

ABSTRACT
Herpes simplex virus 1 (HSV-1) infects humans through stratified epithelia that are composed primarily of keratinocytes. The route of HSV-1 entry into keratinocytes has been the subject of limited investigation, but it is proposed to involve pH-dependent endocytosis, requiring the gD-binding receptor nectin-1. Here, we have utilized the nTERT human keratinocyte cell line as a new model for dissecting the mechanism of HSV-1 entry into the host. Although immortalized, these cells nonetheless retain normal growth and differentiation properties of primary cells. Using short interfering RNA (siRNA) depletion studies, we confirm that, despite nTERT cells expressing high levels of the alternative gD receptor HVEM, HSV-1 requires nectin-1, not HVEM, to enter these cells. Strikingly, virus entry into nTERT cells occurred with unusual rapidity, such that maximum penetration was achieved within 5 min. Moreover, HSV-1 was able to enter keratinocytes but not other cell types at temperatures as low as 7°C, conditions where endocytosis was shown to be completely inhibited. Transmission electron microscopy of early entry events at both 37°C and 7°C identified numerous examples of naked virus capsids located immediately beneath the plasma membrane, with no evidence of virions in cytoplasmic vesicles. Taken together, these results imply that HSV-1 uses the nectin-1 receptor to enter human keratinocyte cells via a previously uncharacterized rapid plasma membrane fusion pathway that functions at low temperature. These studies have important implications for current understanding of the relationship between HSV-1 and its relevant in vivo target cell.
IMPORTANCE The gold standard of antiviral treatment for any human virus infection is the prevention of virus entry into the host cell. In the case of HSV-1, primary infection in the human begins in the epidermis of the skin or the oral mucosa, where the virus infects keratinocytes, and it is therefore important to understand the molecular events involved in HSV-1 entry into this cell type. Nonetheless, few studies have looked specifically at entry into these relevant human cells. Our results reveal a new route for virus entry that is specific to keratinocytes, involves rapid entry, and functions at low temperatures. This may reflect the environmental conditions encountered by HSV-1 when entering its host through the skin and emphasizes the importance of studying virus-host interactions in physiologically relevant cells.
INTRODUCTION
Herpes simplex virus type 1 (HSV-1) gains access to its human host through the epidermis at the oral mucosa, skin, or the cornea, where the majority of the cells are keratinocytes. Hence, although HSV-1 replication has been studied in many cell types in vitro, the role of the human keratinocyte in the HSV-1 life cycle makes it the most physiologically relevant cell type in which to unravel HSV-1 replication strategies important in its natural host. To date, there have been only a few studies on the mechanism of HSV-1 entry into human keratinocytes. While initial studies on other cell types indicated that HSV-1 enters cells by fusion at the plasma membrane following virion attachment to the cell surface receptor (1, 2), it has now become widely accepted that in keratinocytes as well as certain other cell types, the virus particle undergoes endocytosis before fusing with the endocytic membrane internally in the cytoplasm in a pH-dependent fashion (3–6). Crucially, an immunofluorescence study in the HaCaT keratinocyte cell line found that virions colocalized with a marker of fluid-phase endocytosis (3). Other studies, including the use of the inhibitor of endocytosis dynasore and overexpression of a dominant-negative dynamin, have shown that the virus may enter keratinocytes by both endocytosis and plasma membrane fusion (7), suggesting that, unusually, the virus may exploit alternative pathways in this single cell type. To add to the complexity of HSV-1 entry, it has been reported that despite being receptor dependent, HSV-1 endocytosis does not use either clathrin- or caveolin-mediated entry in a range of epithelial cells (8, 9) and can occur in a pH-independent fashion (4), features that may be more indicative of macropinocytosis entry rather than receptor-mediated endocytosis.
HSV-1 is believed to bind keratinocytes through the HveC gD receptor, more commonly known as the adhesion molecule nectin-1 (3, 10). Nectin-1 is expressed in both keratinocytes and neurons (10) and has previously been shown to be highly expressed in the HaCaT keratinocyte cell line (11). Moreover, knockout of nectin-1 from murine keratinocytes greatly reduced HSV-1 entry (12). A second gD receptor, HVEM (HveA), the first HSV-1 receptor identified in a seminal study by Montgomery and coworkers (13), is a member of the tumor necrosis factor receptor family (13), which despite being originally isolated from a HeLa cell cDNA library has been shown to be expressed predominantly on lymphoid cells (14). Furthermore, studies on cultured murine keratinocytes have shown that HVEM is absent from these cells (12). Like HVEM, another potential receptor for HSV-1, in the form of the gB receptor-paired immunoglobulin-like type 2 receptor PILRα (15), has also been shown to be expressed predominantly on cells of the immune system (15, 16).
In the study presented here, we have utilized the human nTERT keratinocyte cell line as a model system for studying HSV-1 entry. The nTERT line is derived from primary human keratinocytes which have been retrovirally induced to express hTERT, the catalytic subunit of the telomerase holoenzyme, and subsequently have spontaneously lost the function of p16INK4a, a tumor suppressor gene which usually inhibits the transition from G1 to S phase. This allows the maintenance of growth for much longer than normal for primary cell types (17). Crucially, nTERT cells are still dependent on epidermal growth factor, are able to differentiate, and remain noninvasive upon transplantation, indicating that they retain many characteristics of primary cells. Moreover, they have been used as a successful model system for other epitheliotropic viruses, such as varicella-zoster virus and human papillomavirus (18, 19), suggesting they are an appropriate system to study HSV-1 infection of relevant human cells.
In this report we show that HSV-1 enters nTERT cells through nectin-1 binding, despite high expression levels of the HVEM receptor. In contrast to other studies, our results show that HSV-1 entry into human keratinocytes occurs only via a direct plasma membrane fusion mechanism. Moreover, infection is initiated rapidly in keratinocytes, with maximal penetration occurring in just 5 min. Unexpectedly, virus penetration in keratinocyte cells was also shown to extend to temperatures as low as 7°C, an event confirmed at the ultrastructural level. Hence, we suggest that HSV-1 has a specific entry route into human keratinocytes that favors the potential environmental conditions that the virus encounters when infecting its human host.
MATERIALS AND METHODS
Cells and viruses.
Vero and HeLa cells were cultured in DMEM supplemented with 10% newborn calf serum (NCS). HaCaT and HFFF2 cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (vol/vol) fetal bovine serum (FBS). nTERT keratinocyte cells (17) were grown in cells maintained in 3:1 DMEM-Ham's F12 supplemented with 10% FBS, 10 ng/ml mouse epidermal growth factor (Serotec), 1 ng/ml cholera toxin (Sigma), 400 ng/ml hydrocortisone (Sigma), 5 mg/ml insulin (Sigma), 5 mg/ml transferrin (Sigma), 13 ng/ml liothyronine (Sigma), and 2 mM l-glutamine (Invitrogen). HSV-1 strain s17 was routinely propagated and titrated in Vero cells in DMEM supplemented with 2% serum. HSV-1 strain sc16, expressing β-galactosidase under the control of the ICP0 promoter inserted into the nonessential US5 locus (110lacZ), was kindly provided by Stacey Efstathiou, University of Cambridge (20). For transmission electron microscopy (TEM) studies, released HSV-1 virions were gradient purified as described previously (21).
Antibodies.
Monoclonal anti-ICP27 and anti-gD were kindly provided by Steve Rice (University of Minnesota) and Tony Minson (University of Cambridge), respectively. Polyclonal anti-VP22 has been described before (22), while monoclonal anti-ICP0 (11060) and anti-α-tubulin were purchased from Santa Cruz Biotechnology and Sigma, respectively.
Adsorption assay.
One-day-old confluent monolayers of cells in 6-well plates were prechilled on ice for 15 min. Cell culture medium was removed and replaced with 200 PFU of virus diluted in 500 μl of ice-cold infection medium. To allow virus to bind but not enter cells, the cells were incubated on ice for the times specified in the figure legends and then washed twice with ice-cold phosphate-buffered saline (PBS). Cells were then overlaid with fresh, warm medium containing 1% human serum and returned to 37°C for 2 days. Cells were fixed and stained with 0.1% crystal violet before plaques were counted.
Penetration assay.
One-day-old confluent monolayers of cells in 6-well plates were prechilled on ice for 15 min. Cell culture medium was removed and replaced with 200 PFU of virus diluted in 500 μl of ice-cold infection medium. Cells were incubated for 1 h on ice before washing with prewarmed/chilled medium (without virus) and incubating at the temperatures stated. After each indicated length of time, cells were placed on ice and treated with 500 μl of ice-cold citrate buffer, pH 3 (23), for 1 min to inactivate bound virus before being washed twice with ice-cold PBS. Cells were then overlaid with fresh, warm medium containing 1% human serum and returned to 37°C for 2 days. Cells were fixed and stained with 0.1% crystal violet before plaques were counted.
siRNAs and transfection.
Two Silencer select, nectin-1-specific short interfering RNAs (siRNAs) were purchased from Thermo Fisher (PVRL1; s11604 and s11605); pooled siRNAs to HVEM were purchased from Insight Biotechnology. siRNAs were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions for reverse transfection. The final concentration of siRNA was 30 nM per set. The Silencer select negative siRNA number 1 (Ambion) was used as a negative control.
qRT-PCR.
Total RNA was extracted from cells using a Qiagen RNeasy kit. Excess DNA was removed from RNA by diluting 1 μg of RNA in 8 μl nuclease-free water (Ambion) followed by the addition of 1 μl 10× DNase reaction buffer (Invitrogen) and 1 μl amplification-grade DNase I (1 U/μl; Invitrogen) and incubating for 15 min at room temperature. DNase was inactivated by a 10-min incubation at 65°C in 25 nM EDTA. Superscript III (Invitrogen) was used to make cDNA according to the manufacturer's instructions using random primers. All quantitative reverse transcription-PCR (qRT-PCR) assays were carried out in 96-well plates using MESA Blue qPCR MasterMix plus for SYBR assay (Eurogentec), using forward (F) and reverse (R) primers for the housekeeping gene RPLP0 (F, ACTCTGCATTCTCGCTTCCT; R, GGACTCGTTTGTACCCGTTG), nectin-1 (F, CTGCAAAGCTGATGCTAACC; R, GATGGGTCCCTTGAAGAAGA), and HVEM (Stratech Scientific Ltd.). Cycling was carried out on a QuantStudio 7 quantitative PCR (qPCR) machine (Applied Biosystems). Relative expression of each receptor was determined using the ΔΔCT method (24), using the housekeeping gene RPLP0 as the reference gene.
β-Gal quantification.
Confluent layers of cells, seeded in 96-well plates, were infected with sc16110lacZ virus at a multiplicity of infection of 2 for 1 h on ice. The inoculum was then removed and replaced with fresh, prewarmed medium, and the cells were incubated for 4 h at 37°C. The medium was removed by aspiration and the cells were washed twice with ice-cold PBS before being lysed and analyzed using the Invitrogen β-galactosidase (β-gal) assay kit. Standards were prepared with β-gal enzyme (Sigma), and absorbance at 420 nm was read on a FLUOstar Omega multimode microplate reader (BMG Labtech, Offenburg, Germany). β-Gal activity was calculated from the standard curve as milliunits of β-galactosidase activity per well.
Electron microscopy.
Cells for electron microscopy (EM) were fixed in 0.5% glutaraldehyde in 200 mM sodium cacodylate buffer for 30 min, washed in buffer, and secondarily fixed in reduced 1% osmium tetroxide, 1.5% potassium ferricyanide for 60 min. The samples were washed in distilled water and stained overnight at 4°C in 0.5% magnesium uranyl acetate, washed in distilled water, and dehydrated in graded ethanol. The samples were then embedded flat in the dish in Epon resin. Resin-filled stubs were placed on embedded cell monolayers and polymerized. Ultrathin sections (typically 50 to 70 nm) were cut parallel or perpendicular to the dish and examined in an FEI Tecnai electron microscope with charge-coupled-device (CCD) camera image acquisition.
SDS-PAGE and Western blotting.
Protein samples were analyzed on 10% polyacrylamide gels and subjected to electrophoresis in Tris-glycine buffer. Gels were transferred to nitrocellulose membrane for Western blot analysis. Western blots were developed using SuperSignal West Pico chemiluminescent substrate (Thermo Fisher Scientific).
Transferrin uptake assay and immunofluorescence.
One-day-old cells seeded on 13-mm coverslips were preincubated at various temperatures for 15 min before being washed once with DMEM without serum. Texas Red-conjugated transferrin (2.5 μg/ml; Invitrogen) in DMEM without serum was then added to cells and incubated at various temperatures for 30 min before washing and fixing with 4% paraformaldehyde in PBS for 20 min, followed by permeabilization with 0.5% Triton X-100 for 10 min. The coverslips were then washed extensively in PBS and mounted in Vectashield containing 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories). Images were acquired using a 63× objective on a Zeiss LSM510 Meta confocal microscope and processed using Adobe Photoshop software.
RESULTS
HSV-1 replicates with faster kinetics in nTERT keratinocytes than Vero cells.
The nTERT cell line has previously been used for studies on human papillomavirus and varicella-zoster virus (18, 19). However, at the beginning of this study the efficiency of HSV-1 infection and replication in these cells was unknown. To establish whether nTERT cells were permissive to HSV-1 infection, these cells were assessed for their ability to support virus replication in a one-step growth curve. Monolayers of nTERT cells were infected with HSV-1 (strain 17) at a multiplicity of 2, and extracellular and cell-associated virus was harvested at the indicated times (Fig. 1A). Progeny virus was first detected at 8 h and increased steadily until 16 h, when production plateaued (Fig. 1A). The level of extracellular virus indicated that virus was released efficiently, demonstrating that HSV-1 can enter and maintain a classical replication cycle in these cells. Interestingly, comparison of HSV-1 one-step growth kinetics in nTERT and Vero cells indicated that progeny virus is produced around 4 h earlier from the nTERT monolayer (Fig. 1B).
FIG 1.

nTERT keratinocyte cell line is permissive to HSV-1 infection. (A) Confluent nTERT cell monolayers were infected with HSV-1 (s17) at a multiplicity of infection of 2. Intracellular and extracellular virus was harvested at the times indicated and titrated onto Vero cells. A representative of three independent experiments is shown as the means ± standard errors of the data. (B and C) nTERT or Vero monolayers were infected with HSV-1 (s17) at a multiplicity of two. Total virus was harvested at the times indicated and either titrated onto Vero cells (B) or processed for Western blotting with antibodies against ICP0, ICP27, VP22, gD, and α-tubulin as a loading control. Lane M, mock infected. (D) Vero and nTERT cells grown on coverslips were infected with HSV-1 (s17) at a multiplicity of 2, fixed at the indicated times, and stained for immunofluorescence with an antibody to ICP0. Cells were imaged under a fluorescence microscope, and the number of ICP0-positive cells per 100 cells was scored. The means ± standard errors of the data from three independent experiments is shown, with data analyzed by the Student t test. ns, not significant. *, P < 0.05.
To determine if earlier production of progeny virus is related to earlier or greater virus protein synthesis, samples of infected Vero and nTERT cells were harvested at representative times up to 8 h after infection and analyzed by Western blotting for two immediate-early proteins (ICP0 and ICP27) and two late proteins (VP22 and gD) using α-tubulin as a loading control (Fig. 1C). All virus proteins tested were detected 2 h earlier in nTERT than Vero cells, with the immediate-early proteins readily detected at just 2 h after infection (Fig. 1C). Nonetheless, maximum levels of viral protein expression in nTERT cells were only slightly enhanced compared to Vero cells. This earlier expression of immediate-early proteins was confirmed by staining cells for ICP0. Counting of ICP0-positive cells in three independent experiments indicated that ICP0 was consistently detectable in over 50% of nTERT cells as early as 1 h after infection (Fig. 1D). In contrast, only around 15% of Vero cells were positive for ICP0 at this time, and it took another 4 h for the proportion of ICP0-positive Vero cells to catch up with nTERT cells. Moreover, titration of our stock virus on Vero and nTERT cells indicated that there was no significant difference in the plaquing efficiency of HSV-1 on these two cell lines that could explain the faster onset of virus infection in the keratinocyte cells (data not shown). Taken together, these results suggest that virus protein expression is induced more rapidly in nTERT cells than Vero cells after infection with the same multiplicity of virus.
HSV-1 enters nTERT keratinocyte cells via the nectin-1 receptor.
Because other studies have indicated nectin-1 is the major receptor used for HSV-1 entry into keratinocytes (3, 10) and nectin-1 has previously been shown to be highly expressed in the human HaCaT keratinocyte cell line (11), we next quantified the level of nectin-1 and HVEM mRNA in both nTERT cells and HeLa cells, the cell type from which both receptors had first been identified (10, 13, 25). As a further control for HVEM mRNA, we included the human THP-1 line where HVEM should be highly expressed (13). Using qRT-PCR, nectin-1 was found to be expressed close to 70-fold higher in nTERT cells than in HeLa cells and THP-1 monocytes (Fig. 2A). Surprisingly, despite HVEM having been originally identified from a HeLa cell cDNA library (13), HVEM mRNA was barely detectable in our HeLa cells by qRT-PCR using conditions where it was detectable in other cell types (Fig. 2B). Moreover, HVEM mRNA was unexpectedly detected in nTERTs at levels around 600-fold higher than in HeLa cells and 8-fold higher than in monocytes (Fig. 2B). This raised the possibility of either or both of these receptors being involved in entry into keratinocytes, so we next depleted each receptor from both HeLa and nTERT cells using siRNAs specific for each. Depletion was shown to be successful in both cell types by qRT-PCR of RNA harvested 48 h after transfection (Fig. 2C). Sixty hours after transfection, cells were infected with a virus expressing β-gal under the control of the IE110 promoter and harvested 5 h later for quantification of β-gal activity, which was used as an indirect measure of virus entry (Fig. 2D). The results show that depletion of nectin-1 from both HeLa and nTERT cells reduced β-gal activity to around 10% of that present in cells transfected with negative siRNA, correlating with the relative depletion of the mRNA for this receptor (Fig. 2D). In contrast, depletion of HVEM had no significant effect on entry into either nTERT cells or HeLa cells (Fig. 2D). Therefore, we conclude that HSV-1 enters both nTERT and HeLa cells primarily via the nectin-1 receptor. To our knowledge, this is the first direct demonstration of a requirement for nectin-1 in HSV-1 entry into HeLa cells.
FIG 2.

HSV-1 entry into nTERT cells requires the gD receptor nectin-1. (A and B) Nectin-1 (A) or HVEM (B) mRNA was quantitated in HeLa and nTERT cells by qRT-PCR using the ΔΔCT method. The housekeeping gene RPLP0 was used to normalize expression of both genes. (C and D) nTERT or HeLa cells were reverse transfected with 30 nM nectin-1, HVEM, or control siRNAs. Seventy-two hours later, cell RNA was extracted from half of the siRNA-transfected cells, and nectin-1 and HVEM mRNA levels were quantitated by qRT-PCR using the ΔΔCT method. (C) The housekeeping gene RPLP0 was used to normalize expression of both genes. The remaining cells were transferred in triplicate to 96-well plates and incubated for a further 16 h. These cells were infected on ice with 110lacZ virus at a multiplicity of infection of 2 for 1 h prior to replacing the inoculum with fresh media at 37°C and incubating for a further 4 h. Cells were lysed and relative β-galactosidase activity measured. (D) A representative experiment is shown as the means ± standard errors of the data relative to the negative siRNA control.
HSV-1 enters nTERT cells with unusual rapidity.
To test the efficiency of HSV-1 binding to nTERT cells in comparison to Vero cells, the rate of virus binding was measured using an adapted form of an adsorption assay (26). Monolayers of Vero or nTERT cells in 6-well plates were prechilled on ice before being infected with 200 PFU of HSV-1 per well, and the infections were maintained on ice to allow binding of virus particles without penetration. After each time point, the inoculum was removed and any unbound virus was washed away before fresh medium was added to the cells that were incubated for 3 days before fixing and staining. Resultant plaques were counted, and the overall rate of adsorption was calculated as the percentage of total adsorption at each time point, where the final plaque count at the 180-min time point on ice was considered 100% (Fig. 3A). In this assay each resultant plaque that was counted originated from a single virus particle that had bound to a cell while on ice. At 5 min after infection, both Vero and nTERT cells had bound approximately 25% of total virus, and the subsequent rate of binding over the next 180 min was almost indistinguishable between the two cell types (Fig. 3A). Hence, both cell types support virus binding with similar characteristics.
FIG 3.

HSV-1 enters nTERT cells more rapidly than Vero cells. (A) Confluent layers of nTERT or Vero cells were infected with 200 PFU of HSV-1 per well of a 6-well plate on ice. After the indicated time, the inoculum was removed and replaced with medium containing 1% human serum. The infection was allowed to develop for 2 days, when plaques were fixed, stained, and counted. Data are represented as percent adsorption, where the final plaque count (at 180 min) is taken as 100%. The means ± standard errors of the data are given from one representative experiment (n = 3). (B) Confluent monolayers of nTERT or Vero cells were infected with 200 PFU of HSV-1 per well of a 6-well plate on ice for 2 h. The inoculum was removed and replaced with medium at 37°C and incubated for the indicated times before treatment with low-pH buffer for 1 min. After treatment, cells were washed and overlaid with medium containing 1% human serum. The infection was allowed to continue for 2 days, when plaques were fixed, stained, and counted. The means ± standard errors of the data are given from one representative experiment (n > 3). Data are displayed as the percentage of virus protected at each time, where the final plaque count (at 60 min postwarming) is taken as 100%. (C) Confluent monolayers of nTERT, Vero, or HFFF2 cells were infected with 200 PFU of HSV-1 and treated as described for panel B.
We next measured the ability of HSV-1 to enter nTERT cells using an adapted penetration assay (23, 27). This assay uses a low-pH buffer to inactivate any bound or extracellular virus particles (28), allowing only virus that has penetrated the cell to be protected from the buffer treatment. To measure the relative efficiency of HSV-1 entry into nTERT cells, both nTERT and Vero cells were prechilled and infected on ice with 200 PFU of HSV-1 for 2 h to allow maximal virus binding without penetration. The cells were then washed and overlaid with fresh, prewarmed medium and warmed to 37°C for increasing times to allow virus penetration to begin. At the indicated times, the cells were treated with low-pH buffer for 1 min to inactivate any bound or extracellular virus particles, fresh medium was added, and cells were incubated at 37°C for plaques to form. Comparison of the percent protection at each time point, taking the final plaque count at 60 min as 100%, revealed a significant difference in the kinetics of HSV-1 penetration into nTERT cells compared to Vero cells (Fig. 3B). In Vero cells the percentage of penetrated virus increased steadily until it plateaued at 100%, 50 min after warming (Fig. 3B, Vero). In contrast, maximal penetration was reached in nTERT cells at just 5 min after warming, indicating that the kinetics of HSV-1 entry is much faster into nTERT cells than Vero cells (Fig. 3B, nTERT).
As the above-described comparison was between a human and a monkey cell line, we wanted to ensure that the dramatic difference in entry rate was cell type specific rather than species specific. To this end, the above-described penetration assay was repeated, this time including the human primary fibroblast cell line HFFF2 (Fig. 3C). In both Vero and nTERT cells, the low-pH protection rates were similar to that seen before (Fig. 3C). However, the percent virus protection rate in HFFF2 cells was comparable to that of Vero cells rather than that in nTERT cells, with the protection rate increasing steadily and plateauing at around 50 min (Fig. 3C). Hence, these data confirm that the rapid virus protection rate identified in nTERT cells is unusual for keratinocyte cells.
HSV-1 enters nTERT cells by rapid plasma membrane fusion.
The penetration assay relies on virus becoming protected from low-pH treatment by being internalized into the cell through penetration. However, given the unexpected results described above, we used EM to examine the cell surface of nTERT cells and determine if there could be a physical explanation for the rapid protection of all virus particles. Vertical sections taken through Vero or nTERT cells fixed in situ revealed that nTERT cells have a very different cell surface profile, with multiple projections on their apical surface compared to the smooth surface of Vero cells (Fig. 4A). This characteristic cell surface was particularly evident when thicker sections were examined (Fig. 4B). We hypothesized that these protrusions are in some way protecting nonpenetrated virions from low-pH disruption, thereby giving us a false high measurement of HSV-1 penetration. Hence, we examined HSV-1-infected nTERT cells by EM at 5 and 20 min after warming to determine the location of virions. Monolayers of nTERT cells were infected on ice with gradient-purified HSV-1 at a multiplicity of infection of 100 to allow maximal virus binding and to synchronize entry. Cells were then shifted to 37°C for 5 or 20 min before immediately fixing on ice and processing for analysis by EM, being prepared either in horizontal or vertical sections (Fig. 5). Examination of the sections revealed the presence of intact virus particles on the surface of the infected keratinocytes (Fig. 5A). Of note, these virions were frequently localized at the base of cell projections, suggesting a preference for binding and entry at these sites. Crucially, capsid profiles containing an electron-dense granular core (nucleic acid) were readily detectable within the cell cytoplasm (Fig. 5B, C, and D, arrows). These profiles were most often identified near the plasma membrane and were always free in the cytosol, suggestive of entry by plasma membrane fusion. In some profiles, dense material could be seen on the underside of the plasma membrane between the membrane and the capsid, which is likely to be tegument proteins deposited after plasma membrane fusion (Fig. 5C, thick arrow). Of 100 internal capsids counted in the sample fixed at 5 min, none were detected in vesicles in the cytosol. This provides evidence that the majority of virus capsids had penetrated keratinocyte cells within 5 min by a route that is likely to involve plasma membrane fusion.
FIG 4.

nTERT keratinocytes exhibit multiple projections on their apical surfaces. Vero and nTERT cells were grown on plastic dishes before fixing and processing for TEM. After embedding, the cell monolayer was sliced in the vertical axis to acquire slices from the top to bottom of cells. Slices of 70 nm (A) or 500 nm (B) were analyzed by TEM. Black arrows indicate where cells were attached to plastic dishes (basal side of cell).
FIG 5.

HSV-1 enters nTERT cells by rapid plasma membrane fusion. Monolayers of nTERT cells were prechilled on ice and infected with 100 PFU per cell of gradient-purified HSV-1 on ice for 1 h. Cells were then shifted to 37°C for 5 min or 20 min before fixing on ice for TEM processing. Representative micrographs show virions attached to the cell surface (A); capsids directly under plasma membrane (B); tegument-like material deposited beside capsids (C); and capsids deeper into the cytoplasm (D). Arrows indicate virions/capsids. Short arrows indicate possible tegument deposited under plasma membrane. Scale bar, 200 nm.
HSV-1 enters nTERT cells at low temperature.
Previous studies have shown that low temperatures inhibit HSV-1 entry into C10 murine melanoma cells (4), a result in agreement with the fact that endocytic uptake requires energy and hence is unable to occur at low temperatures (29). We wished to test the effect of low temperature on HSV-1 entry into nTERT keratinocytes, but to first identify temperatures where endocytosis was inhibited, we utilized Texas Red transferrin to monitor endocytic uptake of the transferrin receptor at different temperatures. Vero and nTERT cells were preincubated at 37°C, 21°C, 15°C, 7°C, or 0°C before being maintained at the same temperature for a further 30-min incubation period in the presence of medium containing Texas Red transferrin. Cells were then washed of surface-bound Texas Red transferrin and fixed for analysis by fluorescence microscopy. At 37°C, as expected, a robust Texas Red signal was detected as scattered punctate domains in the cytoplasm of both nTERT and Vero cells, reflecting transferrin uptake into recycling endosomes (Fig. 6, 37°C). At 21°C this signal was greatly reduced but still detectable in the cytoplasm of both cell types (Fig. 6, 21°C). However, at 15°C very few punctate domains could be identified, while the signal was absent from the 7°C and 0°C samples (Fig. 6). These results indicate that both Vero and nTERT cells lose the ability to endocytose Texas Red transferrin at low temperatures.
FIG 6.
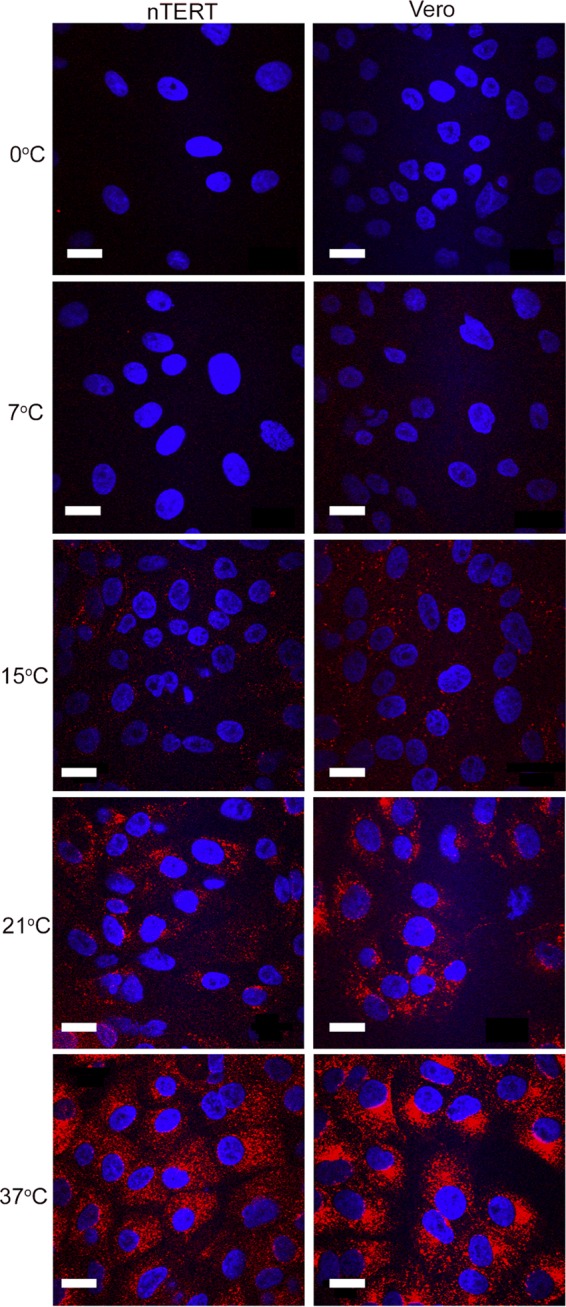
Receptor-mediated endocytosis of transferrin is inhibited at low temperatures. Monolayers of Vero and nTERT cells were incubated at the indicated temperatures for 10 min before the medium was replaced with medium containing Texas Red-conjugated transferrin (2.5 μγ/ml) at the same temperature for a further 30 min. Cells were fixed in 4% paraformaldehyde and nuclei stained with DAPI (blue). Images were acquired on a confocal Zeiss LSM510 META inverted laser-scanning microscope. Scale bar, 20 μm.
To determine if HSV-1 can enter either nTERT or Vero cells at the same temperatures tested for endocytosis, a modified version of the penetration assay was used where cells were infected on ice as before but then shifted to a specific temperature for 1 h before low-pH treatment, with the number of plaques counted at 37°C taken to be 100%. No virus protection from low-pH buffer treatment was seen after incubation for a further hour on ice in either Vero or nTERT cells, in line with the known inability of virus to enter cells at this extreme temperature (Fig. 7A). Lowering of the entry temperature reduced entry into Vero cells by 80% at 21°C, by 90% at 15°C, and by 100% at 7°C (Fig. 7A, Vero). In contrast, virus entry into nTERT cells was reduced by only 10% at 21°C and 25% at 15°C (75%), while at 7°C, over 30% of bound virus survived the low-pH treatment of the penetration assay (Fig. 7A, nTERT). Moreover, the same experiment carried out in the HaCaT keratinocyte cell line revealed that this ability to initiate infection at low temperatures was not specific to nTERT keratinocytes but seemed to be characteristic of human keratinocytes in general (Fig. 7A, HaCaT).
FIG 7.

HSV-1 enters keratinocyte cells at low temperatures. (A) Confluent monolayers of nTERT, HaCaT, or Vero cells were infected with 200 PFU of HSV-1 per well of a 6-well plate on ice for 1 h. The inoculum was removed and replaced with medium at the temperature indicated and incubated at that temperature for 1 h before treatment with low-pH buffer for 1 min. After treatment, cells were washed and overlaid with medium containing 1% human serum. The infection was allowed to continue for 2 days, when plaques were fixed, stained, and counted. The means ± standard errors of the data are given from one representative experiment (n > 3). Data are displayed as the percentage of virus protected (% protection) at each temperature, where the plaque count at 37°C was taken as 100%. (B to D) Monolayers of nTERT cells seeded in 60-mm dishes were prechilled on ice and infected with 100 PFU per cell of gradient-purified HSV-1 on ice for 1 h. Cells were then shifted to 7°C for 60 min before fixing on ice for TEM processing. Vertical sections were analyzed, and representative images are shown with arrows identifying intracellular capsids. Scale bar, 200 nm.
To confirm true virus entry into nTERT cells at 7°C, EM analysis was carried out, revealing that profiles of naked capsids containing nucleic acid were readily detectable within the cell cytoplasm of cells infected at 7°C (Fig. 7B to D, thin arrows). This confirms that HSV-1 is indeed able to penetrate keratinocytes at 7°C by plasma membrane fusion. Under these conditions, intracellular capsids were always observed at the cell periphery and often within cell surface protrusions, which were evident at this low temperature (Fig. 7B to D). These images confirm that virus particles are able to enter the cytosol of keratinocytes and are not merely protected on the extracellular surface after incubation at 7°C. We conclude from these studies that HSV-1 enters human keratinocytes by using the receptor nectin-1 to initiate a rapid plasma membrane fusion pathway that occurs at low temperature, potentially providing an accurate reflection of conditions of virus infection in the human host.
DISCUSSION
The first stage of any successful virus infection involves the entry of the virus into its target cell in the host. Viruses have coevolved with their hosts to exploit efficient routes of entry into cells and to target cell types that provide a favorable environment for their propagation and subsequent transmission. In the case of HSV-1, this route of entry involves the stratified epithelia that make up the epidermis of the skin or oral mucosa. These tissues are comprised mainly of keratinocytes and hence in its natural host, HSV-1 enters and replicates in the keratinocyte. Nonetheless, few studies have focused on how HSV-1 enters or replicates in this cell type, while studies on other human or even animal cells have left an unclear picture for the exact mode of HSV-1 entry into the cell.
In this study, we set about establishing how HSV-1 enters a highly relevant model keratinocyte system, the nTERT cell. This cell line represents a tractable model that is nonetheless representative of primary keratinocytes and has been used successfully for the study of other epitheliotropic viruses (18, 19). Importantly, these cells retain the ability to form stratified epithelia in the form of organotypic rafts in culture. Interestingly, we found that HSV-1 replicates faster and releases progeny earlier and in higher quantities in nTERT cells than in Vero cells, suggesting that the virus is fine-tuned to the biology of the keratinocyte. Our study has revealed three important and novel aspects of HSV-1 entry into keratinocytes that serve to emphasize the importance of studying viruses in their relevant host cell. First, we have found that HSV-1 enters with extreme rapidity, with over 90% of virus inside cells within 5 min. Second, and strikingly, we have shown that, unlike the case in other cells, HSV-1 can enter keratinocytes at temperatures as low as 7°C, with almost complete virus entry occurring at temperatures as low as 21°C. Third, and despite the fact that it is now generally accepted in the literature that HSV-1 enters keratinocytes and many other cell types by endocytosis (3–6), entry at both 37°C and 7°C occurred by direct plasma membrane fusion. Although this is counter to current thinking, it is notable that most studies on endocytosis of HSV-1 have not involved human keratinocytes. In our hands, we found no evidence for endocytosis under any of the conditions that we studied, a fact that was confirmed by showing virus entry into these cells at temperatures at which both receptor-mediated endocytosis and macropinocytosis are known to be inhibited (30, 31) and where Texas Red transferrin failed to be taken up into the cells. The only entry profiles observed under the conditions we examined were naked capsids located either directly beneath the plasma membrane or a short distance inside. It is important to note that in our ultrastructural analyses we have not scraped and pelleted cells prior to fixation for EM but have fixed, embedded, and sectioned cells in situ, so that cells could be precisely oriented in relation to the plastic substrate of the dish in which they have been grown and infected. We suggest that because of the finger-like profile of the normal keratinocyte plasma membrane, cross-sectioning in an unknown orientation could result in virions caught in the extracellular space between these projections potentially appearing similar to virions trapped inside endocytic vesicles. Indeed, our results would fit with a recent, extensive study that showed that HSV-1 entry into four different epithelial cell types did not require low-pH conditions, endosomal maturation, dynamins, or clathrin- or caveolin-dependent uptake (8). These authors found that a dynamic actin cytoskeleton was the only requirement for virus entry, a feature that could be explained by either plasma membrane fusion or macropinocytosis. In our own experiments, we found that at 7°C capsids were always found at the cell periphery, a result that likely reflects either an inability to progress through the cortical actin network or the presence of an unstable microtubule network under these conditions, both of which would block inward transport of capsids to the nucleus and cause accumulation in the cytosol beneath the plasma membrane.
Being an epitheliotropic virus that, during primary infection, infects its host at external sites of the body, such as skin or lips, the ability of HSV-1 to infect skin cells at temperatures below core body temperature has physiological relevance. Indeed, studies have shown that the temperature of the skin can vary depending on the environmental conditions (32, 33), with measurements of the skin varying from 14°C to 33°C depending on the external environment. HSV-1 is not alone in its ability to infect cells at temperatures below that of the human core; for example, the alphavirus Sindbis virus, which is transmitted by mosquitoes, has been shown to infect BHK cells at 15°C (34, 35) while rhinoviruses infect preferentially at 32°C, the temperature of the lining of the nose (36). As the lipid composition of membranes dictates their fluidity at low temperature, it may be the case that keratinocyte membranes have evolved to function optimally at lower temperatures in the extremities of the body, and hence the ability of membranes to fuse would be retained under these conditions. In such a scenario, it is also possible that receptor clustering occurs in the keratinocyte but not the Vero cell membrane at low temperature, thereby facilitating efficient binding and fusion under these conditions.
In addition to low-temperature entry, the extreme rapidity of virus entry that we measured in both nTERT and HaCaT human keratinocytes was an unexpected result that requires further investigation. An obvious explanation would be the presence of a high concentration of receptor at the cell surface, and by the use of qPCR we have shown that both nectin-1 and HVEM mRNAs are expressed at high levels in nTERT cells, with over 600-fold more HVEM mRNA in nTERT than HeLa cells. However, despite the high levels of HVEM, its depletion by siRNA had little effect on HSV-1 entry. In contrast, siRNA depletion indicated that nectin-1 is the major entry receptor for HSV-1 in nTERT cells. Although we cannot rule out a contribution of HVEM to HSV-1 entry, the fact that nectin-1 depletion reduced beta-galactosidase expression from an immediate-early promoter to less than 10% of control cells implies that nectin-1 is indeed the preferred receptor for HSV-1 in these cells. Moreover, this is also the scenario in HeLa cells where there is 60-fold less nectin-1 mRNA present, suggesting that even low levels of nectin-1 can support HSV-1 entry. Together with our results on HeLa and nTERT cells presented here, nectin-1 has now been shown to be the preferred entry receptor over HVEM in a range of different cell types (37–41). As such, it could be argued that the relatively high level of nectin-1 expression in nTERT cells explains the rapid entry phenotype. However, we have also measured the nectin-1 mRNA level in the HaCaT keratinocyte cell type shown here to support rapid HSV-1 entry in a similar fashion and have found that they express around 30-fold less nectin-1 than nTERT cells (data not shown). This suggests that there is more to the process of rapid HSV-1 entry than simply a higher concentration of the relevant gD receptor. For instance, nectin-1 might form local high concentrations in specific cell surface microdomains, as has been suggested in other studies (9). In relation to this, although nectin-1 has been reported to be absent from lipid rafts in other cell types (42), virus gene expression was delayed in keratinocytes depleted of cholesterol (7). As such, further studies such as cholesterol depletion with cyclodextrans would be required to determine if lipid rafts play a role in virus entry into nTERT cells.
Alternatively, despite not requiring HVEM for entry, the high concentration of HVEM in keratinocytes may facilitate rapid cell fusion by strengthening the virion interaction with the cell surface. Likewise, an additional coreceptor for one of the other fusion components may perform a similar function to increase the engagement of the fusion machinery with the plasma membrane. In light of this, we have demonstrated the presence of mRNA for the gB coreceptor pILRα in nTERT cells at levels only slightly lower than those in THP-1 monocytes (data not shown). Interestingly, another study has identified the innate immune scavenger receptor MARCO as a cellular factor that binds glycoprotein C and significantly enhances adsorption of HSV-1 to keratinocytes (43). Although an attractive candidate for a keratinocyte coreceptor for HSV-1 entry, our results here show that despite the vast difference in penetration rate between Vero and nTERT cells, there is no difference in the ability of HSV-1 to adsorb to either cell type, suggesting that MARCO enhancement is unlikely to explain the difference in speed of entry into nTERT cells.
Taken together, our findings provide new insights into the entry and replication of HSV-1 in its physiologically relevant target cell, the human keratinocyte. The use of a naturally infected host cell rather than commonly available laboratory cell types has revealed that HSV-1 uses an unusual plasma membrane fusion pathway that is likely to reflect the biology of the keratinocyte in its natural context, a pathway that now requires further investigation to understand its molecular features and identify new targets for antiviral intervention. Furthermore, as these cells retain the ability to form three-dimensional stratified epithelia representative of human skin, they provide us with a new and excellent model for HSV-1 infection at its site of entry into the host.
ACKNOWLEDGMENTS
We thank Michael Hollinshead, now at University of Cambridge, for assistance with electron microscopy. We also thank Judith Breuer, University College London, for nTERT and HaCaT cells and Stacey Efstathiou, University of Cambridge, for the β-galactosidase-encoding virus used in this study.
Funding Statement
This work was funded by a Wellcome Trust Ph.D. studentship to C.L.S.
REFERENCES
- 1.Wittels M, Spear PG. 1991. Penetration of cells by herpes simplex virus does not require a low pH-dependent endocytic pathway. Virus Res 18:271–290. doi: 10.1016/0168-1702(91)90024-P. [DOI] [PubMed] [Google Scholar]
- 2.Spear PG. 1993. Entry of alphaherpesviruses into cells. Semin Virol 4:167–180. doi: 10.1006/smvy.1993.1012. [DOI] [Google Scholar]
- 3.Nicola AV, Hou J, Major EO, Straus SE. 2005. Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J Virol 79:7609–7616. doi: 10.1128/JVI.79.12.7609-7616.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Milne RS, Nicola AV, Whitbeck JC, Eisenberg RJ, Cohen GH. 2005. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J Virol 79:6655–6663. doi: 10.1128/JVI.79.11.6655-6663.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nicola AV, McEvoy AM, Straus SE. 2003. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J Virol 77:5324–5332. doi: 10.1128/JVI.77.9.5324-5332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nicola AV, Straus SE. 2004. Cellular and viral requirements for rapid endocytic entry of herpes simplex virus. J Virol 78:7508–7517. doi: 10.1128/JVI.78.14.7508-7517.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rahn E, Petermann P, Hsu MJ, Rixon FJ, Knebel-Morsdorf D. 2011. Entry pathways of herpes simplex virus type 1 into human keratinocytes are dynamin- and cholesterol-dependent. PLoS One 6:e25464. doi: 10.1371/journal.pone.0025464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Devadas D, Koithan T, Diestel R, Prank U, Sodeik B, Dohner K. 2014. Herpes simplex virus internalization into epithelial cells requires Na+/H+ exchangers and p21-activated kinases but neither clathrin- nor caveolin-mediated endocytosis. J Virol 88:13378–13395. doi: 10.1128/JVI.03631-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clement C, Tiwari V, Scanlan PM, Valyi-Nagy T, Yue BY, Shukla D. 2006. A novel role for phagocytosis-like uptake in herpes simplex virus entry. J Cell Biol 174:1009–1021. doi: 10.1083/jcb.200509155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618–1620. doi: 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]
- 11.Huber MT, Wisner TW, Hegde NR, Goldsmith KA, Rauch DA, Roller RJ, Krummenacher C, Eisenberg RJ, Cohen GH, Johnson DC. 2001. Herpes simplex virus with highly reduced gD levels can efficiently enter and spread between human keratinocytes. J Virol 75:10309–10318. doi: 10.1128/JVI.75.21.10309-10318.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petermann P, Thier K, Rahn E, Rixon FJ, Bloch W, Ozcelik S, Krummenacher C, Barron MJ, Dixon MJ, Scheu S, Pfeffer K, Knebel-Morsdorf D. 2015. Entry mechanisms of herpes simplex virus 1 into murine epidermis: involvement of nectin-1 and herpesvirus entry mediator as cellular receptors. J Virol 89:262–274. doi: 10.1128/JVI.02917-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Montgomery RI, Warner MS, Lum BJ, Spear PG. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427–436. doi: 10.1016/S0092-8674(00)81363-X. [DOI] [PubMed] [Google Scholar]
- 14.Marsters SA, Ayres TM, Skubatch M, Gray CL, Rothe M, Ashkenazi A. 1997. Herpesvirus entry mediator, a member of the tumor necrosis factor receptor (TNFR) family, interacts with members of the TNFR-associated factor family and activates the transcription factors NF-kappaB and AP-1. J Biol Chem 272:14029–14032. doi: 10.1074/jbc.272.22.14029. [DOI] [PubMed] [Google Scholar]
- 15.Satoh T, Arii J, Suenaga T, Wang J, Kogure A, Uehori J, Arase N, Shiratori I, Tanaka S, Kawaguchi Y, Spear PG, Lanier LL, Arase H. 2008. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 132:935–944. doi: 10.1016/j.cell.2008.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fournier N, Chalus L, Durand I, Garcia E, Pin JJ, Churakova T, Patel S, Zlot C, Gorman D, Zurawski S, Abrams J, Bates EE, Garrone P. 2000. FDF03, a novel inhibitory receptor of the immunoglobulin superfamily, is expressed by human dendritic and myeloid cells. J Immunol 165:1197–1209. doi: 10.4049/jimmunol.165.3.1197. [DOI] [PubMed] [Google Scholar]
- 17.Dickson MA, Hahn WC, Ino Y, Ronfard V, Wu JY, Weinberg RA, Louis DN, Li FP, Rheinwald JG. 2000. Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol Cell Biol 20:1436–1447. doi: 10.1128/MCB.20.4.1436-1447.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones M, Dry IR, Frampton D, Singh M, Kanda RK, Yee MB, Kellam P, Hollinshead M, Kinchington PR, O'Toole EA, Breuer J. 2014. RNA-seq analysis of host and viral gene expression highlights interaction between varicella zoster virus and keratinocyte differentiation. PLoS Pathog 10:e1003896. doi: 10.1371/journal.ppat.1003896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patterson NA, Smith JL, Ozbun MA. 2005. Human papillomavirus type 31b infection of human keratinocytes does not require heparan sulfate. J Virol 79:6838–6847. doi: 10.1128/JVI.79.11.6838-6847.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lachmann RH, Sadarangani M, Atkinson HR, Efstathiou S. 1999. An analysis of herpes simplex virus gene expression during latency establishment and reactivation. J Gen Virol 80(Part 5):1271–1282. doi: 10.1099/0022-1317-80-5-1271. [DOI] [PubMed] [Google Scholar]
- 21.Elliott G, Hafezi W, Whiteley A, Bernard E. 2005. Deletion of the herpes simplex virus VP22-encoding gene (UL49) alters the expression, localization, and virion incorporation of ICP0. J Virol 79:9735–9745. doi: 10.1128/JVI.79.15.9735-9745.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elliott G, O'Hare P. 1997. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell 88:223–233. doi: 10.1016/S0092-8674(00)81843-7. [DOI] [PubMed] [Google Scholar]
- 23.Highlander SL, Sutherland SL, Gage PJ, Johnson DC, Levine M, Glorioso JC. 1987. Neutralizing monoclonal antibodies specific for herpes simplex virus glycoprotein D inhibit virus penetration. J Virol 61:3356–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 25.Warner MS, Geraghty RJ, Martinez WM, Montgomery RI, Whitbeck JC, Xu R, Eisenberg RJ, Cohen GH, Spear PG. 1998. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology 246:179–189. doi: 10.1006/viro.1998.9218. [DOI] [PubMed] [Google Scholar]
- 26.McClain DS, Fuller AO. 1994. Cell-specific kinetics and efficiency of herpes simplex virus type 1 entry are determined by two distinct phases of attachment. Virology 198:690–702. doi: 10.1006/viro.1994.1081. [DOI] [PubMed] [Google Scholar]
- 27.Huang AS, Wagner RR. 1964. Penetration of herpes simplex virus into human epidermoid cells. Proc Soc Exp Biol Med 116:863–869. doi: 10.3181/00379727-116-29392. [DOI] [PubMed] [Google Scholar]
- 28.Desai PJ, Schaffer PA, Minson AC. 1988. Excretion of non-infectious virus particles lacking glycoprotein H by a temperature-sensitive mutant of herpes simplex virus type 1: evidence that gH is essential for virion infectivity. J Gen Virol 69(Part 6):1147–1156. doi: 10.1099/0022-1317-69-6-1147. [DOI] [PubMed] [Google Scholar]
- 29.Larrick JW, Enns C, Raubitschek A, Weintraub H. 1985. Receptor-mediated endocytosis of human transferrin and its cell surface receptor. J Cell Physiol 124:283–287. doi: 10.1002/jcp.1041240217. [DOI] [PubMed] [Google Scholar]
- 30.Lippincott-Schwartz J, Roberts TH, Hirschberg K. 2000. Secretory protein trafficking and organelle dynamics in living cells. Annu Rev Cell Dev Biol 16:557–589. doi: 10.1146/annurev.cellbio.16.1.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pratten MK, Lloyd JB. 1979. Effects of temperature, metabolic inhibitors and some other factors on fluid-phase and adsorptive pinocytosis by rat peritoneal macrophages. Biochem J 180:567–571. doi: 10.1042/bj1800567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benedict FG. 1925. Skin Temperature and Heat Loss. Proc Natl Acad Sci U S A 11:549–552. doi: 10.1073/pnas.11.9.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benedict FG, Miles WR, Johnson A. 1919. The temperature of the human skin. Proc Natl Acad Sci U S A 5:218–222. doi: 10.1073/pnas.5.6.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang G, Hernandez R, Weninger K, Brown DT. 2007. Infection of cells by Sindbis virus at low temperature. Virology 362:461–467. doi: 10.1016/j.virol.2006.12.036. [DOI] [PubMed] [Google Scholar]
- 35.Vancini R, Wang G, Ferreira D, Hernandez R, Brown DT. 2013. Alphavirus genome delivery occurs directly at the plasma membrane in a time- and temperature-dependent process. J Virol 87:4352–4359. doi: 10.1128/JVI.03412-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Papadopoulos NG, Sanderson G, Hunter J, Johnston SL. 1999. Rhinoviruses replicate effectively at lower airway temperatures. J Med Virol 58:100–104. doi:. [DOI] [PubMed] [Google Scholar]
- 37.Shah A, Farooq AV, Tiwari V, Kim MJ, Shukla D. 2010. HSV-1 infection of human corneal epithelial cells: receptor-mediated entry and trends of re-infection. Mol Vision 16:2476–2486. [PMC free article] [PubMed] [Google Scholar]
- 38.Petermann P, Rahn E, Thier K, Hsu MJ, Rixon FJ, Kopp SJ, Knebel-Morsdorf D. 2015. Role of Nectin-1 and herpesvirus entry mediator as cellular receptors for herpes simplex virus 1 on primary murine dermal fibroblasts. J Virol 89:9407–9416. doi: 10.1128/JVI.01415-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kopp SJ, Banisadr G, Glajch K, Maurer UE, Grunewald K, Miller RJ, Osten P, Spear PG. 2009. Infection of neurons and encephalitis after intracranial inoculation of herpes simplex virus requires the entry receptor nectin-1. Proc Natl Acad Sci U S A 106:17916–17920. doi: 10.1073/pnas.0908892106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tiwari V, Oh MJ, Kovacs M, Shukla SY, Valyi-Nagy T, Shukla D. 2008. Role for nectin-1 in herpes simplex virus 1 entry and spread in human retinal pigment epithelial cells. FEBS J 275:5272–5285. doi: 10.1111/j.1742-4658.2008.06655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Galen B, Cheshenko N, Tuyama A, Ramratnam B, Herold BC. 2006. Access to nectin favors herpes simplex virus infection at the apical surface of polarized human epithelial cells. J Virol 80:12209–12218. doi: 10.1128/JVI.01503-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bender FC, Whitbeck JC, Ponce de Leon M, Lou H, Eisenberg RJ, Cohen GH. 2003. Specific association of glycoprotein B with lipid rafts during herpes simplex virus entry. J Virol 77:9542–9552. doi: 10.1128/JVI.77.17.9542-9552.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.MacLeod DT, Nakatsuji T, Yamasaki K, Kobzik L, Gallo RL. 2013. HSV-1 exploits the innate immune scavenger receptor MARCO to enhance epithelial adsorption and infection. Nat Commun 4:1963. [DOI] [PMC free article] [PubMed] [Google Scholar]