
FIGURE 1.
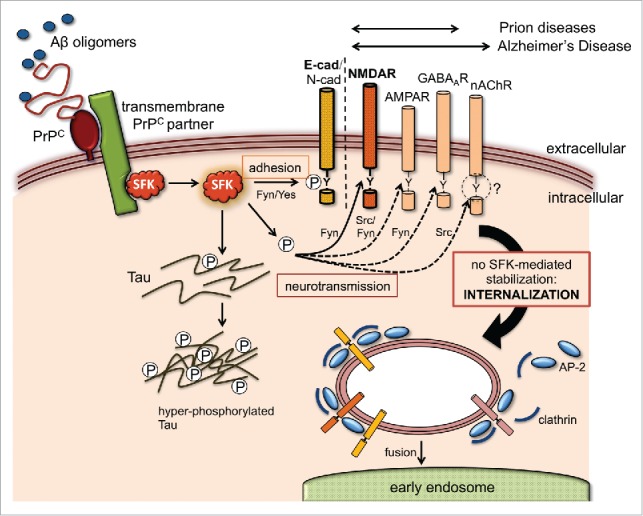
Potential effect of Aβ and PrPC on SFK-regulated trafficking of neuroreceptor and adhesion complexes. Aβ oligomers bind PrPC on neuronal cell surfaces with high affinity, triggering the activation of SFKs, often through a transmembrane PrPC partner (e.g. caveolin, mGluR5, NCAM, EGFR9). Apart from phosphorylating the disease-associated Tau protein and other cytosolic targets, SFKs can modulate the cell surface stability of numerous transmembrane proteins via endocytic tyrosine motifs at their cytoplasmic tails. Some of these, such as neuroreceptor subunits (NMDAR, AMPAR, GABAAR and nAChR) and adhesion proteins (E- and N-cadherin), are directly involved in synaptic function and can potentially explain key pathological features of prion and Alzheimer's disease. SFKs control the trafficking of these transmembrane proteins by directly phosphorylating their endocytic motifs or by targeting components of their tyrosine endocytic machinery like AP-2 and clathrin. These modifications can result in either increased stability at the plasma membrane (gain of function) or protein internalization and degradation (loss of function). To date, experimental evidence for an influence of Aβ and PrP on this mechanism is only available for NMDAR6 and AJ complexes.10 In the case of the nAChR, Src kinase activity controls its trafficking but tyrosine endocytic motifs appear dispensable (as suggested by the question mark). Double arrows above the transmembrane target proteins indicate their relevance to prion or Alzheimer's disease.