. One early study using domain deletion experiments concluded that the last C-terminal domain (Tail domain hereafter) is responsible for HS binding (Fig. 1), which was based on the dramatically reduced binding of the truncated OPG to heparin-Sepharose (7). However, due to the limitations of domain deletion experiments, whether the Tail domain is truly and solely responsible for HS binding can be debated. It is likely that other basic residues, which are abundant in both D1 and D2 domains, also make contribution to HS binding (Fig. 1). Another important feature of OPG is that it exists as both monomer and dimer (3). Interestingly, the dimeric OPG binds heparin significantly better than the monomer (7). Although an earlier study suggests that the dimer is mediated by an intermolecular disulfide bond formed at Cys400 (7), a recent study challenges this view (8). Therefore, it remains to be determined how exactly OPG dimerizes, and what is the relationship between OPG dimerization and HS-OPG interactions.
FIGURE 1.
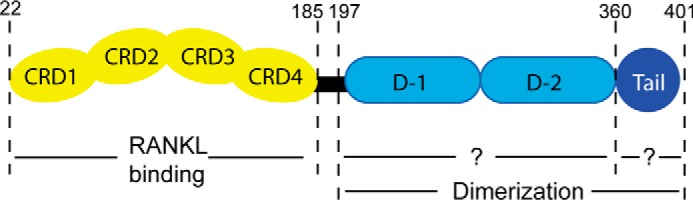
Schematic view of the OPG domain structure. The N-terminal cysteine-rich domains (CRD1–4) are responsible for RANKL binding. The structure and exact function of the two death domains (D1 and D2) and the last C-terminal domain (Tail) are largely unknown. Amino acid numbering is based on mouse OPG.
Like any other HS-binding proteins, the ability to bind HS most likely enables OPG to function in a more refined manner. However, although several studies hinted on how HS might regulate the function of OPG, the exact role of HS in OPG biology remains elusive to this day. One study has shown that myeloma cells are capable of internalizing and depleting OPG in an HS-dependent manner, which may contribute to the reduced OPG levels in the bone marrow of myeloma patients, who often suffer from osteolysis (9). Another study, using an osteosarcoma mouse model, has found that full-length OPG (able to bind HS) is less effective than a truncated OPG (unable to bind HS) in reducing the osteolytic bone lesion, likely due to its reduced bioavailability as a result of binding to peripheral HS (10). In essence, both studies pointed to a role of HS in regulating the bioavailability of OPG in tissue. However, these studies did not explain how HS might regulate the most important function of OPG, which is to inhibit RANKL activity. Another study has attempted to address this question directly and found that exogenously added heparin promoted osteoclast activity in an osteoblast-bone marrow cell co-culture system (11). The authors attribute the effect of heparin in this system to heparin-OPG interactions, which they believe inhibits OPG-RANKL interaction. The limitation of this study is that adding heparin into co-culture will undoubtedly alter the behavior of many HS-binding proteins, which makes clean interpretation difficult.
In this report, we show that HS is required for immobilization of OPG onto the surface of osteoblasts, and that HS forms a stable complex with OPG dimer and also induces dimerization of OPG monomer. We further provided evidence that HS binding induces a global conformational change of OPG, which makes it more compact and less flexible. Most importantly, using three different methods, we were able to show that HS, OPG, and RANKL form a stable ternary complex. We then mapped the exact heparin-binding site of OPG by mutagenesis and created OPG mutants that bind RANKL normally but lack the capacity to bind HS. By comparing the inhibitory effect of the HS-binding deficient OPG to wild-type OPG, we found for the first time that the main function of HS-OPG interactions in osteoclastogenesis is to lower the inhibitory threshold of OPG against RANKL at the surface of osteoblasts.
Results
OPG Binds Osteoblast Cell Surface in a HS-dependent Manner
Based on the high-affinity interaction between heparin and OPG, we predict that osteoblast cell surface HS proteoglycan (HSPG) represents a major binding site of OPG. To directly test this, we used a flow cytometry-based OPG binding assay to determine the binding between OPG and HS at the surface of MC3T3-E1 cell, a widely used murine osteoblast cell line. As shown in Fig. 2A, OPG binds avidly to the surface of MC3T3 cells with an apparent Kd of 1.2 ± 0.05 nm. The binding is almost entirely HS-dependent because removal of cell surface HS with heparin lyase III (HL-III) nearly abolished OPG binding (Fig. 2B). This result suggests that HS expressed by osteoblasts has the capacity to sequester OPG and that HS is the dominant binding partner for OPG at the cell surface.
FIGURE 2.

Binding of OPG to osteoblasts is HS dependent. A, binding of recombinant mouse OPG (mOPG, 20 ng to 1 μg/ml) to MC3T3 cells was determined by a FACS-based binding assay. The bound mOPG were detected by staining with a goat anti-mOPG antibody, followed by anti-goat IgG-Alexa 647. The shaded histogram is from cells stained with only with antibodies. B, binding of mOPG (100 ng/ml) to untreated MC3T3 cells or cells pretreated with heparin lyase III (HL-III). C, the inhibitory effects of heparin-derived octasaccharide (H8), decasaccharide (H10), and dodecasaccharide (H12) on mOPG binding to MC3T3 cells was determined by FACS assay. Oligosaccharides were used at 10 nm to 40 μm. Data are representative of at least three separate assays. D, comparison of the inhibitory effects of H12 and H14. Oligosaccharides were used at 30 nm (narrow line) and 300 nm (thick line).
As an alternative way to demonstrate that OPG binds MC3T3-E1 cells in a HS-dependent manner, we performed a competition assay using oligosaccharides derived from heparin. The result showed that all tested oligosaccharides, including octasaccharide (H8), decasaccharide (H10), and dodecasaccharide (H12) were able to achieve 100% inhibition of OPG binding to cell surface HS (Fig. 2C), which suggests that binding of OPG to the cell surface is purely HS-dependent. Interestingly, among these oligosaccharides, H12 apparently inhibits much more efficiently. We have determined that the H12 (IC50 = 32 nm) inhibits 20-fold better than the H10 (IC50 = 650 nm), whereas H10 only inhibits 5-fold better than the H8 (IC50 = 3200 nm). The substantially increased inhibitory potency of H12 indicates that H12 likely fully occupies the HS-binding site of OPG, whereas H10 only partially occupied the site, resulting in much lower inhibitory potency. To further support this notion, we directly compared the inhibitory capability of the tetradecasaccharide (H14) and H12 on OPG binding (Fig. 2D). Indeed, further increasing the length of the oligosaccharide only marginally increased the inhibitory efficiency, which suggests that H12 is truly the minimal length to occupy the HS-binding site.
HS-OPG Interaction Depends on 2-O-Sulfation
By performing a binding assay using pgsF-17 CHO cells, which lack uronyl 2-O-sulfotransferase activity, we have determined that the sulfate group at the 2-O position of iduronic acid plays a critical role in mediating HS-OPG interactions. Compared with CHO-K1 cells, binding of OPG to pgsF-17 cells was reduced by 93% (Fig. 3A). Because the overall sulfation levels of the HS produced by pgsF-17 cells and CHO-K1 cells are identical (12), this result suggests that the interaction between HS and OPG is highly specific and not simply driven by charge. In addition, we have also found that binding of OPG to pgsD-677 cells, which lack HS but make 3 to 4 times more chondroitin sulfate (13), was also reduced by 95%. This result suggests that chondroitin sulfate cannot substitute the role of HS in mediating OPG binding to cell surface.
FIGURE 3.
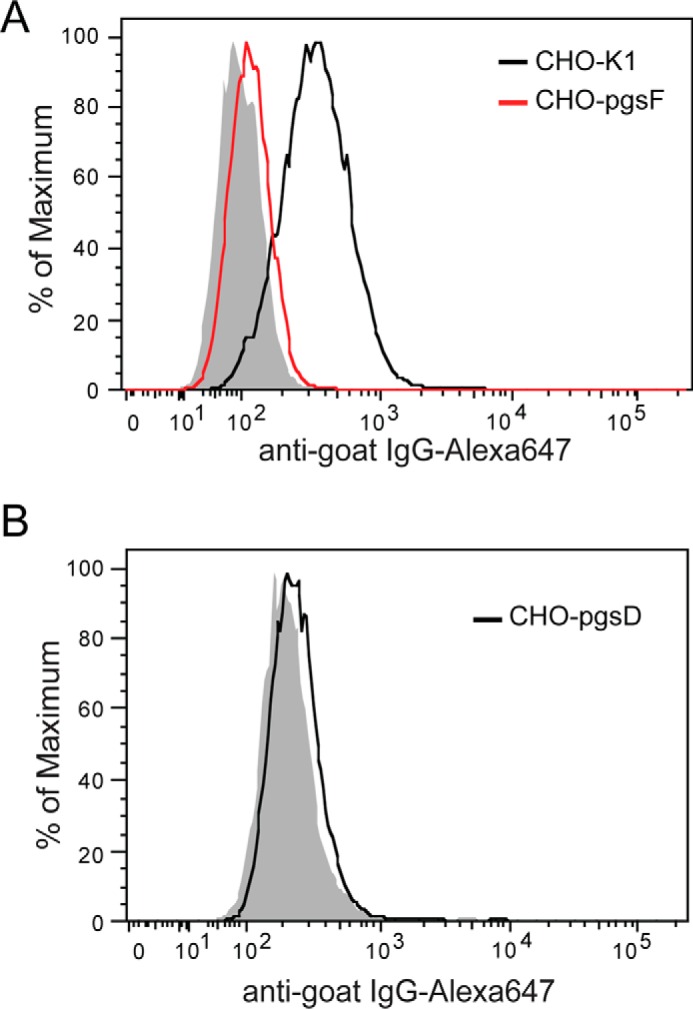
2-O-Sulfation plays a critical role in mediating HS-OPG interactions. A, binding of mOPG (100 ng/ml) to CHO-K1 and pgsF (2-O-sulfotransferase deficient CHO) cells are determined by FACS. B, binding of mOPG (100 ng/ml) to pgsD CHO cells. pgsD cells express no HS but abundant chondroitin sulfate is at the cell surface. Data are representative of at least three separate assays.
HS Induces OPG Dimerization
Because one of the major ways that HS modulates protein function is by inducing oligomerization (14), we tested how HS might change the oligomeric state of OPG. It was reported that the naturally occurring OPG exists as both monomer and dimer, and both species appear to be stable (3, 15). In our hands, the recombinant murine OPG (mOPG) produced in 293F cells contains ∼40% monomers and ∼60% dimers. Both species can be purified to homogeneity based on their differences in heparin-binding affinity and molecular size. When mOPG dimer was incubated with H12 in solution and analyzed by size exclusion chromatography (SEC), we consistently observed a 0.2-ml forward shift compared with the elution position of the free dimer (Fig. 4A). This slight shift in elution position suggests that H12 did not induce the dimer to form higher order oligomers, but it clearly formed a stable complex with the dimer. Next, we tested mOPG monomer-H12 interactions by SEC. Interestingly, we found that H12 promotes dimerization of the monomer. By adjusting the molar ratio between H12 and monomer, we have determined the binding stoichiometry is most likely 1:1 (Fig. 4B), a molar ratio that was required to fully convert monomer to dimer. This result suggests that HS is able to form a stable complex with both dimeric and monomeric OPG, and induces dimerization of the latter.
FIGURE 4.
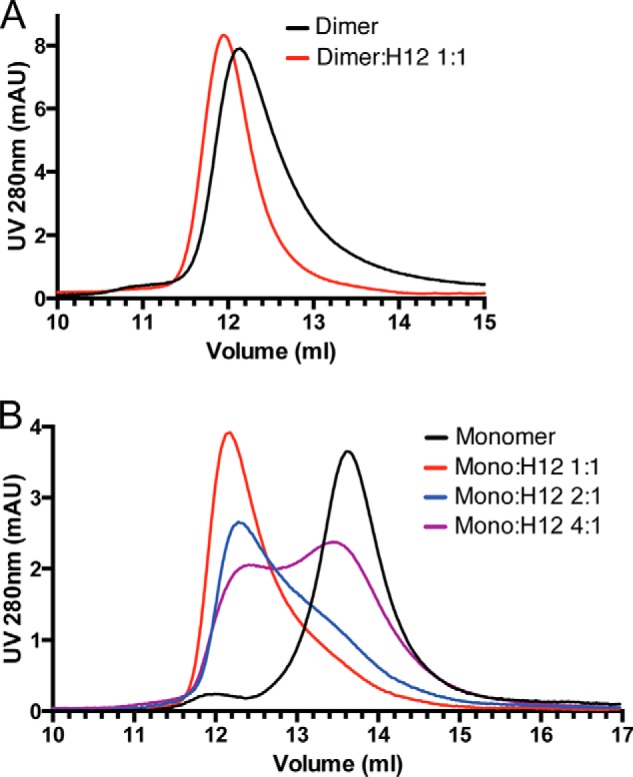
HS induces OPG dimerization. A, HS forms a stable complex with OPG dimer. 40 μg (0.8 nmol) of mOPG dimer was incubated with 3 μg (0.8 nmol) of heparin-derived dodecasaccharide (H12) at room temperature for 2 h and resolved on a SEC column (Enrich650, Bio-Rad). B, HS induces dimerization of the OPG monomer. 20 μg (0.4 nmol) of mOPG monomer was incubated with 0.35 (0.1 nmol), 0.7 (0.2 nmol), or 1.4 μg (0.4 nmol) of H12 at room temperature for 2 h and resolved on a SEC column.
Intermolecular Disulfide Bond Is Dispensable for OPG Dimerization
Although earlier studies have suggested that an intermolecular disulfide bond formed at Cys400 may be responsible for formation of the dimer (7), this view was challenged by a later study, which reported that the C400A mutant remains a dimer as determined by analytical ultracentrifugation (8). Therefore, it remains to be determined how exactly OPG dimerizes. To determine whether Cys400 is truly involved in dimerization, we have prepared both C400A and C400S mutants of OPG. Analysis of both mutants by heparin column showed that they clearly lacked the dimer peak and were purely monomeric (Fig. 5A). This result definitively demonstrated that Cys400 is required for forming the naturally occurring OPG dimer, most likely through a disulfide bridge as reported previously (7), which explains why it is highly stable. Interestingly, whereas the intermolecular disulfide bond is required for forming the naturally occurring dimer, it is not required for forming HS-induced dimer. As shown in Fig. 5B, the monomeric C400A is able to form a stable dimer when incubated with H12. This finding strongly suggests that dimerization mechanisms other than intermolecular disulfide bond must exist, which most likely involves noncovalent interactions between monomers.
FIGURE 5.
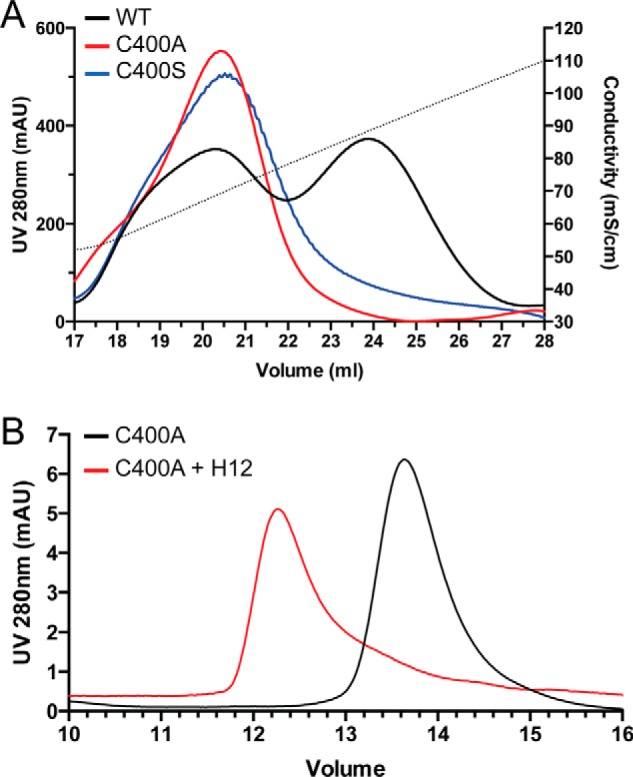
Mechanism of OPG dimerization. A, naturally occurring OPG dimer requires formation of intermolecular disulfide bond. Purification of WT mOPG or C400A and C400S mutants on heparin-Sepharose is shown. The dotted line represents the salt gradient (in conductivity mS/cm). The WT mOPG was eluted as two peaks (low-salt peak is monomer, high-salt peak is dimer), whereas both cysteine 400 mutants were eluted as a single low-salt peak. B, HS-induced OPG dimer does not require intermolecular disulfide bond. 30 μg of C400A monomer was incubated with 2 μg of H12 at room temperature for 2 h and resolved on a SEC column.
HS Induces Dramatic Conformational Change of OPG
One way that HS might regulate the function of OPG is to induce its conformational change. To test this hypothesis directly, we analyzed the solution structures of the dimer and dimer-H12 complex by small-angle X-ray scattering (SAXS). Comparing the scattering curves revealed that the global conformation of the dimer-H12 complex is substantially different from the free dimer, as suggested by the drastically different scattering pattern in almost all regions (Fig. 6A). From the raw scattering curve, a P(r) function (pair-distance distribution function) plots can be derived. P(r) plot is used to describe the paired-set of distances between all of the electrons within the macromolecule, the shape of which is directly determined by the conformation (16). Again, the P(r) plots show substantial differences between the two structures (Fig. 6B). From the P(r) plot, we have determined that the radius of gyration (Rg) of the dimer and dimer-H12 complexes are 61.6 and 55.2 Å, respectively. This 10% drop in Rg value strongly suggests that the complex adopt a much more compact structure than the free dimer. We further analyzed the scattering data with a Kratky plot, which is used to assess the intrinsic flexibility/unfoldedness of macromolecules (16). In general, a globular protein will display a bell-shaped curve, whereas a protein with highly flexible or intrinsically unfolded regions will display an elevated tail in the high q region. The Kratky plot of the free dimer is clearly not bell-shaped and has a highly elevated tail in the high q region, which suggests the existence of highly flexible regions (Fig. 6C). This is largely consistent with the elongated shape of OPG. In contrast, the Kratky plot of the H12-dimer complex is drastically different, displaying a bell-shaped peak and an only slightly elevated tail. This result suggests that H12 binding greatly reduced the flexibility of the dimer, and made part of the protein more globular protein-like. At last, SAXS allows us to answer an important question, which is whether the structures of the H12-dimer (H12-nature dimer) and H12-monomer (H12-induced dimer) complexes are different. As shown in Fig. 6D, it is clear that the two scattering curves completely overlap with each other, which confirms that the structures of the two complexes are indistinguishable. Collectively, our SAXS analysis strongly suggests that HS-OPG interaction substantially alters the structure of OPG, which makes it more compact and less flexible.
FIGURE 6.

HS binding induces conformational change of OPG. A, raw SAXS scattering curves of SEC purified mOPG dimer or dimer-H12 complex collected at 4 mg/ml. B, P(r) function plots of mOPG dimer or dimer-H12 complex. C, Kratky plots of mOPG dimer or dimer-H12 complex. D, raw SAXS scattering curves of SEC purified mOPG dimer-H12 and monomer-H12 complexes collected at 4 mg/ml.
HS-OPG-RANKL Forms a Stable Ternary Complex
To understand the biological function of the HS-OPG interaction in osteoclastogenesis, we sought to determine how HS-OPG interactions might affect OPG-RANKL interactions. Because a couple of previous studies suggested that the HS-OPG interaction might inhibit OPG-RANKL interaction (10, 11), we first examined whether OPG-RANKL interactions are compatible with HS-OPG interactions. A simple way to test this is to examine whether the preformed OPG-RANKL complex can still bind heparin-Sepharose. Much to our surprise, RANKL, which does not bind the heparin column under 150 mm NaCl (Fig. 7A), co-eluted with the OPG dimer by 1.05 m NaCl (Fig. 7B). Co-elution of RANKL with OPG by high salt can only be explained by formation of heparin-OPG-RANKL ternary complex on the Sepharose. Additionally, this result also suggests that OPG-RANKL interactions had no negative effect on the heparin-OPG interaction because the OPG-RANKL complex was eluted by the same salt concentration (1.05 m) as required to elute free OPG dimer (Fig. 7B).
FIGURE 7.

HS, OPG, and RANKL form a ternary complex. A, binding of purified RANKL (60 μg) to heparin-Sepharose. Dotted line represents the salt gradient. B, OPG dimer or purified OPG dimer-RANKL complex were bound to a heparin-Sepharose column and eluted with a salt gradient (dotted line). Fractions (16 to 19 ml, 0.5 ml/fraction) of the OPG-RANKL complex run were resolved by SDS-PAGE and visualized by silver staining. C, OPG monomer (20 μg), RANKL (20 μg), and H12 (2 μg) were incubated in different combinations as indicated for 4 h at 25 °C. The complexes were resolved on an SEC column. The elution positions of the Mr standards BSA, IgY, and ferritin are marked with black triangles (13.5, 12.05, and 11.05 ml, respectively). Two peak fractions of the ternary complex (11.8 ml peak) were visualized by silver stain. D, binding of RANKL or preformed RANKL-OPG complex (100 ng/ml) to MC3T3 cells determined by FACS. RANKL binding was detected by a phycoerythrin (PE)-conjugated anti-mouse RANKL mAb. E, binding of the RANKL-OPG complex to MC3T3 cells shown in double staining dot plot using anti-RANKL-PE and goat anti-mOPG followed by anti-goat IgG-Alexa 647.
As another way to validate the ternary complex can truly be formed, we preformed a binding experiment in solution by mixing H12, OPG monomer, and RANKL. When only the OPG monomer was incubated with RANKL, the OPG-RANKL complex can be formed, which eluted at 12.8 ml on the SEC column (Fig. 7C, red trace). On the other hand, when the OPG monomer was incubated with H12, the monomer was converted to dimer with an elution position of 12.1 ml (black trace). When the three molecules were mixed together, a new peak at position 11.8 ml appeared (blue trace). SDS-PAGE analysis showed clearly that this peak contains OPG and RANKL. Because this peak only appeared when H12 was present, it is reasonable to conclude that this peak represents a stable H12-OPG-RANKL complex. Based on the elution position of the ternary complex on SEC (Fig. 7C), which is between IgY (180 kDa) and ferritin (450 kDa), the molecular mass of the complex is likely in the range of 200 to 300 kDa. This suggests that the complex most likely consists of four molecules of H12 (molecular mass = 14 kDa), four molecules of OPG (as two dimers, ∼200 kDa with N-glycans), and one RANKL trimer (53 kDa). This estimation is consistent with a previous study using analytical centrifugation, where a complex has been determined to contain two OPG dimers and one RANKL trimer (8).
Although we have shown that the ternary complex of heparin (or H12), OPG, and RANKL can be formed, it is important to demonstrate that OPG and RANKL can also form a ternary complex with cell surface HS on osteoblasts. To this end, we performed a binding experiment with either free RANKL or preformed OPG-RANKL complex. As expected, whereas free RANKL displayed no binding, strong binding of RANKL was observed when the OPG-RANKL complex was incubated with MC3T3 cells (Fig. 7D). Because binding of OPG to MC3T3 cells is fully HS-dependent, this result can only be explained by formation of the HS-OPG-RANKL ternary complex at the cell surface. Furthermore, the double staining dot plot (Fig. 7E) shows that cells high in bound RANKL are also high in bound OPG, further suggesting that RANKL is attached to the cell surface through OPG. Collectively, we have provided three lines of evidence that HS, OPG, and RANKL readily form a stable ternary complex.
HS-binding Site Is Located in the D2 and Tail Domains of OPG
To explicitly determine the HS-binding site of OPG, we have performed site-directed mutagenesis of all 30 conserved lysine and arginine residues in D1, D2, and Tail domains and tested the binding of these OPG mutants to heparin column. Altogether, we have identified eight residues (Arg283, Lys350, Lys353, Lys359, Arg366, Lys377, Arg370, and Arg379) that participate in HS binding (Fig. 8A). These residues are located in the D2 and Tail domains, and the majority are concentrated around the junction of D2 and Tail domains. As expected, mutating two to four of these residues together display an additive effect, resulting in a much more substantial reduction in binding to heparin column (Fig. 8A).
FIGURE 8.

The HS-binding site consists of residues from D2 and Tail domains. A, binding of purified WT or mutants (all dimers) to heparin was analyzed by a 1-ml HiTrap heparin-Sepharose column at pH 7.1. The salt concentrations required for elution are plotted. Mutants that show >100 mm reduction in elution salt concentration are marked in bold. B, binding of OPG mutants (R370A/R379A or K359A/R370A/R379A, all dimers, 33 ng/ml) and wild-type mOPG (dimer and monomer, 33 ng/ml) to cell surface HS was determined by FACS.
Next, we used a cell surface binding assay to determine the effect of mutating HS-binding residues on HS-OPG interactions. Interestingly, although the triple mutant (K359A/R370A/R379A) was still able to bind the heparin column, it essentially displayed no binding to cell surface HS (Fig. 8B). Similarly, the double mutant (R370A/R379A) also showed a 98% reduction in HS binding. This result suggests that the identified residues indeed play essential roles in the HS-OPG interaction. The result also demonstrated that by mutating key residues that mediates HS-OPG interactions, we are able to create OPG variants that display no binding to cell surface HS, which will be key tools to interrogate the biological functions of HS-OPG interactions.
HS-OPG Interaction Lowers the Inhibitory Threshold of OPG toward RANKL
To start to understand the biological significance of HS-OPG interactions in osteoclastogenesis, we performed a well established osteoblast/bone marrow macrophage (BMM) co-culture osteoclastogenesis assay (17). In this model, primary calvarial osteoblasts are stimulated with vitamin D3 (10 nm) and dexamethasone (100 nm) to induce expression of macrophage colony-stimulating factor (M-CSF) and RANKL, which drives differentiation of co-cultured BMMs into osteoclasts. The extent of osteoclast differentiation was assessed by staining osteoclasts for tartrate-resistant acid phosphatase (TRAP), and by enzymatic assay of TRAP activity. Using this system, we ask the question how HS-OPG interactions at the cell surface affect the inhibitory threshold of OPG. To diminish HS-OPG interactions, we used the OPG mutant K359A/R370A/R379A in this assay, which showed almost no binding to osteoblast HS (Fig. 8B). It is clear from the inhibition curve that the wild-type (WT) OPG is more potent than the mutant OPG at all concentrations tested except the highest concentration (Fig. 9A). Strikingly, whereas WT OPG inhibited 71 ± 3% of osteoclastogenesis at 1 ng/ml, the triple mutant had to be used at 10 ng/ml to reach the similar extent of inhibition (66 ± 3%), which represents a 10-fold reduction in potency (Fig. 9A, lower dotted line). Similarly, the inhibitory effect of WT OPG at 3 ng/ml is similar to that of the triple mutant at 30 ng/ml (85 ± 4 versus 81 ± 1%, Fig. 9A, upper dotted line). In addition, TRAP staining of the osteoclasts verified that the cells treated with 1 ng/ml of WT OPG and 10 ng/ml of triple mutant gave rise to a similar number of osteoclasts with comparable sizes (Fig. 9, B–D). These results strongly suggest that HS-OPG interactions promote more efficient inhibition of RANKL, which significantly lowers the inhibitory threshold of OPG. As an important control to support this conclusion, we also examined how the triple mutant inhibits osteoclastogenesis of BMMs in monoculture, where no osteoblasts were present and the osteoclastogenesis was induced by exogenously added soluble RANKL (50 ng/ml). As expected, in this system, the triple mutant showed identical inhibitory potency to the WT OPG, both with an IC50 of 100 ng/ml. This result suggests that the HS binding-deficient triple mutants had no inherent defect in inhibiting soluble RANKL. Thus, the superior potency of the WT OPG dimer in the co-culture system can only be explained by its more avid interactions with osteoblast HS, which facilitates immobilization of OPG at the cell surface and consequently promotes OPG-RANKL interactions.
FIGURE 9.

HS-OPG interactions lower the inhibitory threshold of OPG against RANKL. A, inhibition of osteoclastogenesis in osteoblasts/bone marrow macrophage co-culture by WT OPG dimer or triple mutant K359A/R370A/R379A at 1, 3, 10, 30, and 100 ng/ml. The extent of osteoclastogenesis is assessed by an enzymatic assay of TRAP activity in whole cell lysate. n = 3. Error bars represent S.D. * represents p < 0.01 by Student's t test. Data are representative of at least three separate assays. B–E, representative images of differently treated co-culture. Mature osteoclasts are visualized by TRAP staining. The pink multinucleated cells are osteoclasts and the round clear cells are undifferentiated bone marrow macrophages. F, inhibition of osteoclastogenesis in bone marrow macrophage monoculture by WT OPG dimer or triple mutant OPG at 50, 100, 200, and 500 ng/ml. Osteoclastogenesis is induced by recombinant soluble RANKL (50 ng/ml) and M-CSF (20 ng/ml).
Discussion
OPG binds RANKL through its N-terminal cysteine-rich domains (CRD, Fig. 1) and it is clear that the CRDs are sufficient to inhibit RANKL (18, 19). In contrast, the function of the rest of the protein, including two death domains and the Tail domain, is less clear. Through a domain deletion experiment, one study suggests that the Tail domain is responsible for binding to HS (7). However, due to the limitation of the domain deletion, it is unclear whether this domain is solely responsible for HS binding, nor is it known where is the exact HS-binding site. By mutating all 30 conserved basic residues in D1, D2, and Tail domains, we have definitively determined the HS-binding site of OPG. Our result suggests that although four basic residues located in the Tail domain (Arg366, Lys377, Arg370, and Arg379) do make major contributions to HS binding, residues located in D2 domain (Arg283, Lys350, Lys353, and Lys359) are also part of the HS-binding site. Knowing the exact HS-binding residues is highly important because it would allow us to manipulate HS-OPG interactions in a highly specific manner, which can never be achieved by the domain deletion method.
An interesting feature of OPG is that it naturally exists in both monomer and dimer (3, 15). However, the exact mechanism of OPG dimerization remains controversial. The center of the controversy is whether Cys400 is involved in forming intermolecular disulfide bonds. Using heparin affinity and size-exclusion chromatography, we have shown here that Cys400 is essential for the formation of naturally occurring OPG dimer because both C400A and C400S mutants only existed in monomeric form (Fig. 5). It is unclear to us why Schneeweis et al. (8) observed that C400A existed as a dimer. The only plausible explanation is that in contrast to our construct, which is a native OPG without any tag and expressed in mammalian cells, their OPG contains a C-terminal His6 tag and was expressed in insect cells. Our study also discovered a previously unknown mechanism of OPG dimerization, which is through binding to HS (Fig. 4B). We further show that the intermolecular disulfide bond is no longer required for OPG dimerization in the presence of HS, because the C400A mutant (monomer) can also be converted into a stable dimer in the presence of HS (Fig. 5B). This result suggests that a dimerization interface of non-covalent nature must exist, which might be exposed/stabilized as a consequence of HS binding.
The dramatic conformational change of OPG that H12 was able to induce was certainly unexpected. SAXS analysis showed that H12 binding induces the OPG dimer to adopt a substantially more compact structure (Rg reduces from 61.6 to 55.2 Å, Fig. 6B). Consequently, the structure of the OPG-H12 complex becomes much more rigid than the free OPG dimer, which is highly flexible (Fig. 6C). It is highly likely that the increased rigidity of OPG would alter OPG-RANKL interactions. Of note, although H12 binding was able to greatly reduce the flexibility of OPG dimer, the complex still contains some flexible regions, which is indicated by the somewhat elevated tail in the Kratky plot (Fig. 6C). Presumably, the residual flexibility of the OPG-H12 complex is stemmed from the N-terminal CRDs (Fig. 1), which likely remains flexible without bound RANKL. In contrast, the C-terminal D1, D2, and Tail domain likely forms a rigid unit upon H12 binding and dimerization. To the best of our knowledge, only two other proteins, antithrombin and Vaccinia virus complement control protein, have been shown to undergo global conformational change upon HS binding (20, 21). Our discovery also illustrated that SAXS can be a highly valuable tool to probe conformational changes of HS-binding proteins upon HS binding, especially for those multidomain proteins that are difficult to crystalize.
One key finding of our study is that HS, OPG, and RANKL forms a stable ternary complex. This is contrary to a couple of previous studies suggesting that the HS-OPG interaction is incompatible with OPG-RANKL interaction. In one study, exogenously added heparin was found to promote osteoclast activity in an osteoblast-bone marrow cell co-culture system (11). The authors attribute the effect of heparin in this system to heparin-OPG interactions, which they believe inhibit OPG-RANKL interaction. In light of our observation of the ternary complex, and the critical role of osteoblast HS in immobilizing OPG onto the cell surface (Fig. 10), we believe that the increased osteoclast activity they observed could be partially due to the competition between soluble heparin and cell surface HS. In principle, adding heparin to the culture would lower the concentration of bound OPG at the surface of osteoblasts, which would lead to decreased inhibition of membrane-bound RANKL, hence increased osteoclast activation. In addition, because exogenous heparin would bind and affect the functions of any HS-binding proteins that might play a role in the system, the observed effect of heparin on osteoclast activity could well involve proteins other than OPG.
FIGURE 10.

The proposed model by which HS regulates OPG-RANKL interactions. Secreted WT OPG is immobilized on cell surface HSPG through binding to the HS chain, which brings OPG to close proximity to the membrane-bound RANKL and increases the probability of successful engagement between OPG and RANKL. Binding to HS also induces a significant conformation change of OPG, which might further facilitate OPG-RANKL interactions. In contrast, HS binding-deficient mutant OPG can only access membrane-bound RANKL through diffusion, which leads to less efficient inhibition of RANKL.
Another study, based on surface plasmon resonance experiment, concluded that heparin inhibits OPG-RANKL interaction (10). It remains unclear to us why that study reached a conclusion that is different from ours. The main difference between our methods and surface plasmon resonance is that our methods do not involve protein immobilization, a procedure that could potentially introduce artifacts into the assay. To reach the conclusion that HS, OPG, and RANKL forms a ternary complex, we have employed three different analytical methods. We have shown that OPG and RANKL form a complex with heparin immobilized on the Sepharose beads (Fig. 7B), that OPG, RANKL, and heparin-derived oligosaccharide form a stable ternary complex in solution (Fig. 7C), and that OPG and RANKL form a ternary complex with osteoblast cell surface HS (Fig. 7D). Combined, we have gathered compelling evidence that the ternary complex is authentic.
Using an HS-binding deficient mutant of OPG (K359A/R370A/R379A), our study has revealed that HS-OPG interactions play a key role in lowering the inhibitory threshold of OPG toward RANKL. In the osteoblast-BMM co-culture system, RANKL was expressed as a transmembrane juxtacrine factor by stimulated osteoblasts, which drives osteoclastogenesis by interacting with RANK expressed on the surface of BMMs. As a secreted decoy receptor, OPG will need to find a way to migrate back to the cell surface to inhibit RANKL. This can certainly be achieved through free diffusion, but binding to HSPG undoubtedly will be a much more efficient way to get a foothold at the cell surface. Once immobilized by HSPG, OPG is positioned in close proximity to membrane-bound RANKL, which would allow highly efficient inhibition (Fig. 10). The fact that HS, OPG, and RANKL can readily form a stable ternary complex at the cell surface (Fig. 7D) provides strong support for this model. Consistent with this model, the differences in inhibitory potency between WT and triple mutant OPG can only be observed at low concentrations, where the ability to bind HSPG is likely crucial for gaining access to RANKL (Fig. 9A). Of note, the low OPG concentration is likely a norm under physiological conditions, given that the serum OPG concentration in a healthy human was reported to be between 0.2 and 1 ng/ml (22, 23). In contrast, at high OPG concentrations, the rate of free diffusion is likely adequately efficient to bring OPG to cell surface, thus masking the HS-binding defect of the triple mutant (Fig. 9A). It is important to note that immobilization of OPG at the cell surface by HS may not be the only mechanisms that contributes to the exceptional inhibitory potency of OPG. For instance, formation of the ternary complex at the cell surface might facilitate internalization of RANKL along with HSPG and OPG. In principle, such a mechanism would permanently clear RANKL from the cell surface, which would prevent reactivation of RANKL when bound OPG dissociates. Alternatively, the conformational change of OPG that HS elicits could favor more stable binding between OPG and RANKL, which would also enable more efficient inhibition. These possibilities are being actively investigated in our laboratory.
Interestingly, the difference in inhibitory potency between the HS binding-deficient OPG and WT OPG was completely masked in the BMMs monoculture system (Fig. 9C). The key difference between the co-culture and monoculture system is how RANKL is presented. In the co-culture system, RANKL is expressed endogenously and membrane-attached, whereas in the monoculture system, RANKL is supplied exogenously in a truncated soluble form. Thus, in the monoculture system, OPG inhibition of RANKL occurs in the culture medium instead of on the osteoblast surface, which obviates the need for immobilization by HS.
In conclusion, we have identified HS as a multifunctional regulator of OPG-RANKL interactions. HS induces OPG dimerization, alters OPG conformation, and forms a ternary complex with OPG and RANKL. By immobilizing OPG to the osteoblast cell surface, HS enables OPG to inhibit RANKL at a very low concentration. Our data strongly suggest and HS is an integral regulator of the RANKL/RANK/OPG pathway in osteoclastogenesis.
Experimental Procedures
Expression and Purification of Full-length Mouse OPG
Recombinant full-length mouse OPG was produced in 293-freestyle cells (ThermoFisher Scientific). The complete open reading frame of full-length mouse OPG (GE Dharmacon) was cloned into pUNO1 (Invivogen) using NcoI and NheI sites. Transfection was performed using FectoPRO transfection reagent (Polyplus transfection). Purification of mOPG from culture supernatant was carried out using HiTrap heparin-Sepharose column (GE Healthcare) at pH 7.1 (HEPES buffer), followed by gel permeation chromatography on a Superdex 200 column in 20 mm Tris, 150 mm NaCl, pH 7.4 (GE Healthcare). After purification, mOPG was >99% pure as judged by silver staining, and mOPG monomers were fully separated from dimers. Throughout the study, we used the K241A/R242A/R243Q (AAQ) mutant interchangeably with WT mOPG because of its higher expression level. The AAQ mutant performed equivalently as WT mOPG in HS-binding, RANKL-binding, and osteoclastogenesis assays.
Expression and Purification of the Extracellular Domain of Mouse RANKL
Recombinant mouse RANKL was generated in Escherichia coli. The coding sequence of mouse RANKL Lys158-Asp316 was amplified from its cDNA (GE Dharmacon) and was cloned into pET21b (Novagen) using NdeI and XhoI sites. Expression was carried out at 18 °C in Origami-B cells (Novagen) carrying the pGro7 (Takara) plasmid expressing chaperonin proteins GroEL and GroES following an established protocol (24). Purification was carried out using HiTrap SP cation exchange column at pH 6.5 (MES buffer), followed by gel permeation chromatography with HiLoad 16/60 Superdex 200 (GE Healthcare). The tagless soluble RANKL was expressed as a stable trimer and was >99% pure as judged by silver staining.
Flow Cytometry-based Binding Assay
MC3T3-E1 cells were lifted from culture dish using Accutase (Biolegend) and incubated with WT or mutant mOPG (20 ng to 1 μg/ml) in 100 μl of PBS, 0.1% BSA for 1 h at 4 °C. Bound mOPG was stained with goat anti-mouse OPG (400 ng/ml, AF459, R&D Systems) for 1 h at 4 °C, followed by anti-goat IgG-Alexa 647 (1:1000, ThermoFisher Scientific) for 30 min and analyzed by flow cytometry. The apparent binding affinity between OPG and cell surface HS was calculated by using the geometric means of Alexa 647 fluorescence intensity as the binding signal. The non-linear curve fitting was performed in Prism (GraphPad, San Diego, CA) based on one-site specific binding. In some experiments, cells were pretreated with recombinant heparin lyases III (5 milliunits/ml, produced as a recombinant protein in E. coli using a plasmid originally obtained from Dr. Jian Liu, University of North Carolina, Chapel Hill) for 15 min at room temperature prior to binding experiments. Binding of mOPG to CHO-K1 and mutant CHO cells (pgsD and pgsF) were performed in the same manner. For competition experiments, the heparin-derived octasaccharide, decasaccharide, and dodecasaccharide (Iduron, UK) were added (from 100 ng to 10 μg/ml) along with mOPG (100 ng/ml) to MC3T3 cells. The IC50 values were calculated in Prism by fitting the curves to one-site competitive binding model. To obtain the percentage of inhibition, we first divided the raw binding signal (geometric mean of Alexa 647 intensity) of OPG binding in the presence of heparin fragments by the control binding signal (OPG binding without added heparin), and the percentage of inhibition was calculated by subtracting this fraction from 1.
For RANKL binding to MC3T3 cells, soluble RANKL (100 ng/ml) or preformed RANKL-OPG complex (1:1000 dilution from a mixture of 100 μg/ml of RANKL and 100 μg/ml of OPG) were incubated with MC3T3 cells for 1 h at 4 °C. Bound RANKL was stained with an anti-mouse RANKL conjugated with phycoerythrin (500 ng/ml, clone IK22/5, ThermoFisher Scientific).
Analytical Size-exclusion Chromatography
For analyses of mOPG and heparin-derived dodecasaccharide (H12) complex, purified OPG dimer, OPG monomer, or mutant OPG (20–40 μg) were incubated with H12 (0.35–3 μg) in 20 mm Tris, 150 mm NaCl, pH 7.4, at room temperature for 2 h. The OPG-RANKL complex was formed the same way by mixing 20 μg of mOPG and 20 μg of soluble RANKL. For the ternary complex, 20 μg of mOPG monomer was incubated with 2 μg of H12 and 20 μg of RANKL at room temperature for 2 h. All complexes were resolved on an Enrich SEC 650 column (10/300 mm, Bio-Rad) using 20 mm Tris, 150 mm NaCl, pH 7.4, at 4 °C.
Site-directed Mutagenesis
Mouse OPG mutants were prepared using published method (25). Mutations were confirmed by sequencing, and recombinant protein was expressed and purified as described for WT mOPG.
Heparin-Sepharose Chromatography
To characterize the binding of mOPG mutants to heparin, 30 μg of purified dimeric mOPG mutants were applied to a 1-ml HiTrap heparin-Sepharose column (GE Healthcare) and eluted with a salt gradient from 200 mm to 1.4 m NaCl, pH 7.1 (HEPES buffer). The conductivity measurements at the peak of the elution were converted to the concentration of NaCl based on a standard curve. For analysis of OPG-RANKL complex binding to heparin column, 50 μg of Superdex 200-purified mOPG-RANKL complex was applied.
Small-angle X-ray Scattering
Scattering data were collected at the beamline 12.3.1 at the Lawrence Berkeley National Laboratory using a mail-in service. Monodispersed mOPG dimer, mOPG dimer-H12, and mOPG monomer-H12 complexes were purified by SEC and concentrated to 1, 2, and 4 mg/ml in buffer containing 25 mm HEPES, 150 mm NaCl, and 1.5% glycerol, pH 7.1. The samples were shipped on blue ice and the scattering data were collected within 24 h using 30 0.5-s exposures. The raw scattering data that are free from radiation damage (between 15 and 30 exposures) were averaged and used for analysis. SAXS data analysis, including Guinier plot, P(r) function plot, and Kratky plot were performed using Scatter program (version 2.3h, developed by Robert Rambo at the Diamond Light Source, UK). The whole set of experiments were performed twice using two different mOPG preparations with similar results.
Osteoclastogenesis Assay
Co-culture Osteoclastogenesis Assay
Primary osteoblasts were isolated from calvaria of 5–8-day-old WT mice following an established protocol (26). Osteoblasts (5 × 103 cells/well) were seeded in a 96-well plate the day before starting the co-culture. Freshly isolated bone marrow cells (from one WT mouse) were suspended in 10 ml of α-MEM containing 10% FBS and 1× penicillin/streptomycin, 10−7 m dexamethasone, and 10−8 m 1α- and 25-dihydroxyvitamin D3. 100 μl of bone marrow cells were added into each well. In selected wells, WT or triple mutant OPG (K359A/R370A/R379A) were added to 1 to 100 ng/ml. The unattached cells were removed after 24 h in culture and the medium was replaced. The medium was replaced every 2 days thereafter until the appearance of giant osteoclasts, which usually takes 6–7 days. To visualize osteoclasts, the cells were fixed and stained for TRAP activity using the an Leukocyte Acid Phosphatase kit (Sigma). For quantitative measurement of TRAP activity, cells were lysed with 50 μl of lysis buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 1% Nonidet P-40) for 30 min at 4 °C. 10 μl of lysate were then mixed with 50 μl of TRAP assay buffer containing 0.5 m sodium acetate, 10 mm tartrate, and 10 mm p-nitrophenyl phosphate substrate and incubated at 37 °C for 15 min. The reaction was stopped by adding 50 μl of 0.5 n NaOH, and the absorbance at 405 nm was measured by a plate reader.
Monoculture Osteoclastogenesis Assay
Non-adherent bone marrow cells were obtained from mouse long bones as described and cultured in 96-well plates (17). Osteoclastogenesis was induced by adding 20 ng/ml of M-CSF (Peprotech) and 50 ng/ml of soluble RANKL (prepared in house with endotoxin level < 0.1 EU/μg of protein) into the medium. In selected wells, WT or triple mutant OPG were added to 50 to 500 ng/ml. The medium was replaced every 2 days thereafter until appearance of giant osteoclasts, which usually takes 5–6 days. Cells were then lysed and assayed for TRAP activity as described above.
Author Contributions
M. L. and D. X. designed the research, performed the work, analyzed and interpreted data, and wrote the manuscript. S. Y. contributed reagents and revised the paper.
Acknowledgments
We thank Dr. Xue Yuan for help with osteoblasts isolation and setting up co-culture osteoclastogenesis assay. We also thank Dr. Lars Pedersen for reviewing the manuscript. SAXS data collection was conducted at the Advanced Light Source (ALS), a national user facility operated by the Lawrence Berkeley National Laboratory on behalf of the Department of Energy, Office of Basic Energy Sciences, through the Integrated Diffraction Analysis Technologies (IDAT) program, supported by DOE Office of Biological and Environmental Research.
The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- RANKL
- receptor activator of nuclear factor κB ligand
- OPG
- osteoprotegerin
- RANK
- receptor activator of nuclear factor κB
- HS
- heparan sulfate
- HSPG
- heparan sulfate proteoglycan
- SAXS
- small-angle X-ray scattering
- SEC
- size exclusion chromatography
- BMM
- bone marrow macrophage
- TRAP
- tartrate-resistant acid phosphatase
- M-CSF
- macrophage colony stimulating factor
- CRD
- cysteine-rich domain.
References
- 1. Lacey D. L., Timms E., Tan H. L., Kelley M. J., Dunstan C. R., Burgess T., Elliott R., Colombero A., Elliott G., Scully S., Hsu H., Sullivan J., Hawkins N., Davy E., Capparelli C., et al. (1998) Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 93, 165–176 [DOI] [PubMed] [Google Scholar]
- 2. Yasuda H., Shima N., Nakagawa N., Yamaguchi K., Kinosaki M., Mochizuki S., Tomoyasu A., Yano K., Goto M., Murakami A., Tsuda E., Morinaga T., Higashio K., Udagawa N., Takahashi N., and Suda T. (1998) Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. U.S.A. 95, 3597–3602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Simonet W. S., Lacey D. L., Dunstan C. R., Kelley M., Chang M. S., Lüthy R., Nguyen H. Q., Wooden S., Bennett L., Boone T., Shimamoto G., DeRose M., Elliott R., Colombero A., Tan H. L., et al. (1997) Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 89, 309–319 [DOI] [PubMed] [Google Scholar]
- 4. Udagawa N., Takahashi N., Yasuda H., Mizuno A., Itoh K., Ueno Y., Shinki T., Gillespie M. T., Martin T. J., Higashio K., and Suda T. (2000) Osteoprotegerin produced by osteoblasts is an important regulator in osteoclast development and function. Endocrinology 141, 3478–3484 [DOI] [PubMed] [Google Scholar]
- 5. Kearns A. E., Khosla S., and Kostenuik P. J. (2008) Receptor activator of nuclear factor κB ligand and osteoprotegerin regulation of bone remodeling in health and disease. Endocr. Rev. 29, 155–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Théoleyre S., Kwan Tat S., Vusio P., Blanchard F., Gallagher J., Ricard-Blum S., Fortun Y., Padrines M., Rédini F., and Heymann D. (2006) Characterization of osteoprotegerin binding to glycosaminoglycans by surface plasmon resonance: role in the interactions with receptor activator of nuclear factor κB ligand (RANKL) and RANK. Biochem. Biophys. Res. Commun. 347, 460–467 [DOI] [PubMed] [Google Scholar]
- 7. Yamaguchi K., Kinosaki M., Goto M., Kobayashi F., Tsuda E., Morinaga T., and Higashio K. (1998) Characterization of structural domains of human osteoclastogenesis inhibitory factor. J. Biol. Chem. 273, 5117–5123 [DOI] [PubMed] [Google Scholar]
- 8. Schneeweis L. A., Willard D., and Milla M. E. (2005) Functional dissection of osteoprotegerin and its interaction with receptor activator of NF-κB ligand. J. Biol. Chem. 280, 41155–41164 [DOI] [PubMed] [Google Scholar]
- 9. Standal T., Seidel C., Hjertner Ø., Plesner T., Sanderson R. D., Waage A., Borset M., and Sundan A. (2002) Osteoprotegerin is bound, internalized, and degraded by multiple myeloma cells. Blood 100, 3002–3007 [DOI] [PubMed] [Google Scholar]
- 10. Lamoureux F., Picarda G., Garrigue-Antar L., Baud'huin M., Trichet V., Vidal A., Miot-Noirault E., Pitard B., Heymann D., and Rédini F. (2009) Glycosaminoglycans as potential regulators of osteoprotegerin therapeutic activity in osteosarcoma. Cancer Res. 69, 526–536 [DOI] [PubMed] [Google Scholar]
- 11. Irie A., Takami M., Kubo H., Sekino-Suzuki N., Kasahara K., and Sanai Y. (2007) Heparin enhances osteoclastic bone resorption by inhibiting osteoprotegerin activity. Bone 41, 165–174 [DOI] [PubMed] [Google Scholar]
- 12. Bai X., and Esko J. D. (1996) An animal cell mutant defective in heparan sulfate hexuronic acid 2-O-sulfation. J. Biol. Chem. 271, 17711–17717 [DOI] [PubMed] [Google Scholar]
- 13. Lidholt K., Weinke J. L., Kiser C. S., Lugemwa F. N., Bame K. J., Cheifetz S., Massagué J., Lindahl U., and Esko J. D. (1992) A single mutation affects both N-acetylglucosaminyltransferase and glucuronosyltransferase activities in a Chinese hamster ovary cell mutant defective in heparan sulfate biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 89, 2267–2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu D., and Esko J. D. (2014) Demystifying heparan sulfate-protein interactions. Annu. Rev. Biochem. 83, 129–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tsuda E., Goto M., Mochizuki S., Yano K., Kobayashi F., Morinaga T., and Higashio K. (1997) Isolation of a novel cytokine from human fibroblasts that specifically inhibits osteoclastogenesis. Biochem. Biophys. Res. Commun. 234, 137–142 [DOI] [PubMed] [Google Scholar]
- 16. Putnam C. D., Hammel M., Hura G. L., and Tainer J. A. (2007) X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys. 40, 191–285 [DOI] [PubMed] [Google Scholar]
- 17. Yuan X., Cao J., Liu T., Li Y. P., Scannapieco F., He X., Oursler M. J., Zhang X., Vacher J., Li C., Olson D., and Yang S. (2015) Regulators of G protein signaling 12 promotes osteoclastogenesis in bone remodeling and pathological bone loss. Cell Death Differ. 22, 2046–2057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Luan X., Lu Q., Jiang Y., Zhang S., Wang Q., Yuan H., Zhao W., Wang J., and Wang X. (2012) Crystal structure of human RANKL complexed with its decoy receptor osteoprotegerin. J. Immunol. 189, 245–252 [DOI] [PubMed] [Google Scholar]
- 19. Nelson C. A., Warren J. T., Wang M. W., Teitelbaum S. L., and Fremont D. H. (2012) RANKL employs distinct binding modes to engage RANK and the osteoprotegerin decoy receptor. Structure 20, 1971–1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ganesh V. K., Smith S. A., Kotwal G. J., and Murthy K. H. (2004) Structure of vaccinia complement protein in complex with heparin and potential implications for complement regulation. Proc. Natl. Acad. Sci. U.S.A. 101, 8924–8929 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21. Jin L., Abrahams J. P., Skinner R., Petitou M., Pike R. N., and Carrell R. W. (1997) The anticoagulant activation of antithrombin by heparin. Proc. Natl. Acad. Sci. U.S.A. 94, 14683–14688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jono S., Ikari Y., Shioi A., Mori K., Miki T., Hara K., and Nishizawa Y. (2002) Serum osteoprotegerin levels are associated with the presence and severity of coronary artery disease. Circulation 106, 1192–1194 [DOI] [PubMed] [Google Scholar]
- 23. Browner W. S., Lui L. Y., and Cummings S. R. (2001) Associations of serum osteoprotegerin levels with diabetes, stroke, bone density, fractures, and mortality in elderly women. J. Clin. Endocrinol. Metab. 86, 631–637 [DOI] [PubMed] [Google Scholar]
- 24. Xu D., Young J. H., Krahn J. M., Song D., Corbett K. D., Chazin W. J., Pedersen L. C., and Esko J. D. (2013) Stable RAGE-heparan sulfate complexes are essential for signal transduction. ACS Chem. Biol. 8, 1611–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zheng L., Baumann U., and Reymond J. L. (2004) An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 32, e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bakker A. D., and Klein-Nulend J. (2012) Osteoblast isolation from murine calvaria and long bones. Methods Mol. Biol. 816, 19–29 [DOI] [PubMed] [Google Scholar]